
Abstract
Mouse embryonic stem cells (ESCs) can differentiate into a wide range – and possibly all cell types in vitro, and thus provide an ideal platform to study systematically the action of transcription factors (TFs) in cell differentiation. Previously, we have generated and analyzed 137 TF-inducible mouse ESC lines. As an extension of this “NIA Mouse ESC Bank,” we generated and characterized 48 additional mouse ESC lines, in which single TFs in each line could be induced in a doxycycline-controllable manner. Together, with the previous ESC lines, the bank now comprises 185 TF-manipulable ESC lines (>10% of all mouse TFs). Global gene expression (transcriptome) profiling revealed that the induction of individual TFs in mouse ESCs for 48 hours shifts their transcriptomes toward specific differentiation fates (e.g., neural lineages by Myt1 Isl1, and St18; mesodermal lineages by Pitx1, Pitx2, Barhl2, and Lmx1a; white blood cells by Myb, Etv2, and Tbx6, and ovary by Pitx1, Pitx2, and Dmrtc2). These data also provide and lists of inferred target genes of each TF and possible functions of these TFs. The results demonstrate the utility of mouse ESC lines and their transcriptome data for understanding the mechanism of cell differentiation and the function of TFs.
Similar content being viewed by others


Introduction
Pluripotent stem cells, such as embryonic stem cells (ESCs) are able to differentiate into many different cell types in vitro1. Because the ESCs are also immortal and can maintain their pluripotency, they can be inexhaustible research tools for investigating cell differentiation processes. Previously, we had established a “NIA Mouse ESC Bank” of 137 ESC lines, each of which carries a transcription factor (TF) that can be induced in a doxycycline-controllable manner2,3. We also carried out global gene expression profiling of these ESC lines 48 hours after the induction of TFs and demonstrated that these transcriptome data indicate the direction of cell differentiation2,3. In particular, we have validated the cell differentiation into neural lineages, skeletal muscles, hepatocytes, and blood cells4.
To increase the number of manipulated TFs and the coverage of cell types, we have generated ESC lines with 48 new transgenic TFs. With a total of 185 TFs, the “NIA Mouse ESC Bank” covers about 10% of all TFs encoded in the mouse genome5. We have measured the global gene expression profiles of these new ESC lines 48 hours after overexpressing each TF and compared the changes to tissue-specific gene expression profiles and functionally annotated gene sets.
Results
Generation of TF-inducible mouse ESC lines
To generate TF-inducible ESC lines, we used the procedure reported previously2,3. The exogenous copy (transgene) of a TF integrated into the ubiquitously expressing Rosa26 locus2,6 is repressed by doxycycline (Dox), which is added to the culture medium., TF can then be activated by Dox removal (Fig. 1a). A Venus reporter included into the expression vector was used to visualize cells with the transgene induced (Fig. 1a,b). All tested clones showed at least 70% Venus-positive cells after the removal of Dox (Fig. 1b). The majority of the forty-eight new manipulated genes were TFs selected from a set of high-priority genes involved in important functions in mouse ES cells and their differentiation7.

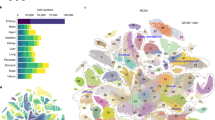
(a) Vector carrying a tetracycline-inducible (Tet-Off) transgenic TF was integrated into stably expressed Rosa26 locus in the genome. (b) Proportion of Venus-positive cells was evaluated by FACS (Dlx4 clone). (c) Scheme of experiment: To activate the transgenic TF, Dox was removed from the media. Forty-eight hours later, RNA was collected from manipulated cells, and gene expression was quantified with microarrays via comparison with control cells that were continuously cultured in Dox+ conditions. (d) Example of a scatterplot comparing gene expression profiles with or without Dox for Dlx2 induction. (e) Principal Component Analysis (PCA) of gene expression change in ES cells after induction of transcription factors; red – 48 new TFs analyzed in this paper; green – 137 TFs analyzed before3. Analysis is based on genes with significant change of expression (FDR ≤ 0.05, change ≥ 1.5 fold). (f) Number of genes with significant change of their expression after the induction of individual transcription factors.
To identify the effect of each TF on the transcriptome of ESCs, we used microarrays for gene expression profiling after 48 hours of culturing cells without Dox. Cells cultured in the presence of Dox were used as a control (Fig. 1c). The 48 hour time point was selected based on time-course experiments with multiple TFs2,8. This interval is sufficient to observe the change of expression in a large set of downstream genes, but short enough to observe a substantial enrichment of direct targets among responding genes. An example of a scatterplot with color-coded upregulated and downregulated genes after induction of Dlx2 is shown in Fig. 1d. Principal Component Analysis indicated that the new set of tested TFs has, in general, weaker effects on the ESC transcriptome as compared to such TFs as Gata2, Gata3, Cdx2, Nrip1, Dlx3, Ascl1, Gbx2, and Klf4 that were tested before (Fig. 1e).
The downstream effect of different TFs on gene expression was highly non-uniform, consistent with our previous studies2,3. Induction of 6 TFs (Barhl2, Dlx2, Myt1, Fezf2, Myb, and Pitx1) caused a substantial shift in the transcriptome: > 1,000 genes changed their expression (FDR ≤ 0.05, fold change ≥ 1.5) (Fig. 1f). By contrast, 16 TFs had relatively minimal effects resulting in a change of expression in < 50 genes. For most TFs, the number of upregulated genes was comparable to the number of downregulated genes. However, some TFs acted mostly as activators (Cdkn1a, Hoxb1, Foxn4, Foxh1, Sall2, Nr4a2, E2f1, Pax6, and Hoxb4) and other TFs acted as repressors (Prdm1, Cphx1, Dmrtc2, Esrrg, Pax3, and Tcf15).
Association of downstream genes of TFs with tissue-specific expression, gene ontology, and phenotypes
To explore the changes in the expression of downstream genes, we compared our microarray data with three databases: (1) GNF database ver. 3 on tissue-specific gene expression9,10; (2) Gene Ontology (GO) annotations11; and (3) Genetic Association Database (GAD) on gene sets associated with mouse phenotypes12. Because the GNF database is quantitative and the other two are qualitative, we used different methods to quantify association: (1) correlation of median-subtracted log-transformed gene expression values3, and (2) parametric analysis of gene set enrichment, PAGE13 (see Methods).
Comparison with the GNF database showed that the induction of individual TFs shifted the transcriptome toward specific differentiation fates. For example, gene expression change toward neural tissues was observed after induction of Myt1, St18, and Isl1; toward mesodermal lineages after induction of Pitx1, Pitx2, Barhl2, and Lmx1a; toward white blood cells after induction of Etv2, Myb, and Tbx6; and toward ovary after induction of Pitx1, Pitx2, and Dmrtc2 (Fig. 2). TFs associated positively with transcriptome changes toward specific lineages often showed a negative association with those toward different cell lineages. For example, effects of Myt1 correlated positively with neural tissues but negatively with blood lineages (Fig. 2). Validation of the cell-differentiation potential of each TF is beyond the scope of this paper because it requires longer experiments (6–14 days) and is specific for each cell lineage4. As an example, however, here we provide information on the capacity of three TFs (Myt1, Isl1, and St18) to facilitate ESC differentiation towards neural fate. ESC clones with transgenic TFs were cultured in Dox− and Dox+ medium (3 days in α MEM and then 3 days in NeuroCult), and then the proportion of cells with neural progenitor marker PSA-NCAM was quantified by FACS (Canto II, Becton Dickinson). Induction of two TFs, Myt1 and Isl1, (in Dox− condition) resulted in a substantial increase in the proportion of PSA-NCAM(+ ) cells as compared to control (Dox+ condition) (Fig. 3a,b), which confirms that these TFs facilitate neural differentiation. The effect of St18 induction was too weak to score positively; it was somewhat higher than in controls (Dox+ ) for the same clone, but did not differ from controls in other two clones.

Correlation of gene expression response to the induction of transcription factors (this study) with tissue-specific gene expression from the GNF ver. 3 database; color shows z-value for correlation significance, white = non-significant correlation (z < 2).

(a) Analysis of the proportion of cells with neural-progenitor marker PSA-NCAM after induction of three TFs (Myt1, Isl1, and St18) for 6 days (3 days in α MEM abd 3 days in neuroCult) by FACS analysis; TFs were induced in Dox− conditions, whereas cells cultured in Dox+ conditions were used as control. (b) Average proportion of PSA-NCAM-positive cells after induction of three TFs in three replications. (c) Rank plot analysis for enrichment of target genes bound by transcription factors (TFs) in promoters (0–0.5 Kb from TSS) or enhancers (0.5–50 Kb from TSS) among genes upregulated after induction of these TFs. Genes were sorted by expression changes and then the proportion of target genes was estimated in a sliding window of 300 genes. Statistical significance was evaluated using PAGE13.
Analysis of GO gene sets showed that Pitx1, Pitx2, and Barhl2 activated genes associated with collagen and skeleton; Myt1, Hoxc9, Fezf2, Glis2, and Esrrg activated synapse-related genes; St18, Isl1, Dlx2, Dlx4. Lhx8, and Lmx1a activated brain- and neuron-related genes; Sall2 activated voltage gated ion channel-related genes; Nkx2-3 and Nkx6-3 activated eye-related genes; Etv2 and Pdx1 activated angiogenesis-related genes; Tbx6 activated somitogenesis genes, Sry activated male sex determination genes, and Lin28 and Tcfap4 activated interferon-related genes (Table 1, Supplementary Table S1). Some of these associations (e.g., for Etv2, Hoxc9, Nkx2-3, Pitx1, Sry, and Tbx6) were the strongest among all 185 tested TFs. Additional information on the function of manipulated TFs was revealed via analysis of gene sets associated with mammalian phenotypes (GAD database). Pitx1, Pitx2, Barhl2, Foxn4, and Hoxb1 activated skeleton-related genes; Msx1 activated muscle and synapse-related genes; Tbx6 activated chorion and heart-related genes; and Dlx2, Lhx8, and Pdx1 activated ear-related genes (Table 1, Supplementary Table S2).
Predicting direct targets regulated by TFs from gene expression change and TF binding
We tested whether genes upregulated after induction of TFs were enriched in the binding of TFs to promoters and/or enhancers, if such information on genome-wide binding (ChIP-seq) was available in the GEO database. Statistically significant enrichment (PAGE method) was detected for four TFs: Etv2, Pitx1, Isl1, and Dlx2, out of ten tested TFs (Fig. 3c). We used ChIP-seq data from mouse Etv214, Pitx115, Isl116, and human DLX217, because there was no ChIP-seq data on mouse Dlx2. The other six TFs tested (Fezf2, Hoxc9, Msx1, Myb, Pitx2, and Prdm1) did not show significant enrichment.
Target genes regulated by Etv2, Pitx1, Isl1, and Dlx2 were identified using the method of Expected Proportion of False Positives (EPFP ≤ 0.5, change ≥ 1.5 fold) (Supplementary Table S3)18. The largest set of regulated targets (N = 190) was found for Etv2; it was enriched in angiogenesis-related genes (GO:0001525), heart tube development-related genes (GO:0035050), and embryonic hemopoiesis-related genes (GO:0035162) (Supplementary Table S3). Regulated targets of Dlx2 (N = 164) were enriched in genes associated with pituitary gland (GO:0021983), odontogenesis (GO:0042476), neurogenesis (for example, GO:0030182), and skeletal system (GO:0001501).
Discussion
Systematic induction of individual TFs in undifferentiated ESCs followed by global gene expression profiling yields a useful resource for cell and molecular biology. It can identify TFs functioning upstream of any given gene, predict functional roles of TFs in cell differentiation, and select genes for potential application in gene therapy and regenerative medicine2,3. Correlation matrices of gene expression profiles between TF-induced ESCs and various tissues/organs can also provide candidate TFs, whose overexpression can induce the differentiation of ESCs into specific cell types, as we have shown in a proof of concept4. Further mining of the microarray results reported here as well as additional experiments with the ES cell lines and their derivatives could yield further insight into gene regulatory networks.
Previously published research provides a positive control for our bioinformatics-based functional analysis of gene expression change after induction of 48 transgenic TFs. For example, functions of Myt1, St18, and Isl1 in neural tissues has been described19,20,21. TFs Pitx1 and Pitx2 are known to be involved in limb development22, consistent with our analysis of their downstream effects associated with mesoderm lineages. Roles of Etv2 in angiogenesis is consistent between our analysis and published research14,23. Association of Myb with thymocytes has also been described24.
By contrast, the effects of some TFs were not anticipated. For example, Barhl2 is known to function in the brain and spinal cord25,26, but in our data, the induction of Barhl2 in mouse ES cells gave non-neural effects similar to Pitx1 and Pitx2. As another example, Tbx6, which is known to determine neural and cardiac cell fate27, rather resulted in gene expression profiles trending toward macrophages (although GO annotations confirmed cardiac tendency as well). These discrepancies may point to additional unexplored functions of the TFs studied. Alternatively, or in addition, however, some effects observed in our experiments could be artifacts associated with the ectopic induction of TFs in the unusual context of ESC cultures in the medium employed. Thus, the unexpected results are both a caveat and a possible indication of new information.
Enrichment of TF binding in genes upregulated after the induction of Etv2, Pitx1, Isl1, and Dlx2 is in accord with the expectation that downstream effects of TFs are likely to be mediated by TF binding to promoters and enhancers of their targets, which is the primary mechanism of their regulatory function. However, we cannot rule out additional effects of TFs, such as binding to other signaling molecules, protein modification, remodeling of chromatin, or indirect effects caused by an initial rapid activation of another TF(s) followed by a cascade of further gene activation.
In general, the preliminary analyses reported here provide indications that the collection of mouse ES cell lines reported here can be a starting point for more extensive attempts to form lineages and even tissues in vitro. As an example, we confirmed the capacity of Myt1 and Isl1 to enhance neural differentiation of ESCs. All transgenic ESC lines are freely available to the research community as a resource. Similar experiments for more regulatory genes (ideally for all TFs, signaling proteins, and non-coding RNA) should give increasingly complete information about selective gene regulation in mammalian systems. The approach can be further expanded via altering culture conditions, possibly including growth factors, or even the activation of multiple TFs simultaneously.
Experimental Procedures
Cell culture and microarray hybridization
ESC lines carrying a tetracycline-regulatable TF were derived from MC1 (129.3) cell line, which was obtained from the expanded frozen stock at Johns Hopkins University, as described previously2,3. ESCs of passage 25 were cultured in the standard LIF+ medium with added Dox+ on a gelatin-coated dish through the experiments. Cells from each cell line were split into six wells and the media was changed 24 hours after cell plating: three wells with Dox+ medium, and three wells with Dox− medium to induce transgenic TFs. Dox was removed via washing three times with PBS at three-hour intervals. The proportion of Venus-p;ositive cells was evaluated by FACS (Canto II, Becton Dickinson). Total RNA was isolated by TRIzol (Invitrogen) after 48 hours, and two replications were used for microarray hybridization. RNA samples were labeled with total RNA by Low RNA Input Fluorescent Linear Amplification Kit (Agilent). We hybridized Cy3-CTP labeled sample from Dox− medium together with Cy5-CTP labeled sample from Dox+ medium (i.e., control) to the NIA Mouse 44K Microarray v3.0 (Agilent, design ID 015087)28. Slides were scanned with Agilent DNA Microarray Scanner. All DNA Microarray data are available in Table S2, at GEO/NCBI (http://www.ncbi.nlm.nih.gov/geo; GSE72350), and at NIA Array Analysis, http://lgsun.grc.nia.nih.gov/ANOVA29.
Neural differentiation of ESCs
For neural differentiation we used α MEM medium for 3 days followed by 3 days in the neuron-specific cell culture media: NeuroCult(TM) Differentiation Kit. NeuroCult(TM) NSC Basal Medium (Mouse), 450 mL (Catalog #05700), NeuroCult(TM) NSC Differentiation Supplements (Mouse), 50 mL (Catalog #05703). Differentiated cells were examined by flow cytometric analysis: after harvesting cells were stained with APC-conjugated PSA-NCAM antibody MoAb (Millteny Biotec) and then subjected to analysis by FACS Canto II (Becton Dickinson).
Statistical analysis of gene expression data
Microarray data was log-transformed (log10) and normalized by feature intensity in control cells (Dox+ , no induction of TFs): x′ i = xi−ci + Median(ci), where xi and ci are log-transformed feature intensities in Dox− and Dox+ , respectively, in the array i. To combine new data with previous microarray results, we used batch normalization based on the median expression value of each gene. For statistical analysis, we used ExAtlas, which estimates the False Discovery Rate (FDR), to account for multiple hypothesis testing30. The response of genes to the induction of TFs was measured as a logratio (i.e., difference between means of logtransformed intensities) between manipulated (Dox− ) and control (Dox+ ) cells. We considered gene expression change as significant if logratio was significantly different from zero (FDR ≤ 0.05) and the change of expression was ≥ 1.5 fold.
Correlation of gene expression changes induced by TF manipulation (i.e., logratio of Dox− vs. Dox+ ) versus tissue-specific gene expression in the GNF database (i.e., logratio of each tissue vs. median) was evaluated using ExAtlas30. The correlation analysis was done using 15,709 genes that were significant in both data sets. Criteria of significance for the GNF database were FDR ≤ 0.05 and change ≥ 2 fold, which is higher than the 1.5 fold threshold used for our data on TF manipulation because the magnitude of gene expression difference between adult tissues was much greater than the magnitude of gene expression change after the induction of TFs. It was sorted first with hierarchical clustering, and then sorted manually.
Comparison of gene expression changes induced by TF manipulation with functionally annotated gene sets (i.e., GO, GAD, and sets of TF targets) was done using Parametric Analysis of Gene set Enrichment, PAGE13, implemented in ExAtlas30. PAGE was applied separately to upregulated genes (25% top genes sorted by logratio of Dox− vs. Dox+ ) and downregulated genes (25% bottom genes sorted by logratio). Sets of genes bound by TFs were identified from published ChIP-seq data for Etv2 (GSM1436364, GSM1436365, GSM1436367, GSM1436367); Pitx1 (GSM1019784, GSM1019786); Isl1 (GSM782848, GSM928985, GSM928986); Fezf2 (GSM1135048-GSM113504); Hoxc9 (GSM766060, GSM766061); Msx1 (GSM657516); Myb (GSM912903); Pitx2 (GSM1162577); Prdm1 (GSM1616574, GSM1616575); and Dlx2 (GSM1208724). Peak coordinates were downloaded from the GEO database or from supplements to publications31,32. For some TFs, we filtered out peaks with low scores (< 100 for Hoxc9, <60 for Pitx1, <8 for Pitx2). If multiple samples were available, we used only matching peaks in at least three samples for Etv2 or two samples for other TFs. ChIP-seq peaks were annotated based on genomic coordinates of RefSeq and ENSEMBL transcripts downloaded from the UCSC database (http://genome.ucsc.edu). Transcripts were scored based on gene symbol (valid symbols were assigned a score of 3, whereas clones and predicted genes were assigned a score of 1) divided by distance from the peak to the transcription start site, TSS (distances < 1Kb were counted as 1Kb). Each peak was associated with one or two highest-score transcripts, and the second transcript was included if its score was >25% of the highest score. TF binding within 0.5 Kb from TSS was classified as a promoter, and binding within 0.5–50 Kb from TSS was classified as an enhancer.
Additional Information
How to cite this article: Yamamizu, K. et al. Generation and gene expression profiling of 48 transcription-factor-inducible mouse embryonic stem cell lines. Sci. Rep. 6, 25667; doi: 10.1038/srep25667 (2016).
Accession codes
References
Martello, G. & Smith, A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol 30, 647–675, doi: 10.1146/annurev-cellbio-100913-013116 (2014).
Nishiyama, A. et al. Uncovering early response of gene regulatory networks in ES cells by systematic induction of transcription factors. Cell Stem Cells 5, 420–433 (2009).
Correa-Cerro, L. S. et al. Generation of mouse ES cell lines engineered for the forced induction of transcription factors. Sci Rep 1, 167, doi: 10.1038/srep00167 (2012).
Yamamizu, K. et al. Identification of transcription factors for lineage-specific ESC differentiation. Stem Cell Reports 1, 545–559, doi: 10.1016/j.stemcr.2013.10.006 (2013).
Kanamori, M. et al. A genome-wide and nonredundant mouse transcription factor database. Biochem Biophys Res Commun 322, 787–793, doi: 10.1016/j.bbrc.2004.07.179 S0006-291X(04)01714-0 (2004).
Masui, S. et al. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res 33, e43, doi: 33/4/e4310.1093/nar/gni043 (2005).
Matoba, R. et al. Dissecting Oct3/4-regulated gene networks in embryonic stem cells by expression profiling. Plos One 1, e26, doi: 10.1371/journal.pone.0000026 (2006).
Nishiyama, A. et al. Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci Rep 3, 1390, doi: 10.1038/srep01390 (2013).
Wu, C. et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol 10, R130, doi: gb-2009-10-11-r13010.1186/gb-2009-10-11-r130 (2009).
Su, A. I. et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 99, 4465–4470, doi: 10.1073/pnas.012025199012025199 (2002).
Dimmer, E. C. et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res 40, D565–570, doi: gkr104810.1093/nar/gkr1048 (2011).
Zhang, Y. et al. Systematic analysis, comparison, and integration of disease based human genetic association data and mouse genetic phenotypic information. BMC Med Genomics 3, 1, doi: 1755-8794-3-110.1186/1755-8794-3-1 (2010).
Kim, S. Y. & Volsky, D. J. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics 6, 144, doi: 1471-2105-6-14410.1186/1471-2105-6-144 (2005).
Liu, F. et al. Induction of hematopoietic and endothelial cell program orchestrated by ETS transcription factor ER71/ETV2. EMBO Rep 16, 654–669, doi: 10.15252/embr.201439939 (2015).
Infante, C. R., Park, S., Mihala, A. G., Kingsley, D. M. & Menke, D. B. Pitx1 broadly associates with limb enhancers and is enriched on hindlimb cis-regulatory elements. Dev Biol 374, 234–244, doi: 10.1016/j.ydbio.2012.11.017 (2013).
Mazzoni, E. O. et al. Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat Neurosci 16, 1219–1227, doi: 10.1038/nn.3467 (2013).
Yan, J. et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154, 801–813, doi: 10.1016/j.cell.2013.07.034 (2013).
Sharov, A. A. et al. Chromatin properties of regulatory DNA probed by manipulation of transcription factors. J Comput Biol 21, 569–577, doi: 10.1089/cmb.2013.0126 (2014).
Cho, H. H. et al. Isl1 directly controls a cholinergic neuronal identity in the developing forebrain and spinal cord by forming cell type-specific complexes. Plos Genet 10, e1004280, doi: 10.1371/journal.pgen.1004280 (2014).
Matsushita, F., Kameyama, T., Kadokawa, Y. & Marunouchi, T. Spatiotemporal expression pattern of Myt/NZF family zinc finger transcription factors during mouse nervous system development. Dev Dyn 243, 588–600, doi: 10.1002/dvdy.24091 (2014).
Yokoyama, A. et al. Identification of myelin transcription factor 1 (MyT1) as a subunit of the neural cell type-specific lysine-specific demethylase 1 (LSD1) complex. J Biol Chem 289, 18152–18162, doi: 10.1074/jbc.M114.566448 (2014).
Marcil, A., Dumontier, E., Chamberland, M., Camper, S. A. & Drouin, J. Pitx1 and Pitx2 are required for development of hindlimb buds. Development 130, 45–55 (2003).
Craig, M. P. et al. Etv2 and fli1b function together as key regulators of vasculogenesis and angiogenesis. Arterioscler Thromb Vasc Biol 35, 865–876, doi: 10.1161/ATVBAHA.114.304768 (2015).
Hu, T., Simmons, A., Yuan, J., Bender, T. P. & Alberola-Ila, J. The transcription factor c-Myb primes CD4+ CD8+ immature thymocytes for selection into the iNKT lineage. Nat Immunol 11, 435–441, doi: 10.1038/ni.1865 (2010).
Ding, Q., Joshi, P. S., Xie, Z. H., Xiang, M. & Gan, L. BARHL2 transcription factor regulates the ipsilateral/contralateral subtype divergence in postmitotic dI1 neurons of the developing spinal cord. Proc Natl Acad Sci USA 109, 1566–1571, doi: 10.1073/pnas.1112392109 (2012).
Juraver-Geslin, H. A., Gomez-Skarmeta, J. L. & Durand, B. C. The conserved barH-like homeobox-2 gene barhl2 acts downstream of orthodentricle-2 and together with iroquois-3 in establishment of the caudal forebrain signaling center induced by Sonic Hedgehog. Dev Biol 396, 107–120, doi: 10.1016/j.ydbio.2014.09.027 (2014).
Gavrilov, S. et al. Tbx6 is a determinant of cardiac and neural cell fate decisions in multipotent P19CL6 cells. Differentiation 84, 176–184, doi: 10.1016/j.diff.2012.04.007 (2012).
Carter, M. G. et al. Transcript copy number estimation using a mouse whole-genome oligonucleotide microarray. Genome Biol 6, R61, doi: gb-2005-6-7-r6110.1186/gb-2005-6-7-r61 (2005).
Sharov, A. A., Dudekula, D. B. & Ko, M. S. A web-based tool for principal component and significance analysis of microarray data. Bioinformatics 21, 2548–2549 (2005).
Sharov, A. A., Schlessinger, D. & Ko, M. S. ExAtlas: An interactive online tool for meta-analysis of gene expression data. J Bioinform Comput Biol, 1550019, doi: 10.1142/S0219720015500195 (2015).
Mould, A. W., Morgan, M. A., Nelson, A. C., Bikoff, E. K. & Robertson, E. J. Blimp1/Prdm1 Functions in Opposition to Irf1 to Maintain Neonatal Tolerance during Postnatal Intestinal Maturation. Plos Genet 11, e1005375, doi: 10.1371/journal.pgen.1005375 (2015).
Tao, Y. et al. Pitx2, an atrial fibrillation predisposition gene, directly regulates ion transport and intercalated disc genes. Circ Cardiovasc Genet 7, 23–32, doi: 10.1161/CIRCGENETICS.113.000259 (2014).
Acknowledgements
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (ZIA AG000656-13). This work was also in part supported by the Keio University Medical Science Fund – The Mitsunada Sakaguchi Laboratory, the CREST program from the Japan Science and Technology Agency (JST), and the Research Center Network for Realization of Regenerative Medicine, Japan Agency for Medical Research and Development (AMED).
Author information
Authors and Affiliations
Contributions
A.N. and H.Y. constructed transgene vectors encoding various mouse transcription factors; K.Y. and M.A. established 48 TF-manipulable mouse ESC lines; Y.P. carried out microarray hybridizations; D.B.D. performed bioinformatics analyses; A.A.S. performed bioinformatic analyses of microarray data and wrote the manuscript; D.S. supervised the project; M.S.H.K. led the project, supervised experiments, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yamamizu, K., Sharov, A., Piao, Y. et al. Generation and gene expression profiling of 48 transcription-factor-inducible mouse embryonic stem cell lines. Sci Rep 6, 25667 (2016). https://doi.org/10.1038/srep25667
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25667
This article is cited by
-
Atlas of regulated target genes of transcription factors (ART-TF) in human ES cells
BMC Bioinformatics (2022)
-
Streamlined computational pipeline for genetic background characterization of genetically engineered mice based on next generation sequencing data
BMC Genomics (2019)
-
Induction of human pluripotent stem cells into kidney tissues by synthetic mRNAs encoding transcription factors
Scientific Reports (2019)
-
Rapid differentiation of human pluripotent stem cells into functional neurons by mRNAs encoding transcription factors
Scientific Reports (2017)
-
Design of a Lentiviral Vector for the Inducible Expression of MYC: A New Strategy for Construction Approach
Molecular Biotechnology (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
