
Abstract
Breast cancer is a heterogeneous disease, at both an inter- and intra-tumoural level. Appreciating heterogeneity through the application of biomarkers and molecular signatures adds complexity to tumour taxonomy but is key to personalising diagnosis, treatment and prognosis. The extent to which heterogeneity exists and its interpretation remains a challenge to pathologists. Using HER2 as an exemplar, we have developed a simple reproducible heterogeneity index. Cell-to-cell HER2 heterogeneity was extensive in a proportion of both reported ‘amplified’ and ‘non-amplified’ cases. The highest levels of heterogeneity objectively identified occurred in borderline categories and higher ratio non-amplified cases. A case with particularly striking heterogeneity was analysed further with an array of biomarkers in order to assign a molecular diagnosis. Broad biological complexity was evident. In essence, interpretation, depending on the area of tumour sampled, could have been one of three distinct phenotypes, each of which would infer different therapeutic interventions. Therefore, we recommend that heterogeneity is assessed and taken into account when determining treatment options.
Similar content being viewed by others

Introduction
Personalised medicine centers on the paradigm of inter-tumoural heterogeneity, by recognizing that each patient’s tumour is a unique disease entity with a definable molecular fingerprint. Increasingly, this concept of uniqueness is being extended to further incorporate those genotypic differences observed within regions of individual tumours and associated metastatic deposits, referred to as intra-tumoural heterogeneity1.
Intra-tumoural heterogeneity in breast cancer has been acknowledged for some time. Its existence may partially explain why breast cancer remains a challenging disease to treat, with significant morbidity and mortality, despite the use of well-established targeted therapies. In the UK, breast cancer is the third most common cause of cancer death and is accountable for 7% of all cancer-related mortality2. Quantifying the extent of intra-tumoural heterogeneity may improve both predictive and prognostic biomarker assays.
One such biomarker with reported heterogeneity in breast cancer is the Human Epidermal Growth Factor Receptor 2 (HER2), a member of the EGF receptor (EGFR) family. For invasive breast cancers that overexpress HER2 protein (reported range between 15–30%)3, trastuzumab offers a highly effective targeted therapy that can ameliorate the prognostic deficit inferred on patients with HER2 gene amplification. Treatment response rates, however, are variable. Optimal treatment requires that assay timings and techniques provide an accurate and true summary of an individual patient’s HER2 status. However, heterogeneous amplification of the HER2 gene is associated with cancer progression and reduced disease free survival4. Examples of discordant HER2 assay results between core biopsy material and resection specimens have been documented. Though, at least one meta-analysis (with data from 646 tumours) showed overall concordance of 97.8%5,6. If HER2 assessment in cores is generally representative of resections then does assessment of heterogeneity show any major differences between these two sample types?
Therefore, we sought to address two issues in this study. The first concerned reliable and reproducible quantification of HER2 heterogeneity in breast cancer. The second, whether HER2 heterogeneity is specific or a reflection of broader molecular heterogeneity.
We reviewed the extent of heterogeneity within tissue samples assessed for HER2 gene amplification in routine diagnostic practice within the Northern Ireland Molecular Pathology Laboratory (NIMPL). We retrospectively quantified heterogeneity within clinical samples using defined heterogeneity indices, correlated these with subjective assessments of cellular heterogeneity and compared heterogeneity across the different sampling methods of needle core biopsy (NCB) versus resection.
To address the second issue we assessed variation in molecular subtype in an index case with striking clonal heterogeneity. To illustrate this concept we analysed primary and metastatic lesions with an array of IHC biomarkers to classify molecular subtype, identify areas of diagnostic discrepancy and infer therapeutic interventions.
Results
A total of 140 cases were available for analysis. Of these, 83 had been clinically categorised as non-amplified (ratio <1.8), 50 as amplified (ratio >2.2), 5 as borderline amplified (ratio 2.0-to-2.2) and 2 as borderline non-amplified (ratio 1.8-to-2.0). The degree of cell HER2/Chr17 ratio dispersion per case across the entire cohort is presented in Fig. 1. Greater dispersion of ratio per case occurs generally as overall average ratio increases. However, HI (HI1 or HI2) defined heterogeneity is greatest in higher ratio non-amplified, borderline and lower ratio amplified cases. Highly amplified cases show the greatest ratio dispersion but are also defined by more uniform amplification. The lowest ratio non-amplified cases show very little ratio dispersion and uniform non-amplification.

Composite image showing cases arranged according to increasing HER2/Chr17 ratios.
The top graph shows the HER2/Chr17 ratio spread of individual cells per case. Non-amplified cases are shown red, borderline blue and amplified green. The bottom graph shows the H1 and H2 scores for the same samples. The samples names highlighted in red indicate cases previously assessed as showing heterogeneity by HER2 IHC.
Non-amplified cases
Sixty-one (73%) cases showed heterogeneity according to HI1. However, all of the non-amplified cases reviewed showed cell-to-cell heterogeneity according to HI2. Forty-four cases (53%) showed 5–50% of tumour cells with a HER2/Chr17 ratio >2.2. Maximum average HER2/Chr17 ratios were higher for cases showing any heterogeneity versus those with uniformly non-amplified cells (1.74 vs 1.2); minimum ratios were similar (0.8 vs 0.77). By the HI2 index 77 (93%) cases demonstrated at least 5% but fewer than 50% cells that were amplified.
Amplified cases
Thirty-seven (74%) cases showed heterogeneity according to HI1. Thirty-six cases had between 5–50% of tumour cells with a HER2/Chr17 ratio <1.8. Purely amplified cases (i.e.HI1 equivalent to 0) had higher maximum, minimum, mean and median HER2/Chr17 ratio compared to cases with any heterogeneity (10.45; 4.14; 6.68; 5.99 vs 7; 2.42; 3.62; 3.24). These cases had HER2/Chr17 ratios ranging from 4.14 to 10.45 compared to 2.42 to 7 for cases showing any heterogeneity. Forty-two (84%) cases showed heterogeneity according to HI2. The purely amplified cases had HER2/Chr17 ratios ranging from 5.2 to 10.5.
Borderline cases
As expected, reported borderline cases (n = 7) displayed high levels of heterogeneity relative to the non-amplified and amplified groups. HI1 indices ranged from 0.23 to 0.6, HI2 from 0.45 to 0.8.
Subjective versus objective heterogeneity
HER2 gene heterogeneity measurements are shown in Fig. 1 compared to subjective assessments of HER2 IHC heterogeneity as recorded during clinical assessment (cases highlighted in red). A total of 28 cases were previously assessed as showing heterogeneity. The majority of these cases clustered either side of ratios indeterminate HER2/Chr17 amplification. As shown, these tended to have higher HI1 and HI2 ratios.
Specimen type
Heterogeneity index values were compared according to sample type tested in order to determine if the nature of the specimen impacted on the degree of heterogeneity shown. After excluding equivocal cases and those where the nature of the sample was not recorded, HI1 and HI2 values were calculated depending on whether they were resection specimens or needle core biopsies (Fig. 2). No substantial differences in spread of data were identified between core and resection specimens.

Box and Whisker plots of average Her2/Chr17 rations between Needle Core Biopsy and Resection or Amplified, Borderline or Non-Amplified Cases.
Box and Whisker plot of HI1 or HI2 scores between Needle Core Biopsy and Resection.
Case example
Sections taken from the primary tumour displayed distinct well-segregated spatial morphological heterogeneity with H&E (Fig. 3). IHC heterogeneity largely conforming to these same areas was observed. Given that distinct clonal areas were readily identified, molecular subtypes could be assigned to adjacent areas in the primary tumour resection and lymph node metastasis.

The 3 distinctly heterogeneous areas are shown in columns A to C with their biomarker profiles beneath for ER, PR, HER2 and Ki67.
The primary tumour contained areas with differing HER2 expression. Moreover, there was evidence of luminal B HER2 positive (LBHP), HER2 enriched (HE) and luminal A (LA) subgroups in separate areas (Fig. 3). The HER2 enriched area harboured intratumoural DCIS with a LBHP phenotype while the LA area contained focal DCIS with a HER2 enriched phenotype. A high degree of heterogeneity was also observed with the additional biomarkers p53 and p-mTOR, EGFR and IGF1R (Supp Figure 1).
The lymph node contained 3 populations of metastatic cells also showing differing HER2 expression (Fig. 4). Two of these were well segregated. However, a third population of morphologically distinct malignant cells with a dispersed pattern was apparent. The lymph node therefore contained different molecular subtypes: LBHP, LBHN and triple negative basal phenotypes. Again varied expression of p53, p-mTOR, EGFR and IGF1R were noted (Supp Figure 2).

The metastatic deposit of tumour in the lymph node is shown.
Overall lymph node biomarker expression of ER, PR, HER2 and Ki67 are shown for each stain in column C. The interface between 2 heterogeneous metastatic areas is shown in column A. A third more dispersed, though morphologically distinct population is shown in column B.
The luminal B HER2 positive group was present in both primary and nodal metastatic tumours. The HER2 enriched phenotype was detected in DCIS and primary tumour but not the nodal deposit. The node contained a triple negative basal phenotype deposit that was not detected in the primary resection specimen.
Discussion
In this current study, using the proposed HI indices, we objectively scored and quantified heterogeneity in HER2 gene amplification. We identified a subset of patients with a sizable proportion of cells which amplification status differed to the overall assigned HER2 status. All non-amplified and most amplified cases showed heterogeneity according to the HI2 index, suggesting that this might be an overly sensitive measure. However, the HI1 index showed high ratio increases, corresponding to the greatest density of subjectively assessed heterogeneous cases. In the age of personalized medicine, the quantification of HER2 heterogeneity through the indices described may facilitate reliable heterogeneity assessment in routine practice to aid prediction, prognostication and future research. As illustrated by the index case, there are implications of HER2 heterogeneity more widely as an indicator of underlying different molecular phenotypes.
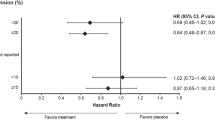
The question invariably asked is if a higher degree of cell-to-cell heterogeneity impacts upon the clinical outcomes of patients? Unfortunately clinical follow-up is limited in the patient cohort. It is intuitive, however, that the proportion of identifiable subdominant clones within a patient’s tissue sample may impact on treatment response and prognosis7. Indeed, a recent publication indicates a significant association with reduced disease-free survival4. Elegant in vivo models8,9,10,11 have demonstrated the importance of subdominant clones within tumours in providing key paracrine signals to influence multiple aspects of tumour biology. The nature of these tumour/subclone interactions are complex and far from being fully elucidated12. However, targeting these minor clones may be an alternate or even complimentary approach if a favorable dominant tumour-promoting microenvironment is indeed reliant on signals from subclones.
Non-amplified cases with a high heterogeneity index, could be candidates for retesting if recurrence or progression occurs as, theoretically, under-recognised previously non-dominant HER2 positive cells become the majority. Determining HER2 heterogeneity in non-amplified breast cores may also indicate regional heterogeneity that would necessitate retesting in resection specimens4. HER2 heterogeneity has been assessed in a number of studies using a range of techniques such as IHC and FISH. This is comprehensibly reviewed in Potts et al.13. In this study the authors develop a measure of HER2 IHC heterogeneity. Our study therefore represent a natural progression of this and we also note similar findings such as highest heterogeneity in cases initially identified as 2+ by IHC. However, we did not note high HI1 scores in cases where the HER2/Chr17 ratio was discordant between assessors. There are counter-arguments that HER2 heterogeneity has little prognostic significance. Patients with highly amplified tumours respond better to anthracycline therapy14 and apparent cell-to-cell heterogeneity may be an artefactual phenomenon15. In the HERA trial all patients received anthracycline therapy and those with borderline and amplified HER2 tumours had no significant differences in prognosis14. This may be due to interactions with targets of anthracyclines, such as TOP2a, that reside in close physical proximity to HER2 on the 17q12-q21 amplicon.
The detailed analysis of our index case reflects the broader molecular heterogeneity, beyond HER2, that may exist within certain breast cancers and metastases. Although an extreme example, it illustrates over the potential shortcomings of the methods used to classify molecular subtypes in breast cancer. The varying molecular profile observed would have consequences for therapy offered (trastuzimab, tamoxifen or neither), as well as outcomes in novel trials. Although most breast tumours do not display such extreme and clear-cut heterogeneous areas, individual tumour variation may occur at an undetectable level using current investigative approaches. The question concerning whether predictive and prognostic biomarkers should be tested on samples beyond the primary tumour is pertinent as exemplified by the differences between the molecular profiles of the primary and metastatic components of this case.
Conclusion
Researchers must be cognizant of biomarker variance in breast cancer specimens when transferring initial laboratory based results to clinical samples. Furthermore, ignoring the possibility of differential biomarker expression across lesional stages (differences in primary tumour and lymph node expression) negates a holistic approach to tumour characterisation and consequent treatment. In addition, these findings emphasize the possibility of erroneously basing treatment decisions on negative, or indeed positive, results obtained at core biopsy.
Materials and Methods
Ethical approval was obtained and tissue acquired through the Northern Ireland Biobank (NIB ref: 12–00017) and all work was carried out in accordance with the approved guidelines. Informed consent was obtained for all samples. For the analysis of HER2 genomic heterogeneity, 140 consecutive cases were reviewed from the NIMPL archives. Basic clinical information for these cases is shown in Table 1. These had been referred for HER2 status determination between October 2012 and September 2013 based on pathological interpretation in 3–4 hospitals served by the NIMPL. All of these cases had HER2 dual-color dual-hapten brightfield in situ hybridization (DDISH) applied and scored in keeping with UK recommendations16. Briefly, FFPE sections were stained with a HER2/CHR17 probe cocktail (Ventana cat no. 800 4422) as previously described17. Ninety-three of these were needle core biopsies, the remainder comprising larger resections. Manual enumeration of the HER2 and chromosome 17 signals was performed in a Clinical Pathological Accreditation (CPA) laboratory within areas of invasive tumour by two scorers (pathologist and clinical scientist) who analysed a total of 40 call nuclei in general (13 cases required additional assessment of 20 nuclei and 4 cases required an additional 40 nuclei to be assessed when discordant results impacted on clinical management). Scoring areas were selected on the basis of the IHC expression and focused on the focal 2+ regions within each sample. The case with striking heterogeneity was identified during routine reporting.
Heterogeneity Index Calculation
The College of American Pathologists (CAP) published a recommendation in 2009, as an extension of their 2007 scoring guidelines, that defines HER2 genetic heterogeneity as the presence of tumour cells with HER2/chromosome 17 (HER2/Chr17) signal ratios of >2.2 in 5–50% of tumour cells analysed in an otherwise non-amplified sample. If >50% cells had a ratio >2.2, this was termed amplified7. We modified this approach to measure heterogeneous cell populations in both non-amplified and amplified specimens: the proportion of cells within predominantly amplified samples that had HER2/Chr17 ratios <1.8 or the proportion of cells within predominantly non-amplified samples that had HER2/Chr17 ratios >2.2. We also applied this calculation to a small number of cases with equivocal HER2 status.
For each sample scored in NI-MPL within the defined time period, all of the archived DDISH HER2 counting tables used for diagnostic purposes were reviewed. Available data included the absolute numbers of HER2 and Chr17 signals per cell used in the analysis. Using these data, 2 different indices were calculated for quantification of heterogeneity.
Heterogeneity Index 1 (HI1)
For cases originally reported as non-amplified, the number of amplified cells was assessed as a measure of heterogeneity. However, cells with borderline values were excluded (ratios 1.8–2.2 inclusive). In each case, the sum of the cells with individual amplified status (based on HER2/Chr17 ratio >2.2) was divided by the total number of cells counted. This calculation provided the HI1 value. A similar principle was applied to originally reported amplified cases wherein the sum of individually non-amplified cells (based on HER2/Chr17 ratio <1.8) was divided by the total number of cells counted.
Heterogeneity Index 2 (HI2)
While using a similar approach to HI1, HI2 also considered individual cells with borderline status. For cases originally reported as non-amplified, borderline cells were considered together with amplified cells. Conversely, for cases originally reported as amplified, borderline cells were considered together with non-amplified cells.
Six cases originally classified as borderline were assessed in a similar manner as above depending on whether they were borderline amplified or borderline non-amplified. The approach to assigning heterogeneity indices is summarized in Fig. 5. The degree of data spread was plotted using the Tukey box plot method.

Summary of methods used to derive HI1 and HI2.
During routine assessment, it is standard practice within our institution to note subjectively the presence of heterogeneity. Heterogeneity indices were compared with cases noted subjectively to have displayed heterogeneity.
Immunohistochemistry
Additional IHC (ER, PR, HER2, Ki-67, p53, EGFR, p-mTOR and IGF1R) was performed on the case with striking heterogeneity. Sections for IHC were cut at 4 microns on a rotary microtome and dried at 37 °C overnight. All IHC was performed on automated immunostainers (Ventana Discovery or Leica BondMaX). Validated and optimised protocols were selected for each biomarker (see Table 2). Antigen binding sites were detected with Omni anti-rabbit or mouse detection system (Ventana cat no. 760–4310 or 760–4311) or a polymer based detection system (Bond cat no. DS 9800). All sections were visualized with DAB, counterstained in haematoxylin and mounted in DPX.
All IHC was interpreted by 2 pathologists (DB and MST). When disagreement on interpretation occurred, cases were reviewed by both pathologists on a multihead microscope to reach a consensus opinion. ER and PR were assessed using the quick score method which considers proportion and intensity of nuclear expression: scores ≤3/8 were considered positive (i.e. ≤1% cells expressing ER)18. HER2 analysis considered membranous expression and was based on the Bond Oracle Test method: scores of 3+ or 2+ with confirmatory ISH were considered positive19. As described elsewhere, p53 interpretation followed a 3 tier system where complete absence and confluent strong positivity were considered aberrant while all intermediary expression was considered non-aberrant17.
EGFR analysis considered membranous expression only. In keeping with the description of interpretation by Chaeng et al., any definite expression was considered as positive20. Ki67 nuclear expression was used to ascertain Ki67 index: a threshold of 14% expression was used to distinguish low and high proliferation indices21. IGF1R membranous expression was almost ubiquitous across all slides and cases and therefore only intensity (weak, moderate, strong) was considered in interpretation. Mild cytoplasmic p-mTOR expression in >5% of malignant cells was considered positive.
Molecular subtype classification of breast cancers using surrogate IHC
The IHC markers (ER, PR, HER2 and Ki67) were used to classify tumour areas as luminal A (LA: ER+, PgR+, HER2−, Ki67 low), luminal B HER2 negative (LBHN: ER+, HER2− and at least one of Ki67 high or PgR−), luminal B HER2 positive (LBHP: ER+, HER2+), HER2 enriched (HE: HER2+, ER− and PgR−) and triple-negative (TN: ER−, PgR−, HER2−)21.
We considered two types of heterogeneity in assessing the example case. The lesions of interest were primary invasive ductal carcinoma (IDC) and lymph node metastatic deposits (LNM). Distinctly separated differences in expression for a particular lesion type were termed spatial heterogeneity (SH). Differences between the lesion types were termed temporal heterogeneity (TH). The presence of heterogeneity was assessed for single biomarkers as well as panels conforming to molecular subtypes.
Additional Information
How to cite this article: Buckley, N. E. et al. Quantification of HER2 heterogeneity in breast cancer – implications for identification of sub-dominant clones for personalised treatment. Sci. Rep. 6, 23383; doi: 10.1038/srep23383 (2016).
References
Salto-Tellez, M. In Principles of molecular diagnostics and personalized cancer medicine (ed. Lynch, H. T. & Tan, D. ) Ch. 18, (Lippincott Williams and Wilkins, 2013).
CRUK. Breast cancer mortality statistics, http://www.cancerresearchuk.org/cancer-info/cancerstats/types/breast/mortality/ (2014), (Accessed: 07/10/2015).
Slamon, D. J. et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 235, 177–182 (1987).
Seol, H. et al. Intratumoral heterogeneity of HER2 gene amplification in breast cancer: its clinicopathological significance. Mod Pathol. 25, 938–948 (2012).
Dekker, T. J. et al. Reliability of core needle biopsy for determining ER and HER2 status in breast cancer. Ann Oncol. 24, 931–937 (2013).
Ricci, M. D. et al. Analysis of the concordance rates between core needle biopsy and surgical excision in patients with breast cancer. Rev Assoc Med Bras. 58, 532–536 (2012).
Vance, G. H. et al. Genetic heterogeneity in HER2 testing in breast cancer: panel summary and guidelines. Arch Pathol Lab Med. 133, 611–612 (2009).
Calbo, J. et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 19, 244–256 (2011).
Cleary, A. S., Leonard, T. L., Gestl, S. A. & Gunther, E. J. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 508, 113–117 (2014).
Marusyk, A. et al. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 514, 54–58 (2014).
Zhang, M. et al. Intratumoral heterogeneity in a Trp53-null mouse model of human breast cancer. Cancer Discov. 5, 520–533 (2015).
Tabassum, D. P. & Polyak, K. Tumorigenesis: it takes a village. Nat Rev Cancer. 15, 473–483 (2015).
Potts, S. J. et al. Evaluating tumor heterogeneity in immunohistochemistry-stained breast cancer tissue. Lab Invest. 92, 1342–1357 (2012).
Dowsett, M. et al. Disease-free survival according to degree of HER2 amplification for patients treated with adjuvant chemotherapy with or without 1 year of trastuzumab: the HERA Trial. J Clin Oncol. 27, 2962–2969 (2009).
Bartlett, A. I. et al. Heterogeneous HER2 gene amplification: impact on patient outcome and a clinically relevant definition. Am J Clin Pathol. 136, 266–274 (2011).
Bartlett, J. M. et al. HER2 testing in the UK: recommendations for breast and gastric in-situ hybridisation methods. J Clin Pathol. 64, 649–653 (2011).
Boyle, D. P. et al. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology. 65, 340–352 (2014).
Hammond, M. E. et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 28, 2784–2795 (2010).
Ellis, I, P. S. & Bobrow, L. Pathology reporting of breast disease: NHSBSP publication no 58, http://www.cancerscreening.nhs.uk/breastscreen/publications/nhsbsp58.html. (2005), (Accessed: 02/01/2012).
Cheang, M. C. et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 14, 1368–1376 (2008).
Goldhirsch, A. et al. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer. Ann Oncol. 24, 2206–2223 (2013).
Acknowledgements
This study was supported financially by Cancer Research UK, the Pathological Society, Breast Cancer Now and Queen’s University Belfast. The samples used in this research were received from the Northern Ireland Biobank which is funded by HSC Research and Development Division of the Public Health Agency in Northern Ireland and Cancer Research UK through the Belfast CR- UK Centre and the Northern Ireland Experimental Cancer Medicine Centre; additional support was received from the Friends of the Cancer Centre. The Northern Ireland Molecular Pathology Laboratory which is responsible for creating resources for the NIB has received funding from Cancer Research UK, the Friends of the Cancer Centre and the Sean Crummey Foundation.
Author information
Authors and Affiliations
Contributions
N.E.B. analysed data and prepared manuscript. D.P.B. identified clinical samples and performed microscopical assessments in heterogeneity cases. C.F. and D.Mc.A. analysed data. P.M. and J.A.J. contributed to preparation of manuscript. M.S.-T., P.M., S.Mc.Q. and J.A.J. scored all of the D.D.I.S.H. cases as part of routine clinical practice. S.Mc.Q. carried out I.H.C., analysed data and contributed to manuscript preparation. M.S.-T. designed the study and oversaw manuscript preparation.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Buckley, N., Forde, C., McArt, D. et al. Quantification of HER2 heterogeneity in breast cancer–implications for identification of sub-dominant clones for personalised treatment. Sci Rep 6, 23383 (2016). https://doi.org/10.1038/srep23383
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep23383
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
