
Abstract
Maternal cigarette smoke exposure (SE) during gestation can cause lifelong adverse effects in the offspring’s brain. Several factors may contribute including inflammation, oxidative stress and hypoxia, whose changes in the developing brain are unknown. Female Balb/c mice were exposed to cigarette smoke prior to mating, during gestation and lactation. Male offspring were studied at postnatal day (P) 1, P20 and 13 weeks (W13). SE dams had reduced inflammatory mediators (IL-1β, IL-6 and toll like receptor (TLR)4 mRNA), antioxidant (manganese superoxide dismutase (MnSOD)) and increased mitochondrial activities (OXPHOS-I, III and V) and protein damage marker nitrotyrosine. Brain hypoxia-inducible factor (HIF)1α and its upstream signalling molecule early growth response factor (EGR)1 were not changed in the SE dams. In the SE offspring, brain IL-1R, IL-6 and TLR4 mRNA were increased at W13. The translocase of outer mitochondrial membrane and MnSOD were reduced at W13 with higher nitrotyrosine staining. HIF-1α was also increased at W13, although EGR1 was only reduced at P1. In conclusion, maternal SE increased markers of hypoxia and oxidative stress with mitochondrial dysfunction and cell damage in both dams and offspring and upregulated inflammatory markers in offspring, which may render SE dams and their offspring vulnerable to additional brain insults.
Similar content being viewed by others


Introduction
Cigarette smoking is a significant risk factor for a number of chronic conditions, such as cerebrovascular and cardiovascular diseases, in addition to respiratory disorders1 and thus remains a major cause of death worldwide2. Despite general education on the risks, smoking during pregnancy and passive smoking during pregnancy are still common in both developed and developing countries3,4 and ~20–45% women smoke during pregnancy in Europe, Australia, South America and South Africa3,4,5. Smoking and second hand smoking in pregnant women may result in placental transfer of toxic agents present in cigarettes and transmit a risk to the developing fetal brain. In addition there are increased risks of developing well-known metabolic, respiratory and behavioural disorders that are recognised in the offspring of first-hand or second-hand smoking mothers (reviewed in6,7,8,9). Nicotine can pass through the placenta and act as a vasoconstrictor, which can reduce uterine blood flow by up to 38%10, leading to deprivation of oxygen and nutrients in the fetus, resulting in hypoxia and undernutrition11. As such, maternal smoking is a known risk factor for intrauterine growth retardation12,13, with adaptive brain structural and functional changes occurring during fetal development14,15,16,17,18. Preterm infants from smoking mothers display significantly smaller frontal lobe and cerebellar volumes after adjustments of confounding factors such as alcohol consumption19. It is likely that maternal smoking alters fetal brain immune function and mitochondrial activity that make such offspring more vulnerable to brain insults.
Oxidative stress is integral to the general inflammatory response20, which occurs due to a metabolic imbalance brought about by excess production of reactive oxygen species (ROS, such as the superoxide anion) and/or a reduced level of host antioxidant defences. Mitochondria are a major site of ROS production during oxidative phosphorylation (OXPHOS) to generate ATP21. During an inflammatory response, there is a high consumption of oxygen and release of the superoxide free radical (O−2) by the mitochondria22, which can, in turn, impair mitochondrial function23 leading to cell and organ impairment. Thus, to protect cell integrity, excessive ROS are removed by antioxidants, including mitochondrial manganese superoxide dismutase (MnSOD). Oxidative stress can also exacerbate associated inflammatory reactions by activating pathways such as c-jun N-terminal kinases and nuclear factor-κ-light-chain-enhancer of activated B cells24. Hence, increased antioxidant levels or activity can significantly reduce the injury size in mice following stroke25. However, if the brain has pre-existing oxidative stress and inflammation, both mitochondrial and cellular function can be affected especially during post-injury repair26,27. Cigarette smoke itself contains a substantial amount of ROS28, which may exceed the baseline antioxidative capacity of the mitochondria to clear both endogenous and exogenous ROS. Indeed, it has been shown that smokers have decreased levels of antioxidants in their serum29. However, it is unclear whether smoking increases brain inflammation and oxidative stress. Therefore, we hypothesise that there may be a causal link between cigarette smoke exposure (SE), increased inflammation, oxidative stress and mitochondrial dysfunction in the brain. The aim of this study was to investigate the impact of continuous maternal cigarette smoke exposure in mice on brain inflammation, mitochondrial function and antioxidant capacity, as well as markers of hypoxia in both mothers and offspring.
Materials and Methods
Maternal cigarette smoke exposure
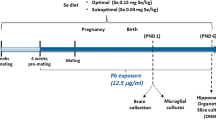
The animal experiments were approved by the Animal Care and Ethics Committee at the University of Technology Sydney (ACEC#2011-313A). All protocols were performed according to the Australian National Health & Medical Research Council Guide for the Care and Use of Laboratory Animals. Virgin female Balb/c mice (6 weeks, Animal Resources Centre, Perth, Australia) were housed at 20 ± 2 °C and maintained on a 12-h light, 12-h dark cycle (lights on at 06:00 h) with ad libitum access to standard laboratory chow and water. After the acclimatisation period, mice were assigned to cigarette SE or sham exposure (SHAM). The SE group was exposed to 2 cigarettes (Winfield Red, ≤16 mg tar, ≤1.2 mg nicotine and ≤15 mg of CO; VIC, Australia) in a perspex chamber (18 L), twice daily for six weeks prior to mating, during gestation and lactation; while the SHAM group was exposed to normal air as previously described30. They were mated with male Balb/c mice (8 weeks) from the same source, which were not exposed to cigarette smoke. The offspring were housed 4–5/cage after weaning and the males were studied at postnatal day (P)1, P20 (weaning) and week 13. The females will be reported separately.
Sample collection
Animals at P1 were sacrificed by decapitation, while animals older than 20 days were killed after anaesthetic overdose (Pentothal®, 0.1 mg/g, i.p., Abbott Australasia Pty. Ltd., NSW, Australia) between 9:00–12:00 h. The mothers were also culled between 9:00–12:00 h (with their last cigarette being at 15:00 h the previous day). Brains were dissected into the left and right hemispheres. The left hemisphere was stored at −80 °C for mRNA and protein analysis, while the right hemisphere was fixed with 4% formalin for immunohistochemical analysis.
Quantitative real-time PCR
Total mRNA was extracted from brain tissues using TriZol reagent (Life Technologies, CA, USA). The purified total RNA was used as a template to generate first-strand cDNA using M-MLV Reverse Transcriptase, RNase H, Point Mutant Kit (Promega, Madison, WI, USA)31. Genes of interest were measured using manufacturer pre-optimized and validated TaqMan primers and probes (Life Technologies, CA, USA). Only the probe sequence is provided by the manufacturer (Table 1). The probes of the target genes were labelled with FAM® dye and those for housekeeping 18 s rRNA were labelled with VIC® dye. Gene expression was standardized to 18 s RNA. The average expression of the control group was assigned as the calibrator against which all other samples are expressed as fold difference.
Western Blotting
The protein levels of early growth response factor (EGR)1, hypoxia-inducible factor (HIF)-1α, manganese superoxide dismutase (MnSOD), translocase of outer membrane (TOM)20 and OXPHOS complex proteins were measured by western blotting. The brain was homogenised using cell lysis buffers for whole protein and mitochondria protein extraction according to manufacturer’s instruction32. Protein samples (40 μg) were separated on NuPage® Novex® 4–12% Bis-Tris gels (Life Technologies, CA, USA) and then transferred to PVDF membranes (Rockford, IL, USA), which were blocked with non-fat milk powder and incubated with the primary antibodies (EGC-1 (1:5000, Santa Cruz Biotechnology), HIF-1α (1:1000, Novus Biologicals); MnSOD (1:1000) & TOM20 (1:2000, Santa Cruz Biotechnology), Mitoprofile Total® OXPHOS complex Rodent WB antibody (1:2500, Abcam)) for overnight and then secondary antibodies (1:2000 for HIF-1α; 1:5000 for MnSOD, TOM20 and OXPHOS complex, goat anti-rabbit or rabbit anti-mouse IgG horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology)) for 1 hour. Protein expression was detected by SuperSignal West Pico Chemiluminescent substrate (Thermo, MA, USA) by exposure of the membrane in FujiFilm (Fujifilm, Tokyo, Japan). Protein band density was determined using Image J software (National Institute of Health, Bethesda, Maryland, USA).
Immunohistochemistry
Formalin fixed brain samples were embedded in paraffin and sectioned (4 μm). Three coronal sections were used from SHAM and SE respectively. They were incubated with primary antibodies against nitrotyrosine (1:400 dilution, Upstate Biotechnology, Temecula, CA) followed by horseradish peroxidase anti-rabbit Envision system (Dako Cytochemistry, Tokyo, Japan). The sections were then counterstained with haematoxylin. Three images of cortex from each section were captured and used for analysis. Calculation of the proportion of area stained positive for nitrotyrosine was then determined using Image J software (National Institute of Health, Bethesda, Maryland, USA). To confirm the antibody specificity, anti-Nitrotyrosine antibody was pre-incubated with 10 mM Nitrotyrosine in PBS for 1 h at room temperature before incubation on the tissue. This yielded no staining (data not shown).
Statistical methods
Results are expressed as mean ± S.E.M. The difference between groups was analysed using unpaired Student t tests (Statistica 9, Statsoft, USA).
Results
Effects of cigarette smoke exposure on the dams
Body parameters
Both SHAM and SE dams had a similar body weight at baseline (17.8 ± 0.2 vs 17.7 ± 0.2 g, n = 10). Before mating, SHAM dams were significantly heavier than the SE dams (18.7 g ± 0.3 vs 16.8 g ± 0.2 g, P < 0.05). When pups were weaned at P20, SE dams (21.9 ± 0.2 g) were 12% lighter than the SHAM dams (24.6 ± 0.4 g, P < 0.01), who also had much higher circulating levels of cotinine, which is a metabolite of nicotine (96.5 ± 33.7 vs. 1.52 ± 0.40 ng/ml in the SHAM, P < 0.05).
Brain inflammatory markers
Brain IL-1β, IL-6 and toll like receptor (TLR)4 mRNA expression were significantly decreased in the SE dams compared with the SHAM dams (P < 0.05, Fig. 1a; P < 0.01, Fig. 2c,e, respectively, n = 6 − 8). The expression of IL-1R and TNF-α mRNA were not different between the groups (Fig. 1).

Brain mRNA expression of inflammatory markers in the SHAM and SE dams (n = 8).
Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. *P < 0.05; **P < 0.01. SE: smoke exposed.

Brain protein levels of MnSOD (a), TOM20 (b) and OXPHOS complexes (CI, CII, CIII, CIV and CV) (c) in the SHAM and SE dams.
Whole gel images of (a–c) in Supplementary Fig. 1. Immunohistochemistry for cortex nitrotyrosine in the dams (d) Scale bar = 50 μm (n = 4). Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. *P < 0.05; **P < 0.01. MnSOD: manganese superoxide dismutase; OXPHOS: oxidative phosphorylation; SE: smoke exposed; TOM20: translocase of the mitochondrial outer membrane.
Oxidative stress markers in the brain mitochondria
Brain mitochondrial MnSOD protein was reduced in the SE mothers (P < 0.01, Fig. 2a, n = 6). TOM20 protein was not different following SE. The protein levels of OXPHOS complexes CI, CIII and CV were significantly higher in the SE mothers compared to SHAM. Brain mitochondrial levels of CII and CIV were very low compared with other members of OXPHOS complexes in both SHAM and SE mothers (Fig. 2c). There was only minimal staining for nitrotyrosine in brains from SHAM mothers and the amount and intensity of staining was greater in the SE mothers. We measured the proportion of area stained positive for nitrotyrosine and this was significantly higher in the SE group (P < 0.01, Fig. 2d). Negative IgG was performed to confirm staining specificity.
Brain hypoxia markers
HIF-1α protein was reduced by 20% (P = NS) in the brains from the SE mothers (Fig. 3a); while its upstream regulator EGR-1 was simular between the groups (Fig. 3b).

Brain protein levels of HIF-1α (a) and EGR1 (b) in the SHAM and SE dams (n = 3).
Whole gel images of (a,b) in Supplementary Fig. 2. Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. EGR1: early growth response factor: HIF-1α: hypoxia-inducible factor; SE: smoke exposed.
Effects of maternal SE on male offspring
Growth
Body weights were not different between the SHAM and SE male offspring at P1 and P20 (Table 2). When the pups reached adulthood at week 13, SE offspring were significantly lighter than the SHAM offspring (P < 0.01, Table 2). Brain weights were smaller in the SE offspring at P1 and week 13 (P < 0.01), however the differences disappeared when expressed as a percentage of body weight (Table 2). Plasma cotinine levels in the SE offspring (7.60 ± 0.33 ng/ml) were double that of the SHAM offspring (3.07 ± 0.10 ng/ml, P < 0.01) at P20.
Brain inflammatory markers
Brain IL-1β mRNA expression was similar between groups at all ages (Fig. 4a–c). IL-1R mRNA expression was significantly increased in the SE offspring at all ages (Fig. 4d,e, P < 0.01; 4f, P < 0.05). IL-6 mRNA was upregulated in the SE offspring only at week 13 (Fig. 4i, P < 0.01). TNFα mRNA expression in the SE offspring was lower at P1 (Fig. 4j, P < 0.05), but not changed at P20 and week 13 (Fig. 4k,l) in comparison to SHAM controls. TLR-4 mRNA expression in the SE offspring was significantly decreased at P1 but increased at week 13 (Fig. 4m,j, P < 0.05) without any change at P20.

Brain mRNA expression of inflammatory markers in the offspring of SHAM and SE mothers at different ages (n = 8).
Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. *P < 0.05; **P < 0.01. SE: smoke exposed.
Oxidative stress markers in the brain mitochondria
At P1, mitochondrial protein levels of both MnSOD and TOM20 were similar between the SHAM and SE offspring (Fig. 5a,d). TOM20 protein was reduced at P20 in the SE offspring, but increased at week 13 (Fig. 6b, P < 0.05). MnSOD levels in SE offspring were reduced at week 13 (Fig. 5c,f, P < 0.05) compared to SHAM controls. OXPHOS complexes CI-V were not different between groups at P1 (Fig. 5f). At P20, brain OXPHOS CI and CV were significantly decreased in the SE offspring (Fig. 5g,P < 0.05); all the OXPHOS complexes CI–V were significantly increased in the SE offspring at week 13 (Fig. 5h,P < 0.01). Brain nitrotyrosine levels were increased in SE offspring at week 13 (Fig. 5d, P < 0.01).

Protein expression of MnSOD (a), TOM20 (b) and OXPHOS complexes (CI, CII, CIII, CIV and CV) (c) in the brain mitochondria in the offspring of SHAM and SE mothers at different ages (n = 3).
Whole gel images of (a–c) in Supplementary Fig. 3. Immunohistochemistry of cortex nitrotyrosine in week 13 offspring (d) Scale bar = 50 μm (n = 3). Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. *P < 0.05; **P < 0.01. MnSOD: manganese superoxide dismutase; OXPHOS: oxidative phosphorylation; SE: smoke exposed; TOM20: translocase of the mitochondrial outer membrane.

Brain protein levels of hypoxia markers in the offspring of SHAM and SE mothers at different ages (a–f) (n = 3).
Whole gel images of (a–c) in Supplementary Fig. 4. Results are expressed as mean ± S.E.M. Data were analysed by student’s unpaired t-test. **P < 0.01. EGR1: Early growth response factor; HIF-1α: hypoxia-inducible factor; SE: smoke exposed.
Brain hypoxia markers
HIF-1α protein was significantly increased at week 13 in the brains of SE offspring (P < 0.05, Fig. 6c); EGR-1 was significantly reduced at P1 (P < 0.01, Fig. 6d), while unchanged at P20 and week13 in the brains of SE offspring in comparison to the offspring from SHAM mothers (Fig. 6e,f).
Discussion
Smoking during pregnancy is considered a major and significant public health issue. A rodent model is commonly used to study the detrimental impact of maternal tobacco exposure on offspring19,33. Administration of nicotine alone is insufficient to reflect the complexity of the cigarette smoke which comprises approximately 3800 constituents33. Here, we have investigated the impact of maternal cigarette smoke exposure on brain inflammatory markers, oxidative stress related mitochondrial activity and markers of hypoxia in both dams and male offspring. There were similar brain changes in both SE mothers and offspring, with respect to reduced anti-oxidative capacity of the brain, which may reduce the ability of mitochondria to scavenge excess ROS generated during increased OXPHOS activity. This is reflected by increased nitrotyrosine levels, a direct product of oxidative stress, in the brains of SE mothers. However, SE mothers and adult SE offspring had quite distinct changes, in markers of brain inflammation and hypoxia, which were lower in the SE mothers, however higher in mature SE offspring. Increased brain oxidative stress and chronic inflammation are evident in certain neurodegenerative diseases, as neurons in the brain are highly sensitive to oxidative stress34,35,36,37. This raises the question of whether the offspring of smoking mothers may have a predisposition to neurodegeneration in adulthood.
Activation of TLRs stimulates the production of major inflammatory cytokines IL-1β and IL-6 in monocytes, which in turn enhances the expression of TLRs via a positive feedback loop38. In this study, TLR4 mRNA expression was downregulated in the SE mothers’ brains, which is consistent with the reduced expression of IL-1β and IL-6 mRNA we observed. The response of TLR4 expression to cigarette smoke has been found to differ between tissues. A thirty minute exposure to cigarette smoke increased TLR4 mRNA expression in gingival epithelial cells39; while TLR4 mRNA expression was reduced in human macrophages and primary monocytes after treatment with cigarette smoke extract40. However, cigarette smoking is often associated with increased inflammatory cytokines such as TNFα, IL-1β and IL-6 in the blood and organs, which are also regulated by EGR141. Acute nicotine administration can increase the expression of TNFα, IL-1β and IL-6 in rat brains42. Khanna and colleagues found that following 30 days of exposure to 4 cigarettes/day in rats, there was a significant increase in brain inflammatory cells43. The difference observed in markers of brain inflammation between the Khanna study and our study may be due to two reasons. Firstly 3R4F research grade cigarettes were used in Khanna’s study, which can contain different chemicals from the commercial cigarettes consumed by the humans in this study. Secondly, nicotine and cotinine clearance is known to increase during pregnancy, which may reduce the overall impact of cigarette smoke exposure44,45, although the change in nicotine metabolism during lactation is unclear. This may affect brain inflammatory response to nicotine and most importantly other chemicals in the cigarette smoke. In addition, in another study, three weeks of treatment with low dose nicotine (<0.5 cigarette/day) was able to reduce inflammatory gene expression in the rat brain46. In the current study, we found that the blood cotinine levels in SE mothers were equivalent to 1–2 cigarettes/day in humans47. Thus, our effect may be more comparable to the low-dose nicotine treatment previously demonstrated in the literature46, which is consistent with our observation of reduced brain expression of inflammatory markers in SE mothers. However, increased brain oxidative stress in the SE dams was also observed increased in the study by Khanna et al.43, suggesting cigarette smoke is a direct cause of oxidative stress regardless of the other responses.
Cotinine levels in P20 offspring in this study are similar to those reported in human infants of continuous smokers48, where chemicals in cigarettes were delivered through the breast milk. Interestingly, the changes of brain inflammatory markers in the SE offspring were somewhat different from their mothers. Only TLR4 mRNA expression at P1 was similar to the SE mothers, which is consistent with a previous study where reduced TLR4 in cord blood was observed in the neonates of smoking mothers39. However, TNF-α mRNA was reduced in P1 offspring. This suggests a differential impact of cigarette smoke exposure on mothers and chemicals delivered through the cord blood to their offspring in utero. This is not surprising as blood nicotine concentration in the fetus is normally higher than in the maternal blood. The different levels of nicotine and potentially other chemicals from cigarettes might be likely to account for the different inflammatory response observed in offspring versus the smoking mothers44,45. Although IL-1β mRNA levels were unchanged in the SE offspring, the persistent increase in IL-1R mRNA observed from birth to adulthood is likely to enhance the inflammatory activity of IL-1β. Surprisingly, at 13 weeks, expression of TLR4, IL-6 and IL-1R mRNA in brain were all increased in SE offspring, which is in contrast to pups at P1 and their SE mothers. This suggests a sustained effect of maternal cigarette smoking in the offspring to change brain inflammatory cytokine production. Microglial activation is known to be increased by low-dose cigarette exposure SE (plasma cotinine levels of 10 ng/ml) in mice49, which may be the reason for increased inflammatory cytokine expression that we observed in the P20 SE offspring. The increase in the inflammatory cytokines at 13 weeks may render them more susceptible to the development of neurodegenerative diseases. Neuroinflammation has been shown to plays a crucial role in the development of neurodegeneration. Rodent studies have shown that smoking can lead to pathological changes and accelerated progression of aging50,51. In murine cortical neurons, an increase in TLR4 can lead to β-amyloid-induced apoptosis through jun N-terminal kinase – and caspase-3-dependent mechanisms52. Injection of IL-1 into rat brain can lead to an elevation of β-amyloid53, which has been shown to play a role in Alzheimer’s Disease (AD). Overexpression of cytokines such as IL-6 can have a neurotoxic effect that leads to neurodegenerative disorders in some individuals54. The elevation of TLR4, IL-1R and IL-6 in adult SE offspring suggests that they might be more vulnerable to diseases such as AD. The incidence of AD is higher among smokers55, which may be transmitted to the offspring due to brain changes by intrauterine SE as we have shown here. However, post-injury functional recovery of neurons is also IL-6 dependent, due to its role in neuronal and glial regeneration56,57,58. In IL-6 knockout mice there was a 60% reduction in neuronal density and decreased sensory function after injury59. Therefore decreased IL-6 mRNA in SE dams might also indicate a compromised ability for recovery when brain injury occurs, which requires further investigation.
Although smoking itself is not considered to be able to cause hypoxia in the brain, maternal smoking is one of the risk factors for intrauterine hypoxia, which can lead to sudden infant death after birth60. This is mainly due to the restriction of placental blood flow caused by nicotine, which can reduce not only nutrients, but also oxygen supply to the growing foetus61. Under normoxic conditions, HIF-1α protein is tightly regulated by oxygen levels. It is maintained at a low level through continuous degradation by the ubiquitin-proteosome pathway62. However, long-term hypoxia and the activation of various signal transduction pathways can prevent HIF-1α degradation62. The expression of HIF-1α protein is organ specific under systemic hypoxia63, where HIF-1α binds to the promoter of TLR4 to upregulate TLR4 expression64. HIF-1α can also initiate various other hypoxia-inducible adaptations by regulating glycolysis, erythropoiesis, angiogenesis and cell proliferation65. Smoking itself has previously been shown to inhibit hypoxia-inducible adaptations in peripheral tissues66. Cigarette smoke exposure SE can also impair the production of HIF-1α as well as the stabilization of HIF-1α protein levels66. HIF-1 has been shown to have complex roles in the brain following injury and depending upon the stimulus and cell type being examined, can be neurotoxic or neuroprotective67,68. Hypoxia-induced angiogenesis is suggested to be inhibited in the smokers due to an impairment in the HIF-1 pathway66, thus smokers are more likely to suffer from more severe injury during stroke, with a worse prognosis compared with the non-smokers69. Here we only observed marginal reduction in brain HIF-1α protein in the SE mothers, which may be due to the low-dose and the relatively short exposure to cigarette smoke. EGR1 regulates the expression of HIF-1α during hypoxia70. Although EGR1 protein was not changed in the SE mothers, it was reduced in the newborn SE offspring. This may be due to a direct suppression by chemicals in the cigarette smoke inhaled by the mothers, which are at higher levels in newly born offspring compared with the mothers. It also needs to be noted that EGR1 is not the only regulator of HIF-1α, therefore the unchanged brain HIF-1α levels we observed in P1 and P20 offspring exposed to cigarette smoke SE may be due to the actions of other factors that regulate HIF-1 function, which is beyond the scope of this study. After birth, brain oxygen is replenished, while the impact of cigarette smoke components in the breast milk on HIF-1α levels also disappeared after weaning. This may lead to higher brain HIF-1α levels at adulthood. However, HIF-1α itself can induce inflammatory responses in the brain71 as we have observed in the adult SE offspring where TLR4 and TNFα are both upregulated. As brain EGR1 levels were unchanged at this age, it may not play a major role in increasing HIF-1α in the SE offspring. Considering the protective effect of HIF-1α, its increase in the brains of SE offspring may be an adaptation to protect against increased oxidative stress in the brain. It has been suggested that under normoxic conditions, increased oxidative stress due to excessive mitochondrial ROS production can increase HIF-1α protein levels72. This impact of oxidative stress is also seen in the brain of SE offspring here. Under basal conditions, 90% of ROS are produced in the mitochondria, mainly by OXPHOS complexes I and III in the electron transport chain73. Complex II is involved in the conversion of metabolic intermediates to complement the action of complexes I and III74. When the activities of both complexes I and III are inhibited, complex II will generate large amounts of superoxide75. Complex IV (known as cytochrome oxidase) is a crucial regulator for OXPHOS, the dysfunction of which leads to reduced ATP levels76; while Complex V converts ADP to ATP74. ROS generated during OXPHOS is both beneficial and detrimental to the cells77. It can activate the antioxidant defence network to prevent damage to the host itself. Thus, ROS are tightly regulated by antioxidant enzymes such as MnSOD78. In the SE offspring, MnSOD was unchanged at P1 and P20, possibly due to the protective effect of the antioxidant-rich breast milk79. Changes in mitochondrial OXPHOS complexes in brains of SE offspring mirror the changes of TOM20 levels, both at weaning and in adulthood. TOM20 imports protein into the mitochondria from the outer mitochondrial membrane80, reflecting changes in energy needs by the mitochondria and body. Reduced OXPHOS complex and TOM20 levels at P20 may be due to a redistribution of nutrients after birth required for the catch-up growth of the other organ systems commonly seen in offspring from smokers. Similar to their mothers, brain mitochondrial complexes I–V and TOM20 in the SE offspring were all increased at 13 weeks, suggesting increased substrate metabolism, which can result in increased ROS production. Conversely, mitochondrial MnSOD levels are low and may not be sufficient to clear excess ROS, resulting in oxidative stress and related tissue damage. Here, we have observed increased levels of nitrotyrosine protein in the brains of SE offspring at 13 weeks. Elevated nitrotyrosine levels are harmful to the brain and is one factor contributing to neurodegenerative diseases in humans81. However, the link between increased brain oxidative stress and any brain dysfunction in the SE offspring remains to be elucidated.
In conclusion, maternal cigarette SE differentially changed brain inflammatory and hypoxia response markers in the mother and offspring. However, oxidative stress and mitochondrial damage were changed in a similar manner in both SE mothers and their offspring, which may predispose them to neurodegeneration in later life.
Additional Information
How to cite this article: Chan, Y. L. et al. Impact of maternal cigarette smoke exposure on brain inflammation and oxidative stress in male mice offspring. Sci. Rep. 6, 25881; doi: 10.1038/srep25881 (2016).
References
World Health Organisation. Global status report on noncommunicable disease. Description of the global burden of NCDs, their risk factors and determinants. (2011) (Date of access: 01/04/2011). Available at http://whqlibdoc.who.int/publications/2011/9789240686458_eng.pdf?ua=1.
World Health Organisation. World health Organisation report on the Glocal tobacco Epidemic, 2011: Warning about the Dangers of Tobacco. (2011) (Date of access: 07/07/2011). Available at http://whqlibdoc.who.int/publications/2011/9789240687813_eng.pdf?ua=1.
Ng, S. P. & Zelikoff, J. T. Smoking during pregnancy: Subsequent effects on offspring immune competence and disease vulnerability in later life. Reprod Toxicol. 23, 428–437 (2007).
Al-Sahab, B., Saqib, M., Hauser, G. & Tamim, H. Prevalence of smoking during pregnancy and associated risk factors among Canadian women: a national survey. BMC Pregnancy Childbirth. 10, 24 (2010).
Ng, S. P. & Zelikoff, J. T. Smoking during pregnancy: subsequent effects on offspring immune competence and disease vulnerability in later life. Reprod Toxicol. 23, 428–437 (2007).
Chen, H. & Morris, M. J. Maternal smoking‒A contributor to the obesity epidemic? Obes Res Clin Pract. 1, 155–163 (2007).
Chen, H., Saad, S., Sandow, S. L. & Bertrand, P. P. Cigarette Smoking and Brain Regulation of Energy Homeostasis. Front Pharmacol. 3, e147 (2012).
Neuman, Å. et al. Maternal Smoking in Pregnancy and Asthma in Preschool Children: A Pooled Analysis of Eight Birth Cohorts. Am J Resp Crit Care. 186, 1037–1043 (2012).
Hernández-Martínez, C., Arija Val, V., Escribano Subías, J. & Canals Sans, J. A longitudinal study on the effects of maternal smoking and secondhand smoke exposure during pregnancy on neonatal neurobehavior. Early Hum Dev. 88, 403–408 (2012).
Bush, P. G. et al. Maternal Cigarette Smoking and Oxygen Diffusion Across the Placenta. Placenta. 21, 824–833 (2000).
Ganapathy, V., Prasad, P. D., Ganapathy, M. E. & Leibach, F. H. Drugs of abuse and placental transport. Adv Drug Deliver Rev. 38, 99–110 (1999).
Lieberman, E., Gremy, I., Lang, J. M. & Cohen, A. P. Low birthweight at term and the timing of fetal exposure to maternal smoking. Am J Public Health. 84, 1127–1131 (1994).
Abel, E. L. Smoking during pregnancy: a review of effects on growth and development of offspring. Hum Biol. 52, 593–625 (1980).
Hanson, M. A. & Gluckman, P. D. Developmental processes and the induction of cardiovascular function: conceptual aspects. J Physiol. 565, 27–34 (2005).
Godfrey, K. M. & Barker, D. J. P. Fetal programming and adult health. Public Health Nutr. 4, 611–624 (2001).
Gluckman, P. D., Cutfield, W., Hofman, P. & Hanson, M. A. The fetal, neonatal and infant environments-the long-term consequences for disease risk. Early Hum Dev. 81, 51–59 (2005).
Barker, D. J. The developmental origins of adult disease. J Am Coll Nutr. 23, 588S–595S (2004).
Barker, D. J. P. Developmental origins of adult health and disease. J Epidemiol Commun H. 58, 114–115 (2004).
Ekblad, M. et al. Maternal smoking during pregnancy and regional brain volumes in preterm infants. J Pediatr. 156, 185–190, e181 (2010).
Gill, R., Tsung, A. & Billiar, T. Linking oxidative stress to inflammation: Toll-like receptors. Free Radic Biol Med. 48, 1121–1132 (2010).
Andreyev, A. Y., Kushnareva, Y. E. & Starkov, A. A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc). 70, 200–214 (2005).
Allen, C. L. & Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 4, 461–470 (2009).
Cano, M. et al. Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radic Biol Med 69, 1–14 (2014).
van der Vaart, H., Postma, D. S., Timens, W. & Ten Hacken, N. H. T. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 59, 713–721 (2004).
Chan, P. H. et al. SOD-1 transgenic mice as a model for studies of neuroprotection in stroke and brain trauma. Ann NY Acad Sci. 738, 93–103 (1994).
Crack, P. J. & Taylor, J. M. Reactive oxygen species and the modulation of stroke. Free Radical Bio Med. 38, 1433–1444 (2005).
Kamel, H. & Iadecola, C. Brain-immune interactions and ischemic stroke: clinical implications. Arch Neurol. 69, 576–581 (2012).
Athanasios, V., Thomais, V. & Konstantinos, F. Tobacco smoke: involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int J Environ Res Pub Health. 6, 445–62 (2009).
Galdston, M., Levytska, V., Schwartz, M. S. & Magnusson, B. Ceruloplasmin. Increased serum concentration and impaired antioxidant activity in cigarette smokers and ability to prevent suppression of elastase inhibitory capacity of alpha 1-proteinase inhibitor. Am Rev Respir Dis. 129, 258–263 (1984).
Al-Odat, I. et al. The impact of maternal cigarette smoke exposure in a rodent model on renal development in the offspring. Plos One. 9, e103443 (2014).
Chen, H., Simar, D. & Morris, M. J. Maternal obesity impairs brain glucose metabolism and neural response to hyperglycemia in male rat offspring. J Neurochem. 129, 297–303 (2014).
Nguyen, L. T. et al. L-Carnitine reverses maternal cigarette smoke exposure-induced renal oxidative stress and mitochondrial dysfunction in mouse offspring. Am J Physiol Renal Physiol. 308, F689–696 (2015).
Seller, M. J. & Bnait, K. S. Effects of tobacco smoke inhalation on the developing mouse embryo and fetus. Reprod Toxicol. 9, 449–459 (1995).
Xie, A., Gao, J., Xu, L. & Meng, D. Shared Mechanisms of Neurodegeneration in Alzheimer’s Disease and Parkinson’s Disease. BioMed Res Int. 2014, 648740 (2014).
Carvalho, K. S. Mitochondrial dysfunction in demyelinating diseases. Semin Pediatr Neurol. 20, 194–201 (2013).
Urrutia, P. J., Mena, N. P. & Nunez, M. T. The interplay between iron accumulation, mitochondrial dysfunction and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 5, 38 (2014).
Michael, T. L. & Beal, M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 443, 787–795 (2006).
Imani Fooladi, A. A., Mousavi, S. F., Seghatoleslami, S., Yazdani, S. & Nourani, M. R. Toll-like receptors: role of inflammation and commensal bacteria. Inflamm Allergy Drug Targets. 10, 198–207 (2011).
Semlali, A., Witoled, C., Alanazi, M. & Rouabhia, M. Whole cigarette smoke increased the expression of TLRs, HBDs and proinflammory cytokines by human gingival epithelial cells through different signaling pathways. PloS one. 7, e52614 (2012).
Sarir, H. et al. Cigarette smoke regulates the expression of TLR4 and IL-8 production by human macrophages. J Inflamm (>Lond). 6, 12 (2009).
Dinarello, C. A. PRoinflammatory cytokines*. Chest. 118, 503–508 (2000).
Razani-Boroujerdi, S. et al. The role of IL-1beta in nicotine-induced immunosuppression and neuroimmune communication. J Neuroimmune Pharm. 6, 585–596 (2011).
Khanna, A., Guo, M., Mehra, M. & Royal Iii, W. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J Neuroimmunol. 254, 69–75 (2013).
Wickstrom, R. Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol. 5, 213–222 (2007).
Benowitz, N. L., Hukkanen, J. & Jacob, P., 3rd . Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 192, 29–60 (2009).
Singh, S. P. et al. Acute and Chronic Nicotine Exposures Modulate the Immune System through Different Pathways. Toxicol Appl Pharm. 164, 65–72 (2000).
Blackford, A. L. et al. Cotinine concentration in smokers from different countries: relationship with amount smoked and cigarette type. Cancer Epidemiol. Biomarkers Prev. 15, 1799–1804 (2006).
Luck, W. & Nau, H. Nicotine and cotinine concentrations in serum and urine of infants exposed via passive smoking or milk from smoking mothers. J Pediatr. 107, 816–820 (1985).
Moreno-Gonzalez, I., Estrada, L. D., Sanchez-Mejias, E. & Soto, C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 4, 1495 (2013).
Ho, Y. S. et al. Cigarette smoking accelerated brain aging and induced pre-Alzheimer-like neuropathology in rats. PloS one. 7, e36752 (2012).
Moreno-Gonzalez, I., Estrada, L. D., Sanchez-Mejias, E. & Soto, C. Smoking exacerbates amyloid pathology in a mouse model of Alzheimer’s disease. Nat Commun. 4, 1495 (2013).
Tang, S. C. et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 213, 114–121 (2008).
Sheng, J. G. et al. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 17, 761–766 (1996).
Mousa, A. & Bakhiet, M. Role of cytokine signaling during nervous system development. Int J Mol Sci. 14, 13931–13957 (2013).
Durazzo, T. C., Mattsson, N. & Weiner, M. W. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 10, S122–S145 (2014).
Vallieres, L., Campbell, I. L., Gage, F. H. & Sawchenko, P. E. Reduced hippocampal neurogenesis in adult transgenic mice with chronic astrocytic production of interleukin-6. J Neurosci. 22, 486–492 (2002).
Valerio, A. et al. Soluble Interleukin-6 (IL-6) Receptor/IL-6 Fusion Protein Enhances in vitro Differentiation of Purified Rat Oligodendroglial Lineage Cells. MMol Cell Neurosci. 21, 602–615 (2002).
Nakanishi, M. et al. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. 25, 649–658 (2007).
Zhong, J., Dietzel, I. D., Wahle, P., Kopf, M. & Heumann, R. Sensory impairments and delayed regeneration of sensory axons in interleukin-6-deficient mice. J Neurosci. 19, 4305–4313 (1999).
Hutter, D., Kingdom, J. & Jaeggi, E. Causes and Mechanisms of Intrauterine Hypoxia and Its Impact on the Fetal Cardiovascular System: A Review. Int J Pediatr. 2010, 9 (2010).
Mantzoros, C. S., Varvarigou, A., Kaklamani, V. G., Beratis, N. G. & Flier, J. S. Effect of birth weight and maternal smoking on cord blood leptin concentrations of full-term and preterm newborns. J Clin Endocrinol Metab. 82, 2856–2861 (1997).
Yoshikawa, T. et al. Up-regulation of hypoxia-inducible factor-1 alpha and VEGF mRNAs in peritoneal dissemination of patients with gastric cancer. Anticancer Res. 26, 3849–3853 (2006).
Stroka, D. M. et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 15, 2445–2453 (2001).
Kim, S. Y. et al. Hypoxic stress up-regulates the expression of Toll-like receptor 4 in macrophages via hypoxia-inducible factor. Immunology. 129, 516–524 (2010).
Imtiyaz, H. Z. & Simon, M. C. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol. 345, 105–120 (2010).
Michaud, S. E., Menard, C., Guy, L. G., Gennaro, G. & Rivard, A. Inhibition of hypoxia-induced angiogenesis by cigarette smoke exposure: impairment of the HIF-1alpha/VEGF pathway. FASEB J. 17, 1150–1152 (2003).
Chu, P. W. Y., Beart, P. M. & Jones, N. M. Preconditioning protects against oxidative injury involving hypoxia-inducible factor-1 and vascular endothelial growth factor in cultured astrocytes. Eur J Pharmacol. 633, 24–32 (2010).
Sharp, F. R. & Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 5, 437–448 (2004).
Shinton, R. & Beevers, G. Meta-analysis of relation between cigarette smoking and stroke. BMJ. 298, 789–794 (1989).
Sperandio, S. et al. The transcription factor Egr1 regulates the HIF-1alpha gene during hypoxia. Mol Carcinog. 48, 38–44 (2009).
Mojsilovic-Petrovic, J. et al. Hypoxia-inducible factor-1 (HIF-1) is involved in the regulation of hypoxia-stimulated expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in astrocytes. J Neuroinflammation. 4, 12 (2007).
Anavi, S., Hahn Obercyger, M., Madar, Z. & Tirosh, O. Mechanism for HIF-1 activation by cholesterol under normoxia: a redox signaling pathway for liver damage. Free Radic Biol Med. 71, 61–9 (2014).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 1757, 509–517 (2006).
Ricci, J. E., Waterhouse, N. & Green, D. R. Mitochondrial functions during cell death, a complex (I–V) dilemma. Cell Death Differ. 10, 488–492 (2003).
Quinlan, C. L. et al. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 287, 27255–27264 (2012).
Kadenbach, B., Huttemann, M., Arnold, S., Lee, I. & Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic Biol Med. 29, 211–221 (2000).
Sena, Laura A. & Chandel, Navdeep S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Molecular Cell. 48, 158–167 (2012).
Lejay, A. et al. Mitochondria: Mitochondrial participation in ischemia-reperfusion injury in skeletal muscle. Int J Biochem Cell Bio. 50C, 101–105 (2014).
Qstrea, E. M., Balun, J. E. & Winkler, R. 616 Serum levels of antioxidants in breastfed vs bottlefed infants: A possible role of breast milk in protecting infants against oxygen toxicity. Pediatr Res. 15, 543–543 (1981).
Yamamoto, H. et al. Dual role of the receptor Tom20 in specificity and efficiency of protein import into mitochondria. Proc Natl Acad Sci. 108, 91–96 (2011).
Butterfield, D. A., Reed, T. & Sultana, R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer’s disease. Free Radic Res. 45, 59–72 (2011).
Acknowledgements
This study was funded by postgraduate support and start-up support to Dr Hui Chen, from the Faculty of Science, University of Technology Sydney. There is no conflict of interest. We would like to thank the Renal Research Group at the Kolling Institute of Medical Research for using their laboratory and consumables, as well as Ms Sue Smith (Kolling Institute of Medical Research) for her support with the immunohistochemistry work.
Author information
Authors and Affiliations
Contributions
H.C., N.J. and S.S. designed the study. Y.L.C., S.S., A.A.Z. and I.A. performed all the experiments. Y.L.C., S.S., C.P., B.O., I.A., A.A.Z., N.J. and H.C. contributed to the writing of the main manuscript text and Y.L.C., S.S. and H.C. prepared Figures 1–6. Y.L.C. prepared Tables 1–2. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chan, Y., Saad, S., Pollock, C. et al. Impact of maternal cigarette smoke exposure on brain inflammation and oxidative stress in male mice offspring. Sci Rep 6, 25881 (2016). https://doi.org/10.1038/srep25881
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25881
This article is cited by
-
Maternal e-cigarette use can disrupt postnatal blood-brain barrier (BBB) integrity and deteriorates motor, learning and memory function: influence of sex and age
Fluids and Barriers of the CNS (2023)
-
Effects of maternal cigarette smoke exposure on the progression of nonalcoholic steatohepatitis in offspring mice
Toxicological Research (2023)
-
Nicotinic regulation of microglia: potential contributions to addiction
Journal of Neural Transmission (2023)
-
Offspring sex affects the susceptibility to maternal smoking-induced lung inflammation and the effect of maternal antioxidant supplementation in mice
Journal of Inflammation (2020)
-
Administration of molecular hydrogen during pregnancy improves behavioral abnormalities of offspring in a maternal immune activation model
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
