
Abstract
In recent years, use of the DNA-intercalating dye propidium monoazide (PMA) in real-time PCR has been reported as a novel method to detect viable bacteria in different types of samples, such as food, environmental, and microbiological samples. In this study, viable cells of Acidovorax citrulli, the causal agent of bacterial seedling blight and fruit blotch, were selectively detected and differentiated from dead cells by real-time fluorescent polymerase chain reaction amplification after the bacterial solution was treated with the DNA-binding dye PMA. The primers and TaqMan probe were based on the A. citrulli genome (Aave_1909, Gene ID: 4669443) and were highly specific for A. citrulli. The detection threshold of this assay was 103 colony-forming units per mL (CFU/mL) in pure cell suspensions containing viable and dead cells and infected watermelon seeds. Application of this assay enables the selective detection of viable cells of A. citrulli and facilitates monitoring of the pathogen in watermelon and melon seeds.
Similar content being viewed by others

Introduction
Acidovorax citrulli, the causal agent of bacterial seedling blight and fruit blotch (BFB), has emerged as a devastating seed-borne pathogen of watermelon and other cucurbits worldwide, which can cause significant economic losses1,2,3,4,5. Owing to its economic importance, A. citrulli is also regarded as an official import plant quarantine bacterium in the USA, China, and Europe. It is well known that the disease is seed-transmitted and that symptomatic fruit generally contain infected seeds6; therefore, the use of pathogen-free seeds and transplants has become an important part of the control strategy. As a result, there is an urgent need for the development of a rapid and accurate method to selectively detect viable, seed-borne A. citrulli from seeds. Several methods of A. citrulli detection have previously been reviewed including isolation on semi-selective media, seriology, and polymerase chain reaction (PCR)-based methods such as classical PCR, real-time PCR, BIO-PCR, and immunomagnetic separation-PCR7,8,9,10,11. Compared to traditional cultivation-based practices, PCR-based methods are rapid and sensitive; however, they are limited by their inability to differentiate between DNA from viable organisms and that from dead microorganisms.
In recent years, the DNA-intercalating agent propidium monoazide (PMA) has been used in PCR to overcome problems related to PCR-based methods. PMA is a derivative of propidium iodide that can selectively penetrate into dead cells with compromised membranes and covalently bind DNA upon exposure to an intense light source. In this new approach, samples are treated with PMA prior to DNA extraction. Thus, PMA can specifically inhibit the PCR amplification of DNA derived from dead cells and enable the differentiation of live and dead cells in a bacterial population12. Correspondingly, its use in combination with real-time PCR has been effectively applied to detect viable cells in many different microbiological species13,14,15,16,17,18,19.
Our objective was to implement the PMA/real-time fluorescent PCR technique for the selective detection of viable, seed-borne A. citrulli. For this purpose, we designed new A. citrulli species-specific primers and TaqMan probe, and evaluated a novel real-time fluorescent PCR method for the specific and sensitive detection of A. citrulli.
Results
Specificity and sensitivity of real time-PCR
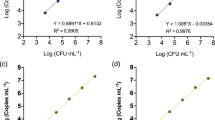
Specificity analyses for the real-time PCR assay showed positive results for all 116 strains of A. citrulli, whereas no reaction was observed for the other 23 bacteria tested including the other closely related Acidovorax species A. avenae, A. konjaci, and A. cattleyae. The results of the real-time fluorescent PCR assay are presented in Supplementary Table S1. The relationship between the concentration of A. citrulli strain tw24, from 108 to 10 CFU/mL, and the Ct value is shown in Fig. 1. Plotting of the log-concentration of the bacteria versus the cycle number yielded a straight-line regression, with a correlation coefficient (R2) of 0.995. The detection limit of the real-time PCR assay with the A. citrulli primers/probe set was approximately 8 × 103 CFU/mL in 10-fold serial dilutions of the pure bacterial cell suspension.

Sensitivity of A. citrulli detection using the TaqMan real-time PCR assay.
DNA was prepared as serial 10-fold dilutions of A. citrulli strain tw24 cell cultures (8 × 108 to 8 × 10 CFU/mL) and used as a template to test the sensitivity of the real-time PCR assay. The standard curve plot, slope, Y-intercept, and R2 are shown.
Effect of PMA treatment on real-time PCR amplification of DNA from viable and dead cells from pure bacterial cultures
To determine whether PMA treatment affected the amplification of viable cell DNA, serial 10-fold dilutions of PMA-treated and non-treated viable A. citrulli at concentrations ranging from 8 × 106 to 8 × 10 CFU/mL were compared. Both curves from the PMA-treated and non-treated viable cells were linear and overlapping (Fig. 2), illustrating that PMA treatment had virtually no effect on DNA amplification for viable cells.

Amplification of DNA from the PMA-treated viable cells by the TaqMan real-time PCR assay.
Standard curve plot of real-time PCR generated using 10-fold serial dilutions of PMA-treated and non-treated A. citrulli strain tw24 cultures (8 × 106 to 8 × 10 CFU/mL).
Additionally, we compared PMA-treated and non-treated viable and dead cells (approximately 8 × 106 CFU/mL) simultaneously to evaluate the efficacy of PMA treatment to selectively detect viable cells in pure cultures. Similar Ct values were observed for the PMA-treated and non-treated viable cells and for non-treated dead cells. In contrast, there was a 10-Ct difference between the PMA-treated dead cells and the other samples (Fig. 3). This result indicates that the PMA/real-time PCR assay effectively detected both live and dead cells and that PMA specifically inhibited DNA amplification from dead cells.

Real-time PCR amplification plots for pure bacterial cultures (106 CFU/mL).
Amplification plot comparison of PMA-treated viable and dead cells with non-treated viable and dead cells using real-time PCR.
Evaluation of PMA/real-time PCR for the detection of viable cells from mixtures of viable and dead cells
Various mixtures of viable cells (8 × 106 to 8 × 10 CFU/mL) and 8 × 106 CFU/mL dead cells were used to evaluate the efficacy of the PMA/real-time PCR assay to selectively detect viable cells in the presence of dead cells. Real-time PCR showed that PMA-treated mixtures displayed a descending trend in Ct that was reciprocal to the number of viable cells, despite the large number of dead cells included in the samples (Fig. 4). In contrast, the Ct values of the untreated mixtures increased only slightly with decreasing numbers of viable cells and remained nearly unchanged in mixtures with fewer viable cells (Fig. 4). These results demonstrate that the Ct values of the PMA-treated mixtures were solely dependent on the amount of DNA from viable cells; thus, the PMA treatment effectively inhibited amplification of DNA from dead cells.

Ct values for viable and dead cell mixtures.
Various mixtures of viable cells (8 × 106 to 8 × 10 CFU/mL) and 8 × 106 CFU/mL dead cells were generated and treated with PMA or left untreated prior to DNA preparation. Each bar represents the average triplicate Ct value.
Application of PMA/real-time PCR to artificially infected seeds
To determine whether the PMA/real-time PCR assay could be successfully applied to detect viable A. citrulli in infected seeds, healthy watermelon seeds inoculated with different concentrations of viable cells (8 × 106 to 8 × 10 CFU/mL), and 8 × 106 CFU/mL dead cells were used. Three sets of seed samples (300 seeds per set) from each treatment group were tested using real-time PCR and PMA/real-time PCR, respectively. The results were consistent with those achieved for bacterial cultures. The Ct values of the PMA-treated seeds increased as the actual number of viable cells increased (Table 1). Conversely, the Ct values of the untreated seeds remained nearly unchanged in the case of fewer viable cells (Table 1). These results demonstrated that PMA inhibited the amplification of dead bacteria and that the PMA/real-time PCR assay selectively detected viable A. citrulli in infected seeds, without interference from the seed matrix.
Application of PMA/real-time PCR to naturally infected seeds
To determine whether the PMA/real-time PCR assay could be successfully applied to actual samples from the field, three naturally infected watermelon seed samples that had been treated with 1% HCl (for sterilization) or left untreated were used. To ensure that the naturally infected seed samples carried viable A. citrulli, we first tested the seed samples using the seedling grow-out method, which defined the incidence of BFB as approximately 16%, 23%, and 29%, respectively. Both HCl treated and untreated samples were then confirmed to either contain or be free of viable A. citrulli by isolation on semi-selective media. The results showed that after approximately 5 days, A. citrulli could be successfully isolated and identified from all three HCl untreated samples on semi-selective media, but not from the three HCl treated samples, indicating that HCl effectively kills pathogens on watermelon seeds. For the three naturally infected watermelon seed samples, similar Ct values were observed for HCl untreated samples by real-time PCR and PMA/real-time PCR, and for HCl treated samples by real-time PCR (Table 2). In contrast, the Ct value of the three HCl treated samples obtained by PMA/real-time PCR were all higher than 37 (Table 2). No reactions were observed from both of the two negative controls. In our practical testing work, we usually consider that the results are negative when the Ct values are higher than 35. These results indicate that the PMA/real-time PCR assay is able to successfully and selectively detect viable A. citrulli in naturally infected seeds, while avoiding the overestimation of bacterial cells that occurs when using classical PCR. Thus, this PMA/real-time PCR assay is suitable for the detection of viable A. citrulli in seed samples from the field.
Discussion
Since the first report of watermelon bacterial fruit blotch caused by A. citrulli in 1990 in China, this disease has been reported in Sinkiang, Ningxia, Inner Mongolia, Hebei, Henan, Shandong, Jiangsu, Nanjing, Guangdong, Fujian, Hainan, Jilin, and Beijing. Economic losses owing to watermelon bacterial fruit blotch result from seed yield reductions and loss of fruit marketability. Effective control of the spread of this disease relies on the availability of sensitive and specific methods that can be used for rapid and accurate detection of the pathogen. Although several methods are available for the detection of A. citrulli, these methods are still accompanied by limitations. Therefore, we developed a method for the selective detection of viable, seed-borne A. citrulli using a combination real-time fluorescent PCR and PMA treatment. Our work demonstrated that this assay could specifically and sensitively detect viable A. citrulli in both pure cultures and infected seeds.
One of the key aspects of our study was the development of an assay using a unique A. citrulli sequence as the sole genetic marker for the target in real-time PCR. Several PCR primers have been designed for A. citrulli based on 16S ribosomal DNA (rDNA), the 16S-23S internally transcribed spacer (ITS) regions of rDNA, the BOX fragment, and hrp gene sequences11. Currently, the most commonly used primer set for the detection of A. citrulli is WFB1/WFB2 (based on 16S rDNA), which is highly sensitive but also amplifies non-target bacterial DNA including that of A. avenae, A. konjaci, A. cattleyae, Comamonas testeroni, and an Acidovorax sp. from Calathea spp.20. Primers SEQID4 and SEQID5 were designed from the 16S-23S ITS region of A. citrulli21. SEQID4 was subsequently modified by removing the nucleotides “TC” from the 5′ end to yield SEQID4m22. Both the original and modified primers are highly specific for A. citrulli but are also susceptible to false positive results. In comparison, primers BX-L1 and BX-S-R2 were designed from a BOX-PCR fragment of A. citrulli and were reported to be highly specific to A. citrulli without compromising detection sensitivity under high-stringency conditions23. The TaqMan real-time PCR assay described in this study offers a remedy for the limitations of the PCR assays mentioned above, as demonstrated by several lines of evidence from the specificity tests. First, the current assay was considered highly specific because it did not amplify template DNA from 23 non target bacterial strains including the closely related Acidovorax species. In addition, the primers allowed detection of all 116 strains of A. citrulli tested, which originated from various locations and from different hosts. Furthermore, the real-time PCR method exhibits several advantages over classical PCR such as providing results in real-time and without the requirement for agarose gel electrophoresis.
Another focus of our study involved differentiating viable A. citrulli cells from dead cells. Several PCR-based methods have been reported for detection of A. citrulli; however, these assays detect total DNA derived from both viable and dead cells. In recent years, PMA and ethidium monoazide (EMA) have been used in PCR to overcome this problem. A combination of EMA and PCR (using primer set WFB1/WFB2) was developed for the selective detection of A. citrulli by Feng et al.24. Both EMA and PMA are able to intercalate double-stranded DNA or RNA and then irreversibly cross-link to the nucleic acids following photoactivation12,25. Some studies have observed that EMA, but not PMA, was able to penetrate into viable cells and was toxic to viable cells12,13. However, the PCR primer sets used in these studies are unable to distinguish A. citrulli from the other closely related Acidovorax species.
In the current study, we have assessed the capability of PMA/real-time PCR for the differentiation of viable A. citrulli from dead cells. Comparison of samples between PMA-treated or non-treated viable or dead A. citrulli cells, or a mixture of viable/dead cells, showed almost no difference between the PMA-treated viable cells and non-treated cells, illustrating that the amplification of DNA from viable cells was not affected by PMA treatment. We also demonstrated that the PMA/real-time PCR assay could efficiently prevent DNA amplification from dead cells. Furthermore, we were able to selectively detect viable cells from mixtures with a large number of dead cells (8 × 106 CFU/mL) using this assay. These results confirmed that the PMA/real-time PCR assay provides a reliable means to determine the presence of viable cells of A. citrulli.
We further applied this PMA/real-time PCR assay for the selective detection of viable A. citrulli in seeds. Because the bacterium is spread by infected seeds, the only method to effectively control the disease is the use of healthy seeds26. Therefore, we applied this assay to selectively detect viable cells of A. citrulli in infected seeds. Our results demonstrated that this assay could positively detect viable cells of A. citrulli from both artificially and naturally infected seeds. However, during the detection process of the sterilized infected seeds using PMA treatment, fluorescence signals were also collected after 37 cycles when there was a high A. citrulli carrier rate. Therefore, we speculate that the amplification of DNA from dead cells was not completely blocked by PMA treatment. A similar phenomenon was also observed by Elizaquivel et al.17. However, according to our usual testing standards, the samples would be judged as negative for Ct values above 35. Therefore, dead cells of A. citrulli on sterilized infected seeds would not be detected as positive findings using the PMA/real-time PCR assay, thus avoiding the overestimation of bacterial cells that occurs when using standard PCR-based methods. As a result, our method should prevent the overestimation of potential disease risk. In addition, we also detected natural infected seeds and those that had undergone sterilization using the traditional agar plating method. The results from this assessment were consistent with those of PMA/real-time PCR, indicating that viable cells of A. citrulli could be isolated from naturally infected seeds, whereas no A. citrulli strains were isolated from the sterilized infected seeds. However, this isolation method required more than 5 days. In contrast, the PMA/real-time PCR assay was simpler and more rapid, requiring only 1 day. Thus, the PMA/real-time PCR assay possesses marked advantages for the detection of viable cells of A. citrulli in seed samples.
In summary, we have developed a new PMA/real-time PCR assay that has been proven to be sensitive and specific for the selective detection of viable cells of A. citrulli from infected seeds. Thus, this method could likely be useful for pathogen detection in seeds or to evaluate seed sterilization.
Materials and Methods
Bacterial strains and culture conditions
We utilized 116 A. citrulli strains and 23 additional strains in our study; the relevant information and sources are listed in Supplementary Table S1. All strains were routinely grown in nutrient broth (NB) for 24–48 h at 28 °C. To enumerate bacterial cells, cultures were serially diluted in 10-fold increments with medium, plated on nutrient agar (NA) plates, and incubated overnight at 28 °C.
Primer design and real-time PCR conditions
To develop stable and specific primers and a probe for the identification of A. citrulli, we evaluated several A. citrulli genes (AAC00-1, NC_008752.1) by BLAST analysis to identify an A. citrulli-specific sequence. A unique sequence (Aave_1909) encoding a hypothetical protein was identified as a specific genetic marker for A. citrulli. Based on this sequence, forward and reverse primers (A. citrulli-FP and A. citrulli-RP) and also a TaqMan probe (A. citrulli-P) were designed using Primer Express 3.0 (Applied Biosystems, Foster City, CA, USA). The expected product size was 121 bp.
A. citrulli-P was labelled at the 5′ and 3′ ends with 6-caboxyfluorescein (FAM) and black hole quencher (BHQ1), respectively. The sequences of the primers and probe were as follows: A. citrulli-FP: 5′-CTGATAATCCTCGGCTCAACAA-3′; A. citrulli-RP: 5′-TGAGCGCATTTCTGACGAG-3′; A. citrulli-P: FAM-AAGAAATACGCCCTCGCCAATCTCC-BHQ1. Oligonucleotides were synthesized by Shanghai Huirui Biotechnology (Shanghai, China).
PCR mixtures contained 10 μL 2× qPCR Master Mix (Shanghai Huirui Biotechnology), 0.8 μL each primer (10 μM), 0.4 μL probe (10 μM), and 1 μL template DNA or pure bacterial culture suspension, and an appropriate volume of ddH2O was added to reach a reaction volume of 20 μL. Amplifications were performed on the ABI Prism 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using the following thermal cycling parameters: 95 °C for 10 min, and 40 cycles at 95 °C for 15 s and 58 °C for 1 min. Fluorescence was measured at the end of each extension step. Threshold cycles were generated automatically by ABI 7900 HT Prism software. All experiments included a non-template control in which 1 μL ddH2O was used rather than the DNA template. Each experiment was performed in triplicate.
Specificity and sensitivity assays
To test specificity of the real-time PCR reaction, 116 strains of A. citrulli from different origins and hosts, 7 closely related Acidovorax strains, and 16 strains from another 9 genera were used (Supplementary Table S1). All strains were grown in NB for 24 to 48 h and 1 μL of culture suspension was used directly for real-time PCR as described above.
To determine the real-time PCR assay sensitivity, the culture of A. citrulli strain tw24 was determined to be equivalent to 8 × 108 CFU/mL by plating and was then diluted in sterile water at 10-fold increments to generate serial dilutions ranging from 8 × 108 to 8 × 10 CFU/mL. Each dilution (1 μL) was used for real-time PCR to generate the standard curve for the detection limit.
Preparation of viable and dead cell mixtures for PMA/real-time PCR
The culture of A. citrulli strain tw24 was grown in NB at 28 °C with shaking at 180 rpm to mid-exponential phase and then adjusted to approximately 8 × 106 CFU/mL by plating. The bacterial suspension was divided into two aliquots. One aliquot was boiled in a water bath for 10 min to kill the cells and then 100 μL of the boiled suspension was spread on an NA plate and incubated at 28 °C for 24–48 h to check cell viability. The other aliquot was used to generate two series of 10-fold dilutions ranging from 8 × 106 to 8 × 10 CFU/mL. One set was used to generate the various samples containing different number of viable cells (8 × 106 to 8 × 10 CFU/mL). The other set was combined with 8 × 106 CFU/mL dead cells to generate the cell mixtures, which consisted of different numbers of viable cells (8 × 106 to 8 × 10 CFU/mL) along with 8 × 106 CFU/mL dead cells. Each sample, containing dead cells, different numbers of viable cells, or cell mixtures, was divided into two aliquots and processed as PMA-treated and untreated cell populations.
PMA treatment and DNA isolation
PMA (Biotium Inc., Hayward, CA, USA) was dissolved in dimethyl sulfoxide to obtain a stock solution of 1 mg/mL and was stored at −20 °C in the dark. PMA (1.5 μL) was added to 500 μL of the viable, heat-killed, and mixed samples described above to final concentrations of 3 μg/mL14,27. The PMA-treated samples were incubated at room temperature in the dark for 5 min with occasional mixing. Subsequently, the samples were exposed to a 650 W halogen light for 5 min to cross-link the intercalated PMA with DNA. DNA was then isolated by boiling in a water bath for 10 min, followed by rapid cooling on ice for 5–10 min. The samples were centrifuged at 8,000 × g for 2–3 min and the DNA-containing supernatant was transferred to a new centrifuge tube for real-time PCR.
Artificially and naturally infected seed assay
Watermelon seeds artificially or naturally infected with A. citrulli were used to evaluate the efficiency of PMA/real-time PCR to distinguish between viable and dead cells in seed samples. The healthy watermelon seeds (Daguoxinxiu, Beijing) used in this assay were first confirmed to be free of A. citrulli by traditional agar plating methods. For artificial inoculation, six clean watermelon seed samples were incubated in a set of mixtures containing different concentrations of viable A. citrulli cells (8 × 106 to 8 × 10 CFU/mL) as well as 8 × 106 CFU/mL dead cells for 1 h, respectively, and then were air-dried. Next, 3 sets of seed samples (300 seeds per set) from each treatment group (watermelon seeds infected with 8 × 106 to 8 × 10 CFU/mL viable cells along with 8 × 106 CFU/mL dead cells) were then tested using real-time PCR and PMA/real-time PCR, respectively.
For the naturally infected seed assay, three infected watermelon seed samples (Daguoxinxing, Hebei; Daguojiaomei, Hebei; Ribenjiaomei, Hebei) were used. All the naturally infected samples were first tested by a seedling grow-out method to determine the incidence of BFB. These were then split into two equivalent samples; one remained untreated, and the other was treated with 1% HCl to kill the pathogen. The infected seeds were soaked in 1% HCl for 10 min and then rinsed with water 4–5 times until the pH value reached 7. Both HCl treated and untreated naturally infected seed samples were tested by traditional agar plating methods to be confirmed as free or containing viable A. citrulli. Next, 300 seeds per treatment group (HCl treated and untreated) of the three naturally infected seed samples were tested using real-time PCR and PMA/real-time PCR, respectively. Tow healthy seed samples that treated with only 1% HCl or treated with only ddH2O were also tested as the negative control.
For traditional isolation, the seeds were soaked in seed extract buffer at 4 °C overnight and then 100 μL of undiluted and 10−1 dilutions of seed wash were spread onto selective media (EBBA) and incubated at 37 °C for 5 days, as described by Zhao et al.28. For real-time PCR and PMA/real-time PCR assays, each set of seed samples (300 seeds per set) was incubated in sterile distilled water for 4 h. Then, 3 mL seed extract was centrifuged at 10,000 × g for 5 min and the pellets were resuspended in 1 mL ddH2O. The suspensions of each sample were split into two equivalent samples and 500 μL was treated with PMA as described above and 500 μL was left untreated. Both PMA-treated and non-treated suspensions were centrifuged at 10,000 × g for 2 min. Pellets were used for DNA extraction using the DNA secure Plant Kit (Tiangen Biotech, Beijing, China) according to the manufacturer’s instructions. Finally, each DNA sample (1 μL) was used for real-time PCR. 1 μL water was uesd to substitute template DNA to serve as non-template control.
Additional Information
How to cite this article: Tian, Q. et al. Selective detection of viable seed-borne Acidovorax citrulli by real-time PCR with propidium monoazide. Sci. Rep. 6, 35457; doi: 10.1038/srep35457 (2016).
References
Isakeit, T., Black, M., Barnes, L. W. & Jones, J. B. First report of infection of honeydew with Acidovorax avenae subsp. citrulli. Plant Dis. 81, 694–694 (1997).
Isakeit, T., Black, M. & Jones, J. Natural infection of citron melon with Acidovorax avenae subsp. citrulli. Plant Dis. 82, 351–351 (1998).
Langston, D. B. Jr., Walcott, R. D., Gitaitis, R. D. & Sanders, F. H. Jr. First report of a fruit rot of pumpkin caused by Acidovorax avenae subsp. citrulli in Georgia. Plant Dis. 83, 199 (1999).
Martin, H., O’Brien, R. G. & Abbott, D. V. First report of Acidovorax avenae subsp. citrulli as a pathogen of cucumber. Plant Dis. 83, 965–965 (1999).
Walcott, R., Langston D. Jr., Sanders, F. H. Jr., Gitaitis, R. D. & Flanders, J. T. Natural outbreak of a bacterial fruit rot of cantaloupe in Georgia caused by Acidovorax avenae subsp. citrulli. Plant Dis. 84, 372–372 (2000).
Rane, K. K. & Latin, R. X. Bacterial fruit blotch of watermelon: Association of the pathogen with seed. Plant Dis. 76, 509–512 (1992).
Schaad, N. W. & Sechler, A. An improved semiselective agar medium for Acidovorax avenae subsp. citrulli. Phytopathology 89, S68–S69 (1999).
Walcott, R. & Gitaitis, R. Detection of Acidovorax avenae subsp. citrulli in watermelon seed using immunomagnetic separation and the polymerase chain reaction. Plant Dis. 84, 470–474 (2000).
Randhawa, P., Pannu, S. & Schaad, N. W. Improved bio-PCR test for detection of Acidovorax avenae subsp. citrulli in watermelon and cantaloupe seeds. Phytopathology 91, S74 (2001).
Bahar, O. et al. New subspecies-specific polymerase chain reaction-based assay for the detection of Acidovorax avenae subsp. citrulli. Plant Pathol. 57, 754–763 (2008).
Feng, J. J. et al. Advances in detection of Acidovorax citrulli, the causal agent of bacterial fruit blotch of cucurbits. Seed Science & Technology 41, 1–15 (2013).
Nocker, A., Cheung, C. Y. & Camper, A. K. Comparison of propidium monoazide with ethidium monoazide for differentiation of live vs. dead bacteria by selective removal of DNA from dead cells. J. Microbiol. Methods 67, 310–320 (2006).
Pan, Y. & Breidt, F. Enumeration of viable Listeria monocytogenes cells by real-time PCR with propidium monoazide and ethidium monoazide in the presence of dead cells. Appl. Environ. Microbiol. 73, 8028–8031 (2007).
Nocker, A., Sossa-Fernandez, P. & Burr, M. D. & Camper, A. K. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl. Environ. Microbiol. 73, 5111–5117 (2007).
Li, B. & Chen, J. Q. Real-time PCR methodology for selective detection of viable Escherichia coli O157:H7 by targeting Z3276 as a genetic marker. Appl. Environ. Microbiol. 78, 5297–5304 (2012).
Sánchez, G., Elizaquível, P. & Aznar, R. Discrimination of infectious hepatitis A viruses by propidium monoazide real-time RT-PCR. Food Environ. Virol. 4, 21–25 (2012).
Elizaquivel, P., Sanchez, G. & Aznar, R. Quantitative detection of viable foodborne E. coli O157:H7, Listeria monocytogenes and Salmonella in fresh-cut vegetables combining propidium monoazide and real-time PCR. Food Control. 25, 704–708 (2012).
Xing-Long, X., Cong-Cong, L., Yang, Q., Yi-Gang, Y. & Hui, W. Molecular monitoring of Escherichia coli O157: H7 sterilization rate using qPCR and propidium monoazide treatment. Lett. Appl. Microbiol. 56, 333–339 (2013).
Feng, J. J. et al. Preliminary study on differentiation of live and dead cell of Pantoea stewartii subsp. stewartii using propidium monoazide combined with real-time fluorescent PCR. Plant Quarantine. 28, 27–32 (2014).
Walcott, R. R. & Gitaitis, R. Detection of Acidovorax avenae subsp. citrulli in watermelon seed using immunomagnetic separation and the polymerase chain reaction. Plant Dis. 84, 470–474 (2000).
Schaad, N. W., Song, W. Y. & Hatziloukas, E. Inventors; The United States of America as represented by the Secretary of Agriculture, assignee. PCR primers for detection of plant pathogenic species and subspecies of Acidovorax. United States patent US 6,146,834. 2000 Nov 14.
Walcott, R. R., Gitaitis, R. D. & Castro, A. C. Role of blossoms in watermelon seed infestations by Acidovorax avenae subsp. citrulli. Phytopathology 93, 528–534 (2003).
Bahar, O. et al. New subspecies-specific polymerase chain reaction-based assay for the detection of Acidovorax avenaesubsp. citrulli. Plant Pathology 57, 754–763 (2008).
Feng, J. J., Kim, J., Liu, X. L., Schaad, N. W. & Li, J. Q. Differentiation of live and dead cell of bacterial plant pathogen in polymerase chain reaction assays using a DNA binding dye. Chem. J. Chin. Univ. 29, 944–948 (2008).
Bolton, P. H. & Kearns, D. R. Spectroscopic properties of ethidium monoazide: a fluorescent photoaffinity label for nucleic acids. Nucleic Acids Res. 5, 4891–4903 (1978).
Rane, K. K. & Latin, R. X. Bacterial fruit blotch of watermelon- association of the pathogen with seed. Plant Dis. 76, 509–512 (1992).
Lou, J. F., Lin, W. T. & Guo, Y. Detection of viable bacterium cells based on propidium monoazide in combination with PCR. J. South China Univ. Technol. 38, 142–146 (2010).
Zhao, T. et al. An improved assay for detection of Acidovorax citrulli in watermelon and melon seed. Seed Science & Technology 37, 78616–13321 (2009).
Acknowledgements
We thank Dr. Baishi Hu, Jianping Yi, Tingchang Zhao, and Lifang Zou for providing bacteria strains. This work was financially supported by the special Fund for Agroscientific Research in the Public Interest (201003066) and the National Science and Technology Support Program (2012BAK11B02).
Author information
Authors and Affiliations
Contributions
Designed the study: Q.T. and W.-j.Z.; performed the experiments: Q.T. and J.H.; strain collection: J.-j.F. and W.-j.Z.; data analysis and discussion: Q.T., J.-j.F., J.H. and W.-j.Z.; drafting of the manuscript: Q.T., J.-j.F., J.H. and W.-j.Z.; revision of the manuscript: Q.T., J.-j.F. and W.-j.Z. All authors have read and approved the final submitted manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tian, Q., Feng, Jj., Hu, J. et al. Selective detection of viable seed-borne Acidovorax citrulli by real-time PCR with propidium monoazide. Sci Rep 6, 35457 (2016). https://doi.org/10.1038/srep35457
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35457
This article is cited by
-
A viability qPCR protocol to assess the efficacy of a heat treatment to sanitize carrot seeds from Candidatus Liberibacter solanacearum
European Journal of Plant Pathology (2023)
-
A real-time PCR assay using locked nucleic acid probe for detection of Acidovorax citrulli
Journal of Plant Diseases and Protection (2022)
-
Sensitive and specific detection of Xanthomonas campestris pv. zinniae by PCR using pathovar-specific primers
European Journal of Plant Pathology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
