
Abstract
Understanding the dynamics of wolf-dog hybridization and delineating evidence-based conservation strategies requires information on the spatial extent of wolf-dog hybridization in real-time, which remains largely unknown. We collected 332 wolf-like scats over ca. 5,000km2 in the NW Iberian Peninsula to evaluate wolf-dog hybridization at population level in a single breeding/pup-rearing season. Mitochondrial DNA (MtDNA) and 18 ancestry informative markers were used for species and individual identification, and to detect wolf-dog hybrids. Genetic relatedness was assessed between hybrids and wolves. We identified 130 genotypes, including 67 wolves and 7 hybrids. Three of the hybrids were backcrosses to dog whereas the others were backcrosses to wolf, the latter accounting for a 5.6% rate of introgression into the wolf population. Our results show a previously undocumented scenario of multiple and widespread wolf-dog hybridization events at the population level. However, there is a clear maintenance of wolf genetic identity, as evidenced by the sharp genetic identification of pure individuals, suggesting the resilience of wolf populations to a small amount of hybridization. We consider that real-time population level assessments of hybridization provide a new perspective into the debate on wolf conservation, with particular focus on current management guidelines applied in wolf-dog hybridization events.
Similar content being viewed by others

Introduction
Hybridization between wild species and their domestic forms is widely perceived as a biodiversity threat. The outcome of this crossbreeding may lead to the introgression of domestic alleles, shaped by artificial selection, into wild populations, with potential negative conservation consequences, such as genetic homogenization, disruption of local adaptation or, ultimately, extinction1,2,3. However, positive effects of such introgression events have also been recently suggested. For example, the black coat colour present in North American wolves (Canis lupus) is the result of a single mutation4 that emerged through past interbreeding with dogs, and exhibits a molecular signature of positive selection, apparently confering advantage in forested habitats5,6.
Hybridization between wolves and dogs is paradigmatic among wild-domestic pairs, attracting a growing attention among managers, researchers and conservationists3,7,8,9. Such attention is justified in the current context of wolf population recovery in human-dominated landscapes10, where encounters between wolves and either free-ranging, feral or pet dogs are expected to be frequent11,12,13. However, wolf-dog hybrids are not confidently identifiable by morphological features alone. Many countries have thus used molecular tools to study and report wolf-dog hybridization events14,15,16,17,18. Most of these studies, however, are based on opportunistic invasive sampling that limits the understanding of the spatial and temporal patterns of hybridization at the population level. Opportunistic sampling requires a wide temporal window to achieve a representative sample size, generally encompassing several generations, thereby increasing the time-lag between population survey, interpretation of the dynamics of hybridization and the implementation of interventions.
Non-invasive samples (NIS) overcome some constraints related to opportunistic sampling, but their effective use in wolf-dog hybridization studies has been so far limited by the high genetic similarity between these two entities. In such cases, a large number of molecular markers is needed to achieve accurate results19,20, but DNA quality and quantity derived from NIS limits the number of markers that can be used21. However, using NIS to detect hybridization is now more appealing since recent studies have shown that a selection of a small number of ancestry informative markers (AIMs) is as efficient as a larger dataset of markers not selected based on their discriminatory power22,23.
The management of wolf-dog hybridization is a controversial issue. First, there is no clear genetic threshold for individual assignment to hybrid classes, which may limit interpretation of what is, in fact, a hybrid. Second, the value and role of contemporary wolf-dog hybrids in ecosystems have not been investigated. Third, in most cases, wild-domestic hybrids are not explicitly addressed in the principal international legal instruments on nature conservation, blurring the boundary for management decisions7,11,24. On top of this, assessments to detect wolf-dog hybridization events at large scales in very short time periods are not available for any wolf population (but see Caniglia et al.25 for an amalgamation of 9 year surveys), which have so far hampered a real-time estimation of hybridization events at the population level and of hybrid density per wolf generation. Monitoring these parameters properly is not only a way to better understand the dynamics of wolf-dog hybridization or to detect alarming situations, but could also help to define possible thresholds for interventions, as well as to evaluate their effectiveness23.
A recent study in NW Galicia, Spain, revealed a dynamic crossbreeding system between Iberian wolves and dogs in a single pack (Barbanza pack23, see Fig. 1). Knowing that packs are commonly formed by related individuals26, and given the dispersal abilities of wolves, individuals from a hybrid pack could move into adjacent territories possibly mating with other wolves and further introgressing dog genes into the wolf population. In this study, we explore this hypothesis presenting the first real-time spatial assessment of wolf-dog hybridization at a population level. To do so, we rely on a systematic sampling survey for the whole wolf distribution in Costa da Morte (NW Galicia) and on a set of 18 AIMs. Our main goals were to i) determine the spatial pattern of hybridization events in a single year at a population level; ii) identify the genetic composition of admixed individuals, inferring their hybrid class; and iii) assess the relatedness among putative hybrids, and between those and the wolves in the area, eventually identifying the pack of origin for the putative hybrids.

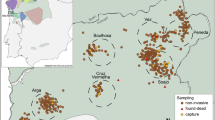
Estimated wolf pack territories (100 km2; see text for details) are denoted by open circles. Each number represents a pack as follows: 1- Muxia; 2- Vimianzo; 3 - Pasarela; 4 - Buxantes; 5- Baiñas; 6 – Ruña; 7 – Carnota; 8 – Negreira; 9 – Cerceda; 10 – Barbanza; 11 – Lousame; 12 – Xesteiras; 13 – Piorneiras. Inset: Location of the study area within the Iberian Peninsula. Arrows indicate the location of hybrids with qi values shifted towards the wolf cluster (LCM045, LCM047, LCM79, LCM122). This figure was produced using ArcGIS (version 10.1 [www.esri.com/software/Arcgis]).
Results
Genotyping and individual identification
From the 332 analyzed samples, MtDNA identification was achieved for 301 samples (91%), comprising 172 samples (57%) with the Iberian wolf haplotype W727, 100 samples (33%) matching to 13 different dog haplotypes (see Supplementary Table S3, for more details on the haplotypes), and 29 samples (10%) corresponding to red fox. A total of 272 samples carrying wolf or dog mtDNA were tested for DNA quality from which 91 were excluded. Four replicates of the remaining 181 samples were genotyped for the 18 AIMs. Finally, 167 samples achieved a consensus genotype with <20% missing data (61% of the 272 initial samples), and were considered for further analysis. Estimated allelic dropout across loci was ADO = 0.033 (0 < ADO < 0.103), and false alleles were FA = 0.005 (0 < FA < 0.016; see Supplementary Table S4 for further information). These values are below the average for similar recent non-invasive genetic studies23,25,28,29, thereby validating the pre-amplification strategy followed in our work.
We identified 130 individual genotypes, with an average of 4% missing data across loci. This dataset had a probability of identity (PID) = 2.58 × 10–15 and a PIDsibs = 2.74 × 10−6, with the expected number of sibling individuals sharing an identical genotype by chance of PIDsibs × sample size = 0.000352, meaning that it is very unlikely that identical genotypes do not identify the same individual. Of the 130 individuals, 109 (84%) were only detected once, and the maximum number of recaptures was five for wolves and two for dogs.
Diversity and differentiation
All loci were polymorphic, showing 2 to 13 alleles per locus for samples from Costa da Morte (CM). Four pairwise combinations of loci showed significant LD in dogs (p < 0.001, corrected for 306 comparisons), whereas none were identified in wolves. Deviations from Hardy-Weinberg expectations were observed in one and four loci in wolves and dogs, respectively (Supplementary Table S5). Iberian wolves exhibited lower genetic diversity than dogs. The mean number of alleles per locus was ANwolf = 3.6 ± 1.8 and ANdog = 6.9 ± 2.8, whereas the mean expected heterozygosity was Hewolf = 0.44 ± 0.24 and Hedog = 0.69 ± 0.14 (Supplementary Table S5). As expected, the use of AIMs allowed a remarkable genetic differentiation between wolves and dogs (FST = 0.335; p < 0.001). Loci that contributed more to this observation were DBar1, Cfx30371 and AHT103, which exhibited FST values > 0.5 (see Supplementary Table S5 for FST values per locus).
Hybridization dynamics in Costa da Morte
Bayesian clustering analysis of simulated genotypes revealed high average assignments to the correct parental class, Qiwolf = 0.998 (minimum qiwolf = 0.964) and Qidog = 0.995 (minimum qidog = 0.894), which allowed us to establish a conservative threshold of qi = 0.910 for the assignment to the wolf population and qi = 0.850 to the dog population. Based on this threshold, we identified as hybrids 100% of the simulated genotypes for generations F1 (qiF1 ≤ 0.709) and F2 (qiF2 ≤ 0.817), 98% and 76% of first generation backcrosses (average qiBxwolf = 0.737, qiBxDog = 0.725) and 64% and 31% of second generation backcrosses (average qi2BxWolf = 0.866; qi2BxDog = 0.841; see Supplementary Fig. S1 for the distribution of q-values for the simulated classes).
Bayesian clustering analysis performed with CM genotypes showed high posterior probabilities to assign reference wolves and dogs (QiWolf = 0.996 and QiDog = 0.995, Table 1). Out of the 130 genotypes considered, we identified 67 wolves (average qi = 0.986), and 56 dogs (average qi = 0.972) (Table 1, Fig. 1). Seven genotypes were partially assigned to both clusters, with qi values shifted towards the wolf cluster for LCM045, LCM047, LCM079, LCM122, and towards the dog cluster for LCM006, LCM068, LCM091 (Table 1). Bayesian credible intervals (BCI, 90%) for these seven hybrids were wide (average range = 0.318) and overlapped with the established thresholds. This is in sharp contrast with non-admixed genotypes, which consistently exhibited narrow BCI values (average range for wolf = 0.044 and dog = 0.106). Considering the percentage of admixed individuals with assignment towards the wolf cluster as a proxy for the rate of dog genome present in the wolf gene pool, we estimate a 5.6% rate of introgression in the sampled wolf population. Hybrid individuals were found in different areas (Fig. 1), occurring within the estimated territory of four different packs (30% of the total number of estimated packs in the study area30). However, five of the seven hybrids appeared within contiguous pack territories.
Considering Bayesian ancestry analysis, only hybrid LCM047, with a genomic content shifted towards the wolf cluster, exhibited a high posterior probability of having a dog ancestry going two generations back (Table 2), likely being a first generation backcross to a wolf. The remaining hybrids showed ambiguous posterior probabilities (LCM122, LCM006) or no evidence of having a pure ancestor in the second population (Table 2). Since this model requires powerful data to overcome the prior population assignment and our dataset has a low power to identify hybrids beyond a first generation backcross, the observed posterior probabilities for these individuals might be evidence for multi-generational backcrosses. Interestingly, dog mtDNA haplotypes were observed in hybrids with genomic content shifted towards dog, while hybrids with genomic content shifted towards wolf showed an Iberian wolf mtDNA haplotype (Table 1).
Relatedness between hybrids and wolves
Pairwise relatedness values between wolves and hybrids with a genomic content shifted towards wolf were higher in pairs sampled in the same area (Fig. 2, Table 3). The mean relatedness of hybrids LCM047, LCM079 and LCM122 to members belonging to the Xesteiras, Muxia and Pasarela packs, respectively, was significantly higher than to the remaining wolf population (Fig. 2, Table 3). Hybrid LCM045 was related to two different packs, Muxia and Barbanza. Interestingly, hybrids LCM045 and LCM079 were the only ones sampled in the same pack territory and presented a pairwise relatedness estimate of r = 0.50 (95%CI: 0.1–0.53), consistent with a parent-offspring or full siblings relationship.

Estimated wolf pack territories (100 km2) are denoted by open circles (see Fig. 1 for pack names). This figure was produced using ArcGIS (version 10.1 [www.esri.com/software/Arcgis]).
Discussion
In this study, we carried out for the first time a real-time assessment of wolf-dog hybridization at a population scale (covering ca. 5,000 km2). We achieved a good compromise among sampling type, relying on NIS, a sampling time restricted to one breeding/pup-rearing period (June-October 2013), and the molecular approach used, which was based on selected AIMs23. We overcame the typical constraint associated with the number of markers that can be genotyped when using NIS by selecting the 18 most informative markers differentiating wolves and dogs in NW Galicia from the set of 52 markers available in our laboratory16,23. We were therefore able to achieve a very high genetic differentiation between species (FST = 0.335), and a reasonable success rate in identifying hybrid classes F1, F2 and first generation backcrosses. Similar results were obtained previously using a smaller panel of 13 AIMs23. However, increasing the number of loci owing to the implemented two-step PCR approach, allowed us to significantly increase the power of individual differentiation (PID and PIDsibs), which is highly valuable for low diversity populations, while decreasing the genotyping errors characteristic of NIS. In fact, the allelic dropout and false allele rates observed in our work were only comparable with studies using invasive sampling (e.g. tissue or blood17,31) or to a recent study using non-invasively collected saliva32. A higher number of loci with lower error rates increases the accuracy of results, and thereby helps to overcome the main limitation associated with using low quality DNA to simultaneously detect both parental species and hybrids.
Bayesian analysis of simulated genotypes indicates that wolves and dogs are assigned with posterior probabilities of qi > 0.96 and qi > 0.90, respectively, despite being generated from reference samples with strict assignments to wolf and dog (qi > 0.98). Furthermore, simulated genotypes do not consider additional standing genetic variation to that present in the reference samples. To account for this bias, we established a conservative threshold of qiwolf > 0.91 and qidog > 0.85, arbitrarily decreasing the threshold values of simulated genotypes by 0.05. This conservative approach ensured detection of pure individuals (wolf or dog), although it could result in underestimating the number of hybrids. Nevertheless, our analysis shows that all F1, F2 and most first generation backcrosses are always correctly assigned as hybrids using our conservative thresholds. Additionally, we observed that the 90% BCI values for hybrids were wider than those found for wolves or dogs. We therefore suggest that the width of the 90% BCIs may be a good proxy for the accurate identification of admixed individuals, as was also suggested by Randi3.
We identified a total of 67 wolves and 56 dogs in a survey area covering 5,000 km2. This accounts for a minimum number of wolves and dogs of 1.34 and 1.12 individuals/100 km2, respectively. However, we believe that the number of dogs might be underestimated because rangers were instructed to avoid dog scats with dog food content while collecting samples. Additionally, dogs were detected in our work throughout the study area in sympatry with wolves (Fig. 1). In fact, free-ranging dogs were often observed during the entire sampling period (authors’ personal observation). This might be interpreted as a measure of human-dominance in the landscape of NW Iberia, and exacerbate the probability of encounters between dogs and wolves, increasing the opportunities for hybridization3,11,16,33,34.
Bayesian clustering analysis revealed a 5.6% rate of introgression in the wolf population of Costa da Morte. Similar rates of introgression have been previously found in other wolf-dog hybridization studies16,17,25,35, although based on invasive sampling and larger temporal scales (but see Caniglia et al.25). Overall, these results suggest that a low prevalence of hybridization might be a general pattern across wolf populations in the human-dominated landscapes of Europe, regardless of the spatial and temporal scales considered. Despite the occurrence of hybridization, a remarkable proportion of the total genetic variation in CM was partitioned between wolves and dogs (FST = 0.335, p < 0.001). Wolves from CM remained genetically distinct from dogs, a general pattern found in European wolf populations16,17,35, suggesting that effective introgression may be lower than that suggested by the percentage of hybridization cases reported. Accordingly, these observations might be indicative of a remarkable resilience by wolf populations to a small amount of hybridization. If true, hybridization might be buffered by the physiological and behavioral differences between wolves and dogs3,36,37, or more likely by the overall cohesion of wolf social structure. In this case, the inferred hybridization events detected in our study could be a consequence of occasional disruptions of wolf social groups26,38,39,40.
Gene flow between wolves and dogs is commonly described as spatially restricted, confined to peripheral wolf ranges or occurring in recently colonized areas14,36,41. However, the NW Iberian Peninsula does not conform with this description42. Additionally, admixed individuals were found scattered in the study area and were potentially associated with at least four different packs (Table 3). Furthermore, the lack of relatedness among hybrids, excepting for the two individuals found within the same pack territory, supports more than a single interbreeding cross in CM. Thus, our observation of hybrids does not conform to offspring dispersion after a single hybridization event. Instead, the most plausible scenario is the occurrence of multiple wolf-dog hybridization events over a large spatial area. Our results show, therefore, a new perspective of hybridization dynamics in European wolf populations.
Wolf-dog hybridization is considered as a matter of conservation concern facing fragmented and isolated wolf populations in Europe9,43,44. However, the legal status of wolf-dog hybrids, and the effectiveness of interventions, are still controversial and debated worldwide7,23,45. In addition, the lack of multidisciplinary information on the behavior and role that hybrids play in nature prevents a proper assessment of the risk of hybridization to species conservation46. The effective identification of hybrids in the field in close-to real-time is of paramount importance for providing base knowledge on the spatial dimension of hybridization, which is essential for subsequent risk assessments. Our study confirms previous studies about the utility of a reliable, accurate and efficient tool to evaluate wolf-dog hybridization23. Current management guidelines state that everything practically possible should be done to remove hybrids from the wild once they have been detected8,43. However, the effectiveness of removing hybrids remains an issue. For example, Godinho et al.23 showed that only 44% of the wolf-dog hybrids detected in a hybrid pack were removed after intervention, and in agreement, the present study has confirmed that hybridization was still present in the same area two years later (Barbanza pack, Fig. 1). It could be argued that more efforts in intervention enforcement could have removed all hybrids from this area. However, the spatial extent of the hybridization events reported here, showing multiple hybridization cases occurring over an area of 5000 km2, illustrates the limitations in the interventions suggested to manage wolf-dog hybridization.
Additional effort is required to understand other aspects of hybridization beyond genetics, such as behavioral or ecological factors, to gain new insights into the mechanisms buffering the impact of hybridization events or the role of hybrids in nature, both of which carry substantial conservation and management implications. Compiling evidence on the effectiveness of interventions, the evolution of hybridization over time and space, or the ecological role of hybrids, is necessary to promote a new debate about the current management guidelines and practices around wolf-dog hybridization events, including the legal basis for wild hybrids.
Methods
Study area and sample collection
Our study area comprises the wolf population of Costa da Morte and surroundings (Galicia, NW Spain), hereafter referred as CM, covering ca. 5,000 km2 where 13 packs have been estimated to occur in recent times30 (Fig. 1). This is a human-dominated landscape mostly characterized by a patchy and heterogeneous landscape, with sparse human settlements (148 people km−2), and wolves feeding frequently on livestock12,42,47. During the 2013 breeding and pup-rearing period48 (April-October), 332 wolf-like scats were collected in CM. All scats compatible with wolf (based on shape, size, content, smell)28 or dog, excluding scats with dog food, were considered as wolf-like scats. In collaboration with rangers from the Regional Government of Galicia, scats were searched for along transects comprising existing paths, forest trails and firebreaks, with sampling focused on areas distant from human settlements30. We sampled a minimum of 10 km per each 10 × 10 km UTM cell, which resulted in a wolf detection probability >0.6 in the study area (based on Bayesian hierarchical-site-occupancy models)49, surveying a total of ca. 750 km. Because we sought to detect hybridization events, we relaxed the criteria used previously to identify a wolf-like scats42, which increased the number of dog and red fox (Vulpes vulpes) scats collected. Scats were georeferenced using a GPS (Fig. 1), and preserved in ethanol 96% at room temperature (20 °C to 25 °C).
DNA extraction, markers and genotyping
Extraction and manipulation of DNA from fecal samples was confined to dedicated rooms with sterile conditions and positive air pressure. DNA extraction from the 332 scats followed Frantz et al.50 using the GuSCN/silica procedure51. Potential PCR inhibitors were further removed using pre-rinsed Microcon® YM-30 centrifugal Filter Units (Millipore, Billerica, MA). Negative controls were included throughout the process to monitor for potential DNA contamination. Species identification was assessed through mitochondrial DNA control region sequences following Vilà et al.52. Successful amplifications were sequenced following the BigDye chemistry (Applied Biosystems). Sequencing products were analyzed on an ABI3130xl genetic analyzer (Applied Biosystems) and aligned using SEQSCAPE 2.5 (Applied Biosystems). Sequences from other species other than wolf or dog were excluded from the study.
Individual identification was based on 18 AIMs. These markers were selected from a panel of 52 markers routinely used in our laboratory16,23 based on FST values between wolves and dogs (15 markers) and probability of identity (3 markers). The set of markers included 17 microsatellites and a 5 bp deletion at intron 3 of the KIT-ligand gene (KITLG) (Supplementary Table S1). All markers were amplified in two-steps using a pre-amplification protocol53. In both steps, AIMs were amplified in four multiplex reactions using the Multiplex PCR Kit (QIAGEN) in a 10 μL final volume. Four replicas of each PCR step were performed. For the first PCR we used approximately 5 ng of DNA and a concentration of 0.2 μM of unlabeled primers. In the second PCR, forward primers were M13-tailed to follow a fluorescent labelling protocol54. Thermocycling conditions are given in Supplementary Table S2. DNA quality was assessed by screening four microsatellites (multiplex MP2, Supplementary Table S1) before proceeding to full genotyping. Samples scored with <40% missing data were selected for further amplifications with the remaining loci. PCR products were separated by size on an ABI3130xl genetic analyzer. Alleles were scored against the GeneScan500 LIZ size standard, using genemapper 4.0 (Applied Biosystems) and checked manually twice.
Data analysis
Two initial replicas of each genotype were used to perform a maximum likelihood estimate of genotyping errors (allelic dropout and false alleles) using pedant 1.055. These estimates were then used to determine the minimum number of replicas needed to minimize genotyping errors using gemini 1.3.056. This number was set to four. Consensus genotypes over four replicas were assembled following Godinho et al.23: (i) heterozygous genotypes were accepted if the same genotype was observed in two independent PCRs; and (ii) homozygous genotypes were accepted if the genotype was observed in three independent PCRs. Samples with more than 20% missing data were removed from the analysis. Identical genotypes were filtered using gimlet 1.3.357 and discarded. The same software was used to evaluate mean allelic dropout and false allele rates across the 18 loci for the whole dataset, and to estimate the probability of identical genotypes being shared by chance (probability of identity, PID and PIDsibs).
Multilocus genotypes were used to estimate nuclear diversity for wolves and dogs (hybrids were excluded, see below for further details on hybrids identification) based on the number of alleles per locus (AN), and the observed (Ho) and expected (He) heterozygosity for each locus using arlequin 3.558. The same software was used to compute the fixation index FIS, estimate departures from Hardy–Weinberg equilibrium (HWE) following Guo and Thompson (1992)59 using a test analogous to Fisher’s exact test58, and to evaluate significance of association between genotypes at pairs of loci in each population (linkage disequilibrium, LD). Statistical significance was adjusted using sequential Bonferroni corrections. Population differentiation was assessed by Fisher’s exact test, analogues of pairwise mean FST60, and an analysis of molecular variance (amova61) using arlequin 3.5.
Bayesian clustering analysis implemented in structure 2.3.462,63 was used to identify wolves, dogs and possible hybrids by assessing individual membership proportions (qi) and their 90% Bayesian Credible Intervals (BCI) to two inferred clusters (K = 2). A panel of 250 Iberian wolves and 230 dogs was used as reference samples to aid the estimation of allelic frequencies. These samples were previously validated using a set of 53 nuclear markers, exhibiting an individual assignment to their putative cluster >98%23. The dog reference panel was increased with an additional set of 17 Can de Palleiro breed samples, a autochthonous Galician breed used also as herding dogs. Bayesian clustering analysis was first implemented using the admixture model with correlated allele frequencies (Usepopinfo activated for reference samples) in 10 independent runs each with 106 MCMC iterations following a burn-in period of 105 iterations, to guarantee similar posterior probabilities of the data in each run. Assumptions about hybrid ancestry were inferred after the use of the model “Use population information to test for migrants”. This model requires prior information on the origin of individuals and assesses the posterior probability of an individual being from i) the a priori assigned population, ii) one of the other populations, or iii) having a recent ancestor (parent, grandparent, great-grandparent) from one of the other populations62. For instance, a high posterior probability value for having a grandparent in the dog population means that the individual is likely a first generation backcross to wolf. Individuals were assigned a priori to the wolf or dog cluster according to qi values determined in the admixture model analysis. Admixed individuals were considered to belong to the population with the higher qi value. The model was run with MIGRPRIOR = 0.1 and GENBACK = 2. This model is not used to distinguish parental from admixed individuals62. Rather, it assumes that the assigned population is usually correct, requiring strong data to contradict this assumption62.
The threshold level to differentiate wolves and dogs from hybrids with our set of AIMs was defined based on the power of the admixture analysis to correctly identify simulated individuals with prior known ancestry. We used the reference wolf and dog samples to generate 100 simulated parental genotypes, and these to generate first (F1) and second (F2) generation hybrids, and first (BxW, BxD) and second (BxW2, BxD2) generation backcrosses to wolves and dogs, respectively, using hybridlab 1.064. Simulated genotypes were analyzed in structure 2.3.4, with the admixture model and correlated allele frequencies without any prior population information. The result of this analysis allowed estimating the percentage of simulated individuals in each hybrid class that were correctly classified as hybrid.
Calculations of pairwise genetic relatedness (r) were performed using the dyadic maximum-likelihood estimator (DyadML65) implemented in coancestry 1.066, with 100,000 bootstraps, and using the combined allele frequencies from CM wolves and hybrids plus wolf reference individuals from Galicia. Estimator selection was based on simulation results (see Appendix S1). Estimations were performed for all wolves and hybrids with a genomic content shifted towards wolf, from CM samples. First, pairwise relatedness was estimated for all pairs of individuals. Second, we compared pairwise relatedness within hybrid individuals. Finally, we compared relatedness between each hybrid and potential wolf pack members. To do this, we linked wolf samples with estimated pack territories. Pack territories were estimated by creating a conservative 100 km2 buffer area centered on the rendezvous sites of known packs in the area67 (Fig. 1). The selection of the size of the buffer area was based on the wolf home range size of adult wolves in the study area30. We clustered all wolf samples within each estimated pack range. We then compared the mean relatedness r of each hybrid-pack dyad using the test for group differences implemented in coancestry 1.0, with 100,000 bootstraps. A significant result would suggest that a given hybrid was more closely related to that pack compared to the rest of the population, indicating a potential natal pack. The spatial distribution of relatedness values between each hybrid-wolf pair was also explored through interpolations using the inverse distance-weighted interpolation algorithm implemented in ArcGIS 10.0 (ESRI Inc., Redlands, CA, USA) to produce a continuous surface of relatedness variation over the study area.
Additional Information
How to cite this article: Pacheco, C. et al. Spatial assessment of wolf-dog hybridization in a single breeding period. Sci. Rep. 7, 42475; doi: 10.1038/srep42475 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Rhymer, J. M. & Simberloff, D. Extinction by hybridization and introgression. Annu. Rev. Ecol. Syst. 27, 83–109 (1996).
Allendorf, F. W., Leary, R. F., Spruell, P. & Wenburg, J. K. The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16, 613–622 (2001).
Randi, E. Detecting hybridization between wild species and their domesticated relatives. Mol. Ecol. 17, 285–93 (2008).
Candille, S. I. et al. A β-Defensin mutation causes black coat colour in domestic dogs. Science 318, 1418–1423 (2007).
Anderson, T., Candille, S. & Musiani, M. Molecular and evolutionary history of melanism in North American gray wolves. Science 323, 1339–1343 (2009).
Hedrick, P. W. Wolf of a different colour. Heredity 103, 435–436 (2009).
Trouwborst, A. Exploring the legal status of wolf-dog hybrids and other dubious animals: international and EU law and the wildlife conservation problem of hybridization with domestic and alien species. Rev. Eur. Comp. Int. Environ. Law 23, 111–124 (2014).
Linnell, J., Salvatori, V. & Boitani, L. Guidelines for population level management plans for large carnivores in Europe. LCIE report, European Comission (2008).
Hindrikson, M. et al. Wolf population genetics in Europe: a systematic review, meta-analysis and suggestions for conservation and management. Biol. Rev, doi: 10.1111/brv.12298 (2016).
Chapron, G. et al. Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346, 1517–1519 (2014).
Lescureux, N. & Linnell, J. D. C. Warring brothers: The complex interactions between wolves (Canis lupus) and dogs (Canis familiaris) in a conservation context. Biol. Conserv. 171, 232–245 (2014).
Llaneza, L. & López-Bao, J. V. Indirect effects of changes in environmental and agricultural policies on the diet of wolves. Eur. J. Wildl. Res. 61, 895–902 (2015).
Millán, J. et al. Patterns of Exposure of Iberian Wolves (Canis lupus) to Canine Viruses in Human-Dominated Landscapes. Ecohealth 13, 1–12 (2015).
Randi, E. & Lucchini, V. Detecting rare introgression of domestic dog genes into wild wolf (Canis lupus) populations by Bayesian admixture analyses of microsatellite variation. Conserv. Genet. 3, 31–45 (2002).
Vilà, C. et al. Combined use of maternal, paternal and bi-parental genetic markers for the identification of wolf-dog hybrids. Heredity 90, 17–24 (2003).
Godinho, R. et al. Genetic evidence for multiple events of hybridization between wolves and domestic dogs in the Iberian Peninsula. Mol. Ecol. 20, 5154–5166 (2011).
Hindrikson, M., Männil, P., Ozolins, J., Krzywinski, A. & Saarma, U. Bucking the trend in wolf-dog hybridization: first evidence from europe of hybridization between female dogs and male wolves. PLoS One 7, e46465 (2012).
Kopaliani, N., Shakarashvili, M., Gurielidze, Z., Qurkhuli, T. & Tarkhnishvili, D. Gene flow between wolf and shepherd dog populations in Georgia (Caucasus). J. Hered. 105, 345–53 (2014).
Vähä, J. & Primmer, C. R. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 15, 63–72 (2006).
VonHoldt, B. M. et al. Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping. Mamm. Genome 24, 80–8 (2012).
Beja-Pereira, A., Oliveira, R., Alves, P. C., Schwartz, M. K. & Luikart, G. Advancing ecological understandings through technological transformations in noninvasive genetics. Mol. Ecol. Resour. 9, 1279–301 (2009).
Randi, E. et al. Multilocus detection of wolf x dog hybridization in italy, and guidelines for marker selection. PLoS One 9, e86409 (2014).
Godinho, R. et al. Real-time assessment of hybridization between wolves and dogs: combining non-invasive samples with ancestry informative markers. Mol. Ecol. Resour. 5, 317–328 (2015).
Stronen, A. V. & Paquet, P. C. Perspectives on the conservation of wild hybrids. Biol. Conserv. 167, 390–395 (2013).
Caniglia, R., Fabbri, E., Galaverni, M., Milanesi, P. & Randi, E. Noninvasive sampling and genetic variability, pack structure, and dynamics in an expanding wolf population. J. Mammal. 95, 41–59 (2014).
Lehman, N. et al. Introgression of coyote mitochondrial DNA into sympatric North American gray wolf populations. Evolution 30, 104–119 (1991).
Valière, N. et al. Long-distance wolf recolonization of France and Switzerland inferred from non-invasive genetic sampling over a period of 10 years. Anim. Conserv. 6, 83–92 (2003).
Broquet, T., Ménard, N. & Petit, E. Noninvasive population genetics: a review of sample source, diet, fragment length and microsatellite motif effects on amplification success and genotyping error rates. Conserv. Genet. 8, 249–260 (2006).
Da Silva, M. J. F. et al. Assessing the impact of hunting pressure on population structure of Guinea baboons (Papio papio) in Guinea-Bissau. Conserv. Genet. 15, 1339–1355 (2014).
Llaneza, L., García, E. J., Palacios, V. & López-Bao, J. V. Wolf monitoring in Galicia, 2013–2014. Report to the Spanish Ministry of Agriculture, Food and Environment, Spain (2014).
Queirós, J., Godinho, R., Lopes, S., Gortazar, C. & Alves, P. C. Effect of microsatellite selection on individual and population genetic inferences: an empirical study using cross‐specific and species‐specific amplifications. Mol. Ecol. Resour. 15, 747–760 (2015).
Lobo, D., Godinho, R., Álvares, F., López-Bao, J. V. & Rodríguez, A. A new method for noninvasive genetic sampling of saliva in ecological research. PLoS One 10, e0139765 (2015).
Blanco, J. C., Reig, S. & de la Cuesta, L. Distribution, status and conservation problems of the wolf Canis lupus in Spain. Biol. Conserv. 60, 73–80 (1992).
Leonard, J., Echegaray, J., Randi, E. & Vilà, C. Free-Ranging Dogs and Wildlife Conservation (ed Gompper, M. E. ) Ch. 7, 70–184 (Oxford University Press, 2014).
Verardi, A., Lucchini, V. & Randi, E. Detecting introgressive hybridization between free-ranging domestic dogs and wild wolves (Canis lupus) by admixture linkage disequilibrium analysis. Mol. Ecol. 15, 2845–55 (2006).
Vila, C. & Wayne, R. Hybridization between wolves and dogs. Conserv. Biol. 13, 195–198 (1999).
Bohling, J. H. & Waits, L. P. Assessing the prevalence of hybridization between sympatric Canis species surrounding the red wolf (Canis rufus) recovery area in North Carolina. Mol. Ecol. 20, 2142–56 (2011).
Rutledge, L. Y. et al. Protection from harvesting restores the natural social structure of eastern wolf packs. Biol. Conserv. 143, 332–339 (2010).
Rutledge, L. Y., White, B. N., Row, J. R. & Patterson, B. R. Intense harvesting of eastern wolves facilitated hybridization with coyotes. Ecol. Evol. 2, 19–33 (2012).
Bohling, J. H. & Waits, L. P. Factors influencing red wolf–coyote hybridization in eastern North Carolina, USA. Biol. Conserv. 184, 108–116 (2015).
Blanco, J. C. & Cortés, Y. Ecología, censos, percepción y evolución del lobo en España: análisis de un conflicto. SECEM, Málaga (2002).
Llaneza, L., López-Bao, J. V. & Sazatornil, V. Insights into wolf presence in human-dominated landscapes: the relative role of food availability, humans and landscape attributes. Divers. Distrib. 18, 459–469 (2012).
Boitani, L., Blanco, J. C., Bjarvall, A., Breitenmoser, U. & Farago, S. Action Plan for the Conservation of Wolves (Canis lupus) in Europe. Council of Europe (2000).
Boitani, L. et al. Key actions for large carnivore populations in Europe. Report to DG Environment, European Commission, Bruxelles (2015).
VonHoldt, B. M. et al. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2 (2016).
Bohling, J. H. Strategies to address the conservation threats posed by hybridization and genetic introgression. Biol. Conserv. 203, 321–327 (2016).
López-Bao, J. V., Sazatornil, V., Llaneza, L. & Rodríguez, A. Indirect Effects on Heathland Conservation and Wolf Persistence of Contradictory Policies that Threaten Traditional Free-Ranging Horse Husbandry. Conserv. Lett. 6, 448–455 (2013).
Mech, L. D. The wolf: The ecology and behavior of an endangered species (eds Mech, L. D. ) (University of Minnesota Press, 1970).
Jiménez, J. et al. Multimethod, multistate Bayesian hierarchical modeling approach for use in regional monitoring of wolves. Conserv. Biol. 30, 883–893 (2016).
Frantz, A. C. et al. Reliable microsatellite genotyping of the Eurasian badger (Meles meles) using faecal DNA. Mol. Ecol. 12, 1649–1661 (2003).
Boom, R. et al. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28, 495–503 (1990).
Vilà, C. et al. Mitochondrial DNA phylogeography and population history of the grey wolf Canis lupus . Mol. Ecol. 8, 2089–2103 (1999).
Smith, M. J. et al. Multiplex preamplification PCR and microsatellite validation enables accurate single nucleotide polymorphism genotyping of historical fish scales. Mol. Ecol. Resour. 11, 268–77 (2011).
Blacket, M. J., Robin, C., Good, R. T., Lee, S. F. & Miller, A. D. Universal primers for fluorescent labelling of PCR fragments - an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 12, 456–463 (2002).
Johnson, P. C. D. & Haydon, D. T. Software for quantifying and simulating microsatellite genotyping error. Bioinform. Biol. Insights 1, 71 (2007).
Valière, N. & Berthier, P. GEMINI: software for testing the effects of genotyping errors and multitubes approach for individual identification. Mol. Ecol. Notes 2, 83–86 (2002).
Valière, N. GIMLET: a computer program for analysing genetic individual identification data. Mol. Ecol. Notes 2, 377–379 (2002).
Excoffier, L. & Lischer, H. E. I. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Cockerham, C. C. & Weir, B. S. Covariances of relatives stemming from a population undergoing mixed self and random mating. Biometrics 40, 157–64 (1984).
Michalakis, Y. & Excoffier, L. A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142, 1061–1064 (1996).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–59 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol. Ecol. Notes 7, 574–578 (2007).
Nielsen, E. E., Bach, L. A. & Kotlicki, P. Hybridlab (Version 1.0): a Program for Generating Simulated Hybrids From Population Samples. Mol. Ecol. Notes 6, 971–973 (2006).
Milligan, B. G. Maximum-likelihood estimation of relatedness. Genetics 163, 1153–67 (2003).
Wang, J. COANCESTRY: a program for simulating, estimating and analysing relatedness and inbreeding coefficients. Mol. Ecol. Resour. 11, 141–5 (2011).
García, E. et al. Primeros datos sobre la ecología espacial del lobo en Galicia. Abstract book III Iberian Wolf Congresshttps://www.signatus.org/docs/III_Congreso_Ib%C3%A9rico_del_Lobo_-_Resumen_ponencias.pdf (date of access: 17/06/2016) (2012).
Acknowledgements
We thank the Regional Government of Galicia for the administrative and logistical support and all the rangers for their incredible work collecting samples within the framework of the Spanish wolf monitoring program, promoted by the Spanish Ministry of Agriculture, Food and Environment and the Autonomous Communities. We are grateful to the Editor S. Urbanelli and two anonymous reviewers for their very helpful comments on this manuscript, and K.P. Koepfli for linguistic editing and proofreading. We thank D. Castro and S. Lopes for lab assistance and P. Perú, A. Rivadulla and V. Sazatornil for field assistance. This work was partially supported by Portuguese funds through the Foundation for Science and Technology (FCT; project PTDC/BIA-EVF/2460/2014 to R.G.). R.G. and J.V.L.B were supported by research contracts from FCT and the Spanish Ministry of Economy and Competitiveness (IF/00564/2012 and JCI-2012-13066, respectively). This is scientific paper no. 14 of the Iberian Wolf Research Team (IWRT).
Author information
Authors and Affiliations
Contributions
J.V.L.B. and R.G. designed the study. J.V.L.B., E.J.G., F.J.L., L.L. and V.P. conducted the fieldwork. R.G. designed and supervised molecular work. C.P. performed the lab work and analysed the genetic data. C.P., R.G. and J.V.L.B. wrote the manuscript. All authors discussed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pacheco, C., López-Bao, J., García, E. et al. Spatial assessment of wolf-dog hybridization in a single breeding period. Sci Rep 7, 42475 (2017). https://doi.org/10.1038/srep42475
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42475
This article is cited by
-
Relatedness-based mate choice and female philopatry: inbreeding trends of wolf packs in a human-dominated landscape
Heredity (2024)
-
The persistence of the critically endangered Asiatic cheetah relies upon urgent connectivity protection: a landscape genetics perspective
Conservation Genetics (2023)
-
A reduced SNP panel to trace gene flow across southern European wolf populations and detect hybridization with other Canis taxa
Scientific Reports (2022)
-
Genetic diversity and population structure of the grey wolf (Canis lupus Linnaeus, 1758) and evidence of wolf × dog hybridisation in the centre of European Russia
Mammalian Biology (2021)
-
A standardized approach to empirically define reliable assignment thresholds and appropriate management categories in deeply introgressed populations
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
