
Abstract
Microbial secondary metabolites are valuable resources for novel drug discovery. In particular, actinomycetes expressed a range of antibiotics against a spectrum of bacteria. In genus level, strain Allosalinactinospora lopnorensis CA15-2T is the first new actinomycete isolated from the Lop Nor region, China. Antimicrobial assays revealed that the strain could inhibit the growth of certain types of bacteria, including Acinetobacter baumannii and Staphylococcus aureus, highlighting its clinical significance. Here we report the 5,894,259 base pairs genome of the strain, containing 5,662 predicted genes, and 832 of them cannot be detected by sequence similarity-based methods, suggesting the new species may carry a novel gene pool. Furthermore, our genome-mining investigation reveals that A. lopnorensis CA15-2T contains 17 gene clusters coding for known or novel secondary metabolites. Meanwhile, at least six secondary metabolites were disclosed from ethyl acetate (EA) extract of the fermentation broth of the strain by high-resolution UPLC-MS. Compared with reported clusters of other species, many new genes were found in clusters, and the physical chromosomal location and order of genes in the clusters are distinct. This study presents evidence in support of A. lopnorensis CA15-2T as a potent natural products source for drug discovery.
Similar content being viewed by others

Introduction
Natural products represent a leading source for drug candidates, especially in the anti-bacterial field1,2. Actinomycetes, regarded as a monophyletic branch of bacteria, are fungi-like bacteria forming long filaments; most importantly, these microbes tend to produce secondary metabolites such as polyketides and non-ribosomal peptides3,4. Many of these compounds have been successfully isolated and transformed into useful drugs, antibiotics, or chemotherapeutic agents5,6. Accordingly, there are more than 22,000 known microbial secondary metabolites, 70% of which are produced by actinomycetes, 20% from fungi, 7% from Bacillus spp. and 1–2% by other bacteria7.
There has been a large push towards research on the anti-bacterial mechanisms of these microbes, which can be traced to the discovery of streptomycin. Presently, researchers have been especially keen to explore new drugs by improving or activating the metabolites of microorganisms. Olano, et al.8 applied genetic approaches to bioactive secondary metabolites producing actinomycetes to improve the activity of relevant enzymes. Ito, T8 modified this method to improve efficacy and reduce the toxicity of actinomycete metabolites by creating new analogs using mutasynthesis. In addition, Yassin and Mankin found 77 new targets after random mutation in the RNA of a large ribosomal subunit9. Another difficulty for experimental work to resolve is that culture conditions can influence the expression of gene clusters in microbes.
At present, most genes and pathways involved in synthesizing various antibiotics have been identified, for example, the DOBISCUIT database collects around one hundred diverse antibiotics biosynthetic gene clusters. Moreover, Blin, et al. summarized previous findings on gene clusters related to different kinds of antibiotics and secondary metabolites, and automated the identification of the gene clusters encoding secondary metabolite production10. These two resources and alike facilitate comparison of potential antibiotics-producing gene clusters. However, “Me-Too” drugs may not help much11. The unremitting emergence of antibiotics-resistance also draws attention to new antibiotics source from exotic origins, such as mangrove12, and marine salterns7. Therefore, microbes from those environments may have great potential for novel antibiotics discovery7,13,14,15. It is particularly notable that microorganisms existing under extreme environmental conditions should exhibit distinctive features and abilities, and thus should be the most likely to yield unique and powerful bioactive compounds.
Schroeckh et al. found that Aspergillus nidulans, when co-cultured with bacteria, could produce several novel metabolites16. The capacity of secondary metabolic biosynthesis of microorganisms is always greater than that observed by fermentation4. Microbial genome analysis is an effective way of novel natural product producing gene discovery, due to clustering of biosynthetic genes of secondary metabolites e.g. polyketides, in genome. Beyond the known gene clusters, exploring the properties of the numerous orphan genes is regarded as the biggest challenge to finding new bioactive metabolites17. After genome sequencing, Udwary et al. found that approximately 9.9% of the marine actinomycete Salinispora tropica genome is related to natural product assembly, and 17 novel biosynthetic loci4. Hu et al. characterized two new cryptic sesquiterpene synthases in the Streptomyces clavuligerus genome by combining genetic and biochemical techniques18 while Soror et al. characterized an unusual member of the hormone-sensitive lipase family of esterases in Streptomyces coelicolor A3 (2)19. Lautru et al. found that in Streptomyces coelicolor M145 several gene clusters encoding new non-ribosomal peptide synthetase(NRPS) systems are not associated with known secondary metabolites; furthermore, they isolated and determined the structure of a new tris-hydroxamate tetrapeptide iron chelator coelichelin using a genome mining approach and guided by substrate predictions20.
To the best of our knowledge, strain Allosalinactinospora lopnorensis CA15-2T is the first new actinomycete in genus level found in the Lop Nor region of the Xinjiang province of China, which is famous for its high temperature, salinity and drought21. Antimicrobial assays revealed that the strain could inhibit the growth of certain types of bacteria, including Acinetobacter baumannii and Staphylococcus aureus, thus, using a genomics approach to unveil and explore the potential abilities of the type strain of the new species and new genus shall guide subsequent experiments for the discovery of novel natural bioactive compounds for helping human tackle bacterial infections.
Results
Culture of strain CA15-2T and its Antimicrobial Assay
Better growth of strain A.lopnorensis CA15-2T on R2A with 5% NaCl than without NaCl were observed at 28 °C pH 7.5 for 10 days (Figure S1) Antimicrobial assay shows that concentrated sample of ethyl acetate extract from the fermentation broth of strain A. lopnorensis CA15-2T exhibits different degrees of inhibitory activity against two fungi and eight bacteria, in particular Acinetobacter baumannii 2799 and Acinetobacter baumannii ATCC19606 (Fig. 1).

(A) Candida albicans CCTCC AY93025; (B) Acinetobacter baumannii 2799; (C) Acinetobacter baumannii ATCC 19606; (D) Escherichia coli ATCC 25922; (E) Escherichia coli ATCC 2800; (F) Klebsiella pneumonia ATCC 700603; (G) Klebsiella pneumonia ATCC 10031; (H) Staphylococcus aureus ATCC 25923; (I) Staphylococcus aureus 2641; (J) Cryptococcus laurentii CCTCC AY91013; (K) Enterococcus faecalis ATCC 29212; (L) Enterococcus faecalis ATCC 33186; (M) Pseudomonas aeruginosa 2774; N: Pseudomonas aeruginosa ATCC 27853.
Genome assembly and annotation
Deep sequencing based on enzymatic digestion DNA fragmentation, yielded 1,945,282 raw reads (average read length =137 bp). After removing short, low quality and suspected-plasmid reads, 260 contigs of more than 5.8 Mb were obtained. The average coverage was 45.1 fold and the G + C content was approximately 69.61%, consistent with the result (69.60%) obtained by reverse-phase HPLC22. Another round of sequencing with DNA fragmentation by sonication generated 3,317,091 raw reads (average read length =222 bp), which were subsequently assembled into 1,456 contigs. In order to obtain a better assembly result, we combined the sequencing reads of the two different DNA fragmentation methods, resulting in a total of 5,200,564 reads, or 1,002,084,924 base-pairs (average read length 179.43 bp). The combined reads were assembled into 233 contigs of 5,897,123 bp, and an average G + C content of 69.61% (Table 1). The raw data and the total shotgun assembly (TSA) were submitted and archived in the GenBank under the accession number LAJC00000000 and SRS881470, under the BioProject nr PRJNA278354 and biosample nr SAMN03418058. Detailed information on sequencing and assembly is shown in Table 1. Specifically, more than 5,000 ORFs were predicted, with 5,549 protein-coding genes subjected to further annotation analysis (Figure S2). A total of 4,717 putative protein-coding genes had homologs identified in the nr database, with 2,987 sequences assigned to 22 functional categories by egg NOG classifications (Figure S3, A–V). The majority of these protein sequences were found to involve, in general function terms, energy and transcription. Fifty-seven RNA coding sequences were detected, including 5S, 16S and 23S rRNAs and the remaining consists of tRNAs (Table 2). The functions of remaining 832 genes cannot be detected by similarity-based search methods, suggesting that A. lopnorensis CA15-2T may carry a novel gene pool.
Phylogenetic and comparative analysis
Phylogenetic results based on 16S rRNA showed that A. lopnorensis CA15-2T, together with Nocardiopsis alba ATCC BAA2165, Nocardiopsis dassonvillei DSM43111, and Thermobifida fusca YX, are located at the same branch of the tree (Figure S4), which suggests that these species should have a close phylogenetic relationship to other species on the tree. The phylogeny constructed by house-keeping genes is in accordance with the 16S rRNA gene sequences (Fig. 2). Orthology analysis showed that 4,378 ortholog families are shared between at least two of the genomes from N. alba ATCCBAA2165, N. dassonvillei DSM43111, T. fuscaYX or A. lopnorensis CA15-2T; 1,718 (27.6%) orthologs were shared by all four species (Fig. 3). Moreover, there were 570 (13%) families shared among all of the Nocardiopsis species, 127 (2.7%) families that only belonged to the strain A. lopnorensis CA15-2T. For those genes involved in the orthologous family, all four species had similar numbers (approximately 1,600) of single-copy orthologous genes (Fig. 4). In addition, 311 unique paralogous genes, belonging to 127 unique families, were found in A. lopnorensis CA15-2T, by contrast, N. dassonvillei has 128 unique paralogous genes, N. alba has 138 ones and T. fusca has only 26 unique paralogous genes, The larger amount of unique paralogous A. lopnorensis CA15-2T might associated with its unequalled features of adaptation to extreme environments and capacity of potential novel secondary metabolites biosynthesis.

Phylogenetic tree based on maximum likelihood method of a muti-alignment of ten housekeeping genes, respectively. The number of boostrap replication was set to 500.

Numbers indicate the number of gene families in each cluster. The Venn diagram was created with R software.

Cumulative histogram displaying the proportion of different types orthologs genes among N. alab, N. dassonvillei, T. fusca and A. lopnorensis CA15-2T.
Four genes encoding the enzymes for ectoine (1,4,5,6,tetra-2-methyl-4-pyrimidonecarboxylic acid) synthesis were identified. The four putative ectoine biosynthetic genes are located very close one after the other (Table 3) and the order of these four genes is highly similar to those of other halophiles of actinomycete, e.g. Streptomyces coelicolor A3 (2), Nocardia farcinica IFM10152 and T. fusca YX23, which provides evidence for the molecular basis of adaptation to a saline environment (Figure S1).
Antibiotics-producing Capability Assessment
Our antimicrobial assay revealed that A. lopnorensis CA15-2T inhibit the growth of specific bacteria, and gene cluster prediction also indicated that it may contain novel gene clusters for antibiotics synthesis. Here, a comparative analysis of A. lopnorensis CA15-2T with 10 diverse antibiotic-producing bacteria, as well as 10 non-antibiotic-producing bacteria was conducted to assess the capacity of producing antibiotics, and a new measurement Q was proposed, in which not only matched similarity (identity value) and length (alignment length) were taken into account, but also the gene amount was used for normalization, in terms of the higher the gene number, the greater the probability of gene sets of this species matching the sequences of gene clusters involved in antibiotic production. The results indicated that the majority of Q values of the antibiotic-producing group were markedly larger than those of non-antibiotics-producing group (Tables 4 and 5). It should be noted that some gene clusters of biosynthetic antibiotics (e.g. Avermectin, Myxothiazol) showed no obvious difference between the antibiotics-producing and non-antibiotic-producing groups merely based on a count of the hit number of gene/PKS genes during the blast search (Table S1,S2), whereas exhibited obvious differences in Q values (Tables 4 and 5). Furthermore, statistical test repeated measures ANOVA for the comparison of antibiotics-producing and non-antibiotic-producing groups showed that the difference in Q value between the antibiotics-producing and non-antibiotic-producing groups is statistically significant. Concretely speaking, multivariate test indicated that the selection of gene cluster did not affect the efficiency of Q-value (P value = 0.001, Table S3), and test of Between-Subjects Effects showed that Q value between the antibiotics-producing and non-antibiotic-producing groups is significantly different (P value ≤0.05, Table S4). On the other hand, twenty one bacteria including ten antibiotics-producing bacteria and ten non-antibiotic-producing bacteria as well as A. lopnorensis CA15-2T was classified using principal component analysis (PCA) on Q value, which illustrated that A. lopnorensis CA15-2T tend to cluster with antibiotics-producing group (Figure S5). Similarly, cluster and phylogenetic analysis of Q value revealed that the strain A. lopnorensis CA15-2T, and the antibiotics-producing species, were placed in the same group (Figure S6,S7), pointing toward the potential presence of the antibiotics-producing capacity in the strain A. lopnorensis CA15-2T.
Detection of gene clusters involved in Antibiotics & Secondary Metabolites
A total of 17 secondary metabolic biosynthesis gene clusters were identified in A. lopnorensis CA15-2T, which are predicted for polyketide, non-ribosomal peptide, siderophore, terpenoid and other products (Table 6). The combined length of these gene clusters was estimated at 412 kb. Specifically, the analysis shows that the genome of A. lopnorensis CA15-2T may contain at least six kinds of type I PKS gene clusters (Table 6), in which three core structures were identified using anti SMASH prediction. In the present study, the analysis revealed that four of the above PKS I gene clusters are likely to be involved in the biosynthesis of simocyclinone, apoptolidin, nigericin and oxazolomycin. The simocyclinone biosynthesis gene cluster of A. lopnorensis CA15-2T contains 35 ORFs (Fig. 5), five of which encode modular PKS. These modular PKSs contain 13 catalytic domains in which acyl-chain elongation involves acyl carrier protein (ACP), β-ketoacyl-ACP reductase (KR), β-ketoacyl-ACP synthase (KS), acyltransferase (AT) and dehydratase (DH). In addition, the simocyclinone cluster of A. lopnorensis CA15-2T is relatively similar to the existing simocyclinone cluster of Nocardiopsis dassonvillei subsp. dassonvillei DSM 43111 in aspects of amino acid sequence (Figure S8,S9), ORF order as well as composition, particularly with respect to several homologous putative proteins (white-striped arrows in Fig. 5) that may play a role in the synthesis of the simocyclinone-derived primer unit, which indicates that these clusters may be derived from donor microorganisms via horizontal gene transfer. Another type I PKS gene cluster may produce nigericin-like compound with high similarity to Ktedonobacter racemifer DSM 44963. The type I PKS cluster contains 19 ORFs, of which 5 encode modular PKS. Likewise, both clusters are composed of numerous hypothetical unknown function proteins (Fig. 6), and the majority of the key PKS genes share high amino acid similarity (Figure S10). Furthermore, an apoptolidin-like PKS I biosynthetic gene cluster was found in the A. lopnorensis CA15-2T genome. Comparison of both clusters indicates that apoptolidin-like cluster has several novel members that are not present in the Streptomyces bingchenggensis BCW-1 cluster (Fig. 7), including oxidoreductase, short-chain dehydrogenase and phosphopantetheine-binding domain-containing protein. However, the apoptolidin cluster of Streptomyces bingchenggensis BCW-1 has more polyketide synthase than the A. lopnorensis CA15-2T cluster. In addition to the abundance of the diverse PKS cluster, A. lopnorensis CA15-2T harbors a non-ribosomal peptide synthetase (NRPS) pathway. The NRPS cluster is composed of 36 ORFs, of which 8 encode key PKS modular with 13 domains. Compared with the most-similar known cluster of Streptomyces sp. W007 contig 00127, the cluster of A. lopnorensis CA15-2T has several unique ORFs including Hydrolase, Ketoacyl synthase and transcriptional regulators, which indicates that A. lopnorensis CA15-2T may have the potential of synthesizing an oxazolomycin-analog (Fig. 8).

The most similar gene cluster from Nocardiopsis dassonvillei is shown, with related genes depicted in the same color to highlight intracluster gene rearrangements. Predicted core structure displayed in the lower right corner.

The most similar gene cluster from Ktedonobacter racemifer is shown, with related genes depicted in the same color to highlight intracluster gene rearrangements. Predicted core structure displayed in the lower right corner.

The most similar gene cluster from Streptomyces bingchenggensis is shown, with related genes depicted in the same color to highlight intracluster gene rearrangements. Predicted core structure displayed in the lower right corner.

The most similar gene cluster from Streptomyces sp. W007 is shown, with related genes depicted in the same color to highlight intracluster gene rearrangements. Predicted core structure displayed in the lower right corner.
Secondary metabolites Identified Directly by Mass Spectrometric Analysis
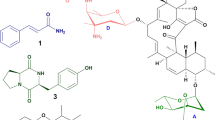
Mass spectra of the ethyl acetate (EA) extract of the fermentation broth of the strain CA15-2T generally exhibited a large number of discrete ions ranging from m/z 50 to 1200 both in positive and in negative ionisation modes. From UPLC-MS profiles, a total of 92 metabolic substances were identified by UNIFI workstation according to the mass-to-charge ratio of molecular ions and their corresponding fragment ions. Amongst them, six compounds, possessing antibacterial, antifungal or antitumor activity, were concerned (Fig. 9: compound 1–6). They affiliate to four groups: diketopiperazines (DKPs) group24 (compound 1–3), phenoxazine derivatives25 (compound 4), alpha-pyrone group26 (compound 5), and pyranonaphthoquinone (PNQ) group27 (compound 6). Concretely speaking, three diketopiperazines (DKPs) compounds (1–3) produced by a deep-sea-derived Nocardiopsis alba SCSIO 03039 formerly reported did not showed any antibacterial activities against four indicator strains Escherichia coli ATCC 25922, Staphylococcus aureus ATCC 29213, Bacillus subtilis SCSIO BS01 and Bacillus thuringiensis SCSIO BT0124. The antibacterial efficacy of Phenoxazine derivative (4) produced by a marine derived strain, Nocardiopsis sp. 236 against M. smegmatis was tested, yet it didn’t show any antibacterial activity28. Nocardiopyrones A (5) produced by Nocardiopsis alkaliphila, an alkalophilic actinomycete showed weak antibacterial activities against Pseudomonas aeruginosa, Enterobacter aerogenes and Escherichia coli with MIC values of 20, 40 and 40 μM and showed no activity against both Staphylococcus aureus and Candida albicans with more than 100 μM of MIC values26. Griseusins G (6) produced by Nocardiopsis sp. YIM DT266, also an alkalophilic actinomycete, showed strong inhibitory activity against Staphylococcus aureus ATCC 29213, Micrococcus luteus, and Bacillus subtilis with MIC values of 0.8, 1.47 and 1.33 μg/ml, respectively29. Antimicrobial results of the six known compounds identified by UPLC-MS revealed that only Griseusins G contributed inhibitory activity against Gram-positive bacteria, such as Staphylococcus aureus, but none showed any activities against Gram-negative bacteria, such as Escherichia coli, Klebsiella pneumonia, especially Acinetobacter baumannii, against which the biggest transparent inhibition zone was produced by the strain A. lopnorensis CA15-2T (Fig. 1B,C). The marked conflict between MS elucidation and antimicrobial assay implies the existence of novel antibiotic substance.

The chromatographic separation was performed on an ACQUITY UPLC HSS T3 column (2.1 × 100 mm). Detailed characterization of separated metabolites was executed with a QTof mass spectrometer outfitted with a high transmission efficiency collision cell (Xevo G2-XS QTof, Waters, Milford, MA). Two ESI ionization modes in MS analysis, positive (A) and negative (B), were carried out respectively to give the diverse molecular properties in the EA extract. Structures and mass-to-charge ratios* of the six antibiotics or antibiotic substances (compounds 1–6) show upon their specific retention times correspondingly. *For the six antibiotics or antibiotic substances: compounds 1–5 in Positive ionization mode (Nocardiopyrones A: [M + NH4]+; others: [M + H]+); compound 6 in Negative ionization mode (Griseusins G: [M-HCOO]−).
Materials and Methods
Strains and Growth Media
Strain A. lopnorensis CA15-2T was deposited in Institute of Medicinal Biotechnology, Chinese Academy of Medical Sciences & Peking Union Medical College and maintained on R2A (Difco, BD, USA) slants containing 5% (w/v) NaCl at 4 °C or as a 20% (v/v) glycerol suspension at −20 °C. Growth features of strain A. lopnorensis CA15-2T were evaluated on R2A medium with 5% NaCl and without NaCl at 28 °C, pH 7.5 for 10 days. Both the seed medium and fermentation broth was tryptic soy broth (TSB; BactoTM, BD, USA) supplemented with 5% (w/v) NaCl. Two fungi strain, Candida albicans CCTCC AY93025 and Cryptococcus laurentii CCTCC AY91013 were obtained from China Center for Type Culture Collection in Wuhan City, Hubei Province of China and were propagated in Sabouraud Dextrose Agar (SDA, Oxoid, UK) or Sabouraud Dextrose Broth (SDB, Oxoid, UK). Eight standard strains including Acinetobacter baumannii ATCC 19606, Escherichia coli ATCC 25922, Klebsiella pneumonia ATCC 10031 and ATCC 700603, Staphylococcus aureus ATCC 25923, Enterococcus faecalis ATCC 33186 and ATCC 29212, Pseudomonas aeruginosa ATCC 27853 were obtained from American Type Culture Collection (ATCC, Rockville, MD). Four drug-resistant strains: Acinetobacter baumannii 2799, Escherichia coli 2800, Staphylococcus aureus 2641 and Pseudomonas aeruginosa 2774 were obtained from Beijing Hospitals. Mueller-Hinton broth (MHB; Oxoid, UK) was used as the cultivation medium of all 12 bacteria strains.
Antimicrobial Assay
Antimicrobial assay were carried out by paper-disc agar diffusion method30. The stock culture of strain CA15-2T was inoculated into 250 mL Erlenmeyer flasks containing 50 mL of TSB (BD) supplemented with 5% (w/v) NaCl as seed culture medium, the seed culture was harvested after 5 days incubation at 37 °C on a rotary shaker at 180 rpm and then 50 mL of culture broth were transferred to 1 L of producing medium with the same ingredients as seed culture medium in 5 L Erlenmeyer flasks. Four sets of 5 L Erlenmeyer flasks were incubated in the same culture condition as the seed culture. Four liters of the fermentation broth were harvested after incubation for 10 days and further centrifuged at 8000 rpm for 10 min at room temperature. Four liters of supernatant were extracted with the same volume of ethyl acetate and the organic layer was further concentrated to dryness under reduced pressure, and then finally was dissolved in 2 mL methanol as sample for test. Forty microlitres sample were loaded on sterilized filter paper with a diameter of 6 mm. After dryness, filter papers were covered on the agar plate containing different fungi or bacteria as indicator strain and antimicrobial activities were observed and recorded after 24 hours incubation at 37 °C and the zones of inhibition were measured.
Mass Spectrometric Analysis
Concentrated sample of EA extracts was analyzed on ACQUITYTM Ultra Performance Liquid Chromatography system (Waters, USA). The chromatographic separation was performed on a ACQUITY UPLC HSS T3 column (2.1 × 100 mm I.D., 1.8 μm). The mobile phase consisted of water containing 0.1% formic acid (A) and acetonitrile (B). The gradient condition was 1–99% B at 0–10 min, 99% B at 10–11 min, 99-1% B at 11–13 min. The flow rate was 0.45 mL/min.
Detailed characterization of each peaks separated by UPLC was performed with a XevoTM G2-XS QTof (Waters, Manchester, UK), quadrupole and orthogonal acceleration time-of-flight tandem mass spectrometer. In negative ion mode capillary voltage was set to 2.5 kV, source temperature to 120 °C, desolvation temperature to 500 °C, and sample cone voltage to 40 V. In positive ion mode capillary voltage was set to 3.0 kV, source temperature to 120 °C, desolvation temperature to 500 °C, and sample cone voltage to 40 V. In both modes, mass spectra were acquired at a speed of 0.2 s/scan and the scanning delay of 0.01 s during analysis. In the MSE experiments, collision energy was set to scan between 6 eV and 15–45 eV. The analysis results were acquired with the lockspray to guarantee accuracy Leucine-enkephalin was used as the lockmass at a concentration of 400 ng/mL and flow rate of 10 μL/min. Data acquisition and analysis were controlled by Waters UNIFI V1.71 software. The scan rang in MS and MS/MS modes were over a range of 50–1200 m/z.
Genome Sequencing, Assembly and Annotation
The genomic DNA library was constructed using the Ion Xpress Plus Fragment Library Kit (Life Technologies) after enzymatic digestion. The library was sequenced using the Ion Torrent platform. Since DNA fragmentation bias can lead to uneven coverage, we constructed another library with the sonication fragmentation method that was also sequenced using the Ion Torrent platform. Genome assembly of the pooled sequencing reads was performed by the Newbler program, after removing short, low-quality reads. Glimmer (ver. 3.0)31 and Prodigal32 were used for gene prediction. The latter was used to minimize prediction errors by Glimmer for high GC prokaryotes33. The prediction results of longer gene length were adopted if the location sites of genes overlap by the two prediction methods. Transfer RNAs (tRNAs) were predicted by tRNAscan34, and ribosomal RNAs (rRNAs) by RNAmmer35. All the putative protein-coding genes were searched against the non-redundant protein sequence (NR) database of the National Center for Biotechnology Information (NCBI) using BLASTP36 for annotation. The functional category was assigned by searching against eggNOG 3.037.
Phylogeny and Genome Comparison
To demonstrate a phylogenetic evolutionary relationship with other species, the complete 16S rRNA gene sequence of A. lopnorensis CA15-2T was retrieved from the annotation results. Lin Guo et al. have demonstrated that this species is a new member of family Nocardiopsaceae based on phylogenetic analysis using 16S rRNA sequence22. In this study, we added nine affinitive species of A. lopnorensis CA15-2T which had been deposited in the NCBI database to further verify the branches. These species are Nocardiopsis alba ATCC BAA 2165 (Biosample accession: SAMN02603688), Nocardiopsis dassonvillei DSM43111 (Biosample accession: SAMN02598425), Thermobifida fuscaYX (Biosample accession: SAMN02598543), Nocardioides sp. JS614 uid58149 (Biosample accession: SAMN02598280), Streptomyces coelicolor A3 (2) uid57801 (Biosample accession: SAMEA1705940), Actinobacillus succinogenes 130Z uid58247 (Biosample accession: SAMN02598296), Actinoplanesmissouriensis431 uid158169, Actinoplanes sp. SE50 110 uid162333 (Biosample accession: SAMN02603081) and ActinosynnemamirumDSM 43827 uid58951 (Biosample accession: SAMN00001904). Sequence alignment was done using MUSCLE38. The aligned 16S rRNA gene sequences were used to reconstruct the phylogeny using Maximum Likelihood (ML) algorithm by MEGA39. For cases of multiple 16S rRNA gene copies in the genome, the longest sequence was chosen for phylogenetic analysis. In addition, 10 house-keeping genes (acsA, aroE, dnaE, guaA, gyrB, mutL, ppsA, pyrC, recA and rpoB)40 were concatenated to reconstruct the phylogeny for comparison with the one by 16S rRNA. Three species with A. lopnorensis CA15-2T in the same branch of the phylogenetic tree (Fig. 2) were used for further comparative analysis by the Ortho MCL software package41, which was used to identify ortholog, inparalog and co-ortholog groups. Finally, overlapping of the ortholgous families were visualized in a Venn diagram created by the R software package.
Identification and assessment of antibiotics-producing of gene clusters involved in secondary metabolites
The assembled contigs of A. lopnorensis CA15-2T were submitted to the anti SMASH2.0 server to search for potential secondary metabolic biosynthetic gene clusters10. Type I pks gene clusters, in which core structures were identified in anti SMASH detection were extracted for comparison with known gene clusters of other species using BLAST36. Multiple amino acid alignment of core proteins was performed by ClustalW42 and depicted by boxshade (http://www.ch.embnet.org/software/BOX_form.html). On the other hand, in order to assess the antibiotics-producing capacity of A. lopnorensis CA15-2T, 15 gene clusters involved in synthesizing the known diverse antibiotics were randomly extracted from the DOBISCUIT database43 (Table S5) to be compared with the genes of A. lopnorensis CA15-2T using BLAST, respectively. Furthermore, 10 gene sets from 10 known antibiotic-producing bacteria, which were picked up according to literature retrieval44,45,46,47,48,49,50 (Table S6), were also compared with the 15 gene clusters, respectively. Ten non-antibiotics-producing bacteria were retrieved from the NCBI database as the control group (Table S6), and the corresponding gene sets of these bacteria were also compared with the 15 gene clusters, respectively. It should be noted that none of research investigated which bacteria could not produce antibiotics, in such case; we select ‘specific’ bacteria as non-antibiotics-producing bacteria which might not to be expected to produce antibiotics, e.g. pathogenic bacteria, acid-tolerant, halo tolerant or heat-resistant bacteria. In order to evaluate the antibiotics-producing capacity of these ten antibiotic-producing bacteria and ten non-antibiotics-producing bacteria, as well as A. lopnorensis CA15-2T, index Q value was developed, in which the identity and alignment length were taken into account, and gene amount was used for normalization, defined as:

where Ij is the best-matched identity value to the blast result (jth species compared with the ith gene cluster), L is the best-matched alignment length of the blast result, and Ni is the gene number of the ith gene cluster and Pj is the gene amount of the jth species. The larger Q, the greater probability of the jth species contains the ith gene cluster. Moreover, Repeated Measures ANOVA test and Principal Component Analysis was used to test the significance of difference between antibiotics-producing group and non-antibiotics-producing group.
Conclusion
Combining genome-scale physiologic, genetic, and metabolic investigation approaches will expand our knowledge pertaining to the exploitation of novel natural active compounds, and may help us to establish strategies to develop these natural products for human use. In this study, we sequenced the genome of a novel actinobacteria strain, A. lopnorensis isolated from the rhizophere of Tamarisk in the Lop Nor region, an extreme environment with hot, drought, and high salinity conditions. From the secondary metabolites predicted with the assembled genome, we identified 17 candidate gene clusters, of which 4 are novel; the remaining 13 clusters refer to type I polyketide synthase (PKS I) systems, nonribosomal peptide synthetases (NRPS) and ectoine. Regarding the known gene clusters, the majority of the genes, and several new members, were found in clusters of the 13 detectable secondary metabolites. Some genes’ loci and directions changed significantly compared to those in reported clusters, and some new members joined these clusters. The findings reflect the flexibility and evolutionary diversity of clusters of secondary metabolites, and also bring new insights for future research on the secondary metabolites of actinomycetes.
Discussion
Bacteria are rich resources for the production of secondary metabolites, which represent a great diversity of economically important chemicals. Actinomycetes, one of the most-important bacterial species, have been, and continue to be, a leading source for new drug discovery1. In recent years, the majority of actinomycetes-based screening programs at large pharmaceutical companies encountered hurdles due to high costs and frequent re-discovery of known compounds, leading to the redundancy6. The discovery of new strains, from extreme environments, has become a hot topic because it provides opportunities for the discovery of novel chemical structures and scaffolds with new biological functions. In the present study, A. lopnorensis CA15-2T was isolated from the Lop Nor region. Lop Nor, which translates to “Lop Lake” in Mongolian, was a former salt lake, and is now completely dried-up. Few forms of life, except some special microbial populations, can exist in such extreme environment51,52. Culture experiment shows that A. lopnorensis CA15-2T grew well in the medium with 5% NaCl. These microorganisms have evolved into halophilic bacteria and archaea with the development of hypersaline habitats, e.g. evaporation of marine saltwater. Generally speaking, microorganisms of this group may accumulate highly water-soluble organic compounds, which are often named ‘compatible solutes’53, to maintain osmotic equilibrium with surrounding medium; thus, they can keep their cytoplasm and the cell’s interior remains basically unchanged. The majority of compatible solutes are the amino acid derivatives glycine-betaine and ectoine in halophilic bacteria52. In this study, ectoine biosynthetic gene cluster was detected using whole genome sequencing. The metabolic pathway of Ectoine had been worked out by previous studies54,55. Briefly, aspartate-semialdehyde is transaminated to 2,4-diaminobutyric acid (DABA) by catalyzing with DABA transaminase (EctB), and DABA is then transferred to Nγ-acetyl-L-2,4-diaminobutyric by DABA-Nγ-acetyltransferase (EctA). Finally, ectoine synthase (EctC) catalyzes the cyclic condensation of Ng-acetyl-L-2, 4-diaminobutyric acid, which leads to the formation of ectoine. Ectoine hydroxylase (EctD) can convert some of the ectoine to 5-hydroxyectoine under certain stress conditions (Figure S11). The identification of Ect ABCD genes based on deep sequencing facilitate us to understand the adaptation of A. lopnorensis CA15-2T to a salty environment.
Concerning antimicrobial experiments, A. lopnorensis CA15-2T showed a wide-ranging antimicrobial activity, in particular effectively inhibiting the growth of Acinetobacter baumannii, which provides clues to the possible presence of secondary metabolites as antibacterial substances. Acinetobacter baumannii belonging to Nonfermentative Gram-negative bacilli, has aroused wide concern in clinical and microbiology research, due to the spread of multidrug-resistant (MDR) bacillary strains among critically ill, hospitalized patients, sometimes in form of epidemics56,57. Multidrug-resistant Acinetobacter baumannii has been recognized to be among the most difficult to treat gram-negative bacilli58. Hence, discovery of novel antibiotics in A. lopnorensis CA15-2T undoubtedly brings new hope for fighting against Acinetobacter baumannii. Mass spectrometric analysis reveals 6 compounds with antifungal or antitumor activity. However, only Griseusins G among these compounds demonstrated inhibitory activity against Gram-positive bacteria (i.e., Staphylococcus aureus). None of them showed any activities against Gram-negative bacteria (i.e., Acinetobacter baumannii). In contrast to the antibiotic action merely against Gram-positive bacteria by known compound, findings from antimicrobial assay may infer a novel, yet antibiotic-related gene pool, and compounds encoded by them, in Allosalinactinospora lopnorensis CA15-2T. Normally, secondary metabolites of a microorganism are catalyzed by a series of enzyme-encoding genes that are usually organized and located in a specific section of the genome, also known as gene clusters. This feature allows us to identify potential biosynthetic clusters based on genome mining approach, e.g. basic homology sequence search BALST against existing gene clusters. To date, the genome-guided approach has been used in studies of microbial natural products4. Complete genome sequencing has provided unparalleled access to secondary metabolite producing genes, especially using high-throughput sequencing techniques, which have been widely used to successfully discover natural products in microorganisms for several years1,4. Furthermore, whole genome analysis in microorganisms is increasingly being performed by diverse research, clinical and public health laboratory groups59. In this study, the genome of A. lopnorensis CA15-2T was sequenced using the Ion Torrent PGM platform. Two different DNA fragmentation methods were used to construct a DNA library. Although high G + C content, multicopy segments in the genome and sequencing errors might cause frequent termination of contig assembly, which is not easily filled by conventional sequencing methods, two sequences could provide sufficient sequence and complementary information for further analysis. Through biosynthetic cluster detection, 17 known or predicted gene clusters were figured out, majority of clusters were involved in production of antibiotic substances, including several well-known compounds synthetic pathways like simocyclinone, apoptolidin and oxazolomycin. Concretely speaking, simocyclinone is active against gram-positive bacteria and some studies have showed that it may process distinct cytostatic activities against human tumor cell lines60,61. In addition, Ruth H. Flatman et al. demonstrated that it may inhibit an early step of the gyrase catalytic cycle by preventing binding of the enzyme to DNA62. Apoptolidin has been proved to be a selective cytotoxic macrolide compound63, which can be produced by the polyketide synthesis gene cluster of Streptomyces bingchenggensis BCW-1. Some studies showed that apoptolidin exhibit selective activity against cells, and it could induce cell death in malignant cell lines when applied together with LDH inhibitor oxamate64,65. Furthermore, oxazolomycin that might be synthesized by NRPS cluster in A. lopnorensis CA15-2T, has been proven to show diverse and important antibacterial, antitumor and anti-human immunodeficiency virus activity66.
Remarkably, the genes’ loci and directions changed, and some orphan genes were found in some of these clusters, which implies A. lopnorensis CA15-2T probably produces novel antibiotics. Given the transcriptional status and common co-existence of transcriptional regulators in gene clusters of secondary metabolites, the loci of genes might influence the final assembly of the secondary metabolic complex, because different transcription factors may regulate the expression level of genes. Likewise, the direction of genes may regulate expression in transcription levels. Moreover, metabolic substance’s gene cluster might be silenced by a specific regulation mechanism in genome67, and just expressed under particular conditions, which might be one of reasons that we could not identify the specific products from MS analysis. Another probably reason is that A. lopnorensis CA15-2T has the capacity of producing other classes of antimicrobial substances, such as antimicrobial peptides, which could not be detected by MS analysis. Actually, another major class of antimicrobial agents is composed of ribosomally synthesized and post-translationally modified peptides (RiPPs), which serve as a promising alternative to conventional antibiotics biosythesized by polyketides or non-ribosomal pathways68,69. In order to test this hypothesis, genome scanning was carried out using BAGEL3 server70. The results revealed that A. lopnorensis CA15-2T genome might encode two RiPPs: Linear azol(in)e-containing peptide (LAP) (Figure S12) and Lasso peptide (Figure S13). LAPs have been widely identified by mining publicly available genome sequences. According to previous studies68,69, the critical components in LAP biosynthesis are the inactive precursor peptide and the heterotrimeric synthetase complex comprised of a dehydrogenase and cyclodehydratase, all of which were identified in A. lopnorensis CA15-2T (Figure S12). In addition, some LAPs have been proven to exhibit antibiotic activity, e.g. Goadsproin produced by Streptomyces sp. TPA058469. Lasso peptides, the most extraordinary RiPPs, are regarded as promising antibacterial peptides due to their specific knotted structure71, the lasso fold that gives them stability against heat, chemical attack and proteases72. Previous studies69 on the biosynthesis of Lasso peptide have revealed that at least four encoding genes are essential for its formation. Three of them were detected in the genome of A. lopnorensis CA15-2T except for the gene encoding transporter (Figure S13), which may transport the lasso peptide out of cell. One possible reason is that incomplete genome assembly of A. lopnorensis CA15-2T leading to the interruption of gene cluster, or the gene of A. lopnorensis CA15-2T is so distinct with the existing ones that we could not identify it based on sequence similarity search. Majority of Lasso peptides are enzyme inhibitors or receptor antagonists, more importantly, some of them have been proven to possess antibacterial activity against either Gram-negative bacteria or Gram-positive bacteria69. This finding may provide the clue to interpret that A. lopnorensis CA15-2T has the capability of inhibiting against Acinetobacter baumannii. Certainly, further experimental investigation is requisite to validate the truth of these RiPPs in A. lopnorensis CA15-2T, e.g. proteomics approach. In conclusion, this study demonstrated that A. lopnorensis CA15-2T has enormous potential to antibiotic development. Despite efforts and progress made towards gene function identification in the field of analytical chemistry, it’s still not easy to connect the detected genotypes with chemotypes, indicating the identification of these metabolites remains a big challenge.
Additional Information
How to cite this article: Huang, C. et al. Genome-guided Investigation of Antibiotic Substances produced by Allosalinactinospora lopnorensis CA15-2T from Lop Nor region, China. Sci. Rep. 6, 20667; doi: 10.1038/srep20667 (2016).
References
McAlpine, J. B. et al. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J Nat Prod 68, 493–496 (2005).
Berdy, J. Bioactive microbial metabolites. J Antibiot (Tokyo) 58, 1–26 (2005).
Gontang, E. A., Gaudencio, S. P., Fenical, W. & Jensen, P. R. Sequence-based analysis of secondary-metabolite biosynthesis in marine actinobacteria. Appl Environ Microbiol 76, 2487–2499 (2010).
Udwary, D. W. et al. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci USA 104, 10376–10381 (2007).
Hong, K. et al. Actinomycetes for marine drug discovery isolated from mangrove soils and plants in China. Mar Drugs 7, 24–44 (2009).
Zotchev, S. B. Marine actinomycetes as an emerging resource for the drug development pipelines. J Biotechnol 158, 168–175 (2012).
Subramani, R. & Aalbersberg, W. Marine actinomycetes: an ongoing source of novel bioactive metabolites. Microbiol Res 167, 571–580 (2012).
Ito, T. [Biosynthetic study of actinomycetes-metabolites for creating novel analogs]. Yakugaku Zasshi 133, 1007–1015 (2013).
Yassin, A. & Mankin, A. S. Potential new antibiotic sites in the ribosome revealed by deleterious mutations in RNA of the large ribosomal subunit. J Biol Chem 282, 24329–24342 (2007).
Blin, K. et al. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41, W204–212 (2013).
Gagne, J. J. & Choudhry, N. K. How many “me-too” drugs is too many? Jama 305, 711–712 (2011).
Hong, K. [Actinomycetes from mangrove and their secondary metabolites]. Wei Sheng Wu Xue Bao 53, 1131–1141 (2013).
Shi, Y., Zhang, X. & Lou, K. [Endophyte microbial community in Achnatherum inebrians]. Wei Sheng Wu Xue Bao 52, 1297–1308 (2012).
Subramani, R., Kumar, R., Prasad, P., Aalbersberg, W. & Retheesh, S. T. Cytotoxic and antibacterial substances against multi-drug resistant pathogens from marine sponge symbiont: Citrinin, a secondary metabolite of Penicillium sp. Asian Pac J Trop Biomed 3, 291–296 (2013).
Ballav, S., Kerkar, S., Thomas, S. & Augustine, N. Halophilic and halotolerant actinomycetes from a marine saltern of Goa, India producing anti-bacterial metabolites. Journal of bioscience and bioengineering 119, 323–330 (2015).
Schroeckh, V. et al. Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA 106, 14558–14563 (2009).
Gross, H. Strategies to unravel the function of orphan biosynthesis pathways: recent examples and future prospects. Appl Microbiol Biotechnol 75, 267–277 (2007).
Hu, Y., Chou, W. K., Hopson, R. & Cane, D. E. Genome mining in Streptomyces clavuligerus: expression and biochemical characterization of two new cryptic sesquiterpene synthases. Chem Biol 18, 32–37 (2011).
Soror, S. H., Rao, R. & Cullum, J. Mining the genome sequence for novel enzyme activity: characterisation of an unusual member of the hormone-sensitive lipase family of esterases from the genome of Streptomyces coelicolor A3 (2). Protein Eng Des Sel 22, 333–339 (2009).
Lautru, S., Deeth, R. J., Bailey, L. M. & Challis, G. L. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat Chem Biol 1, 265–269 (2005).
Liu, B. et al. [Biodiversity and functional enzymes of cultured halophilic archaeon in Lop Nur region]. Wei Sheng Wu Xue Bao 51, 1222–1231 (2011).
Guo, L. et al. Allosalinactinospora lopnorensis gen. nov., sp. nov., a new member of the family Nocardiopsaceae isolated from soil. Int J Syst Evol Microbiol 65, 206–213 (2015).
Schwibbert, K. et al. A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581 T. Environ Microbiol 13, 1973–1994 (2011).
Zhang, Q. et al. New diketopiperazine derivatives from a deep-sea-derived Nocardiopsis alba SCSIO 03039. J Antibiot (Tokyo) 66, 31–36 (2013).
Imai, S., Shimazu, A., Furihata, K., Hayakawa, Y. & Seto, H. Isolation and structure of a new phenoxazine antibiotic, exfoliazone, produced by Streptomyces exfoliatus. J Antibiot (Tokyo) 43, 1606–1607 (1990).
Wang, Z. et al. New pyran-2-ones from alkalophilic actinomycete, Nocardiopsis alkaliphila sp. Nov. YIM-80379. Chem Biodivers 10, 281–287 (2013).
Brimble, M. A., Duncalf, L. J. & Nairn, M. R. Pyranonaphthoquinone antibiotics-isolation, structure and biological activity. Nat Prod Rep 16, 267–281 (1999).
Lu, C. H., Li, Y. Y., Wang, H. X., Wang, B. M. & Shen, Y. M. A new phenoxazine derivative isolated from marine sediment actinomycetes, Nocardiopsis sp. 236. Drug Discov Ther 7, 101–104 (2013).
Ding, Z. G. et al. Griseusins F and G, spiro-naphthoquinones from a tin mine tailings-derived alkalophilic Nocardiopsis species. J Nat Prod 75, 1994–1998 (2012).
Murray, P. R. et al. Manual of Clinical Microbiology (6th edn). Trends in Microbiology 3, 449–449 (1995).
Delcher, A. L., Harmon, D., Kasif, S., White, O. & Salzberg, S. L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27, 4636–4641 (1999).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC bioinformatics 11, 119 (2010).
Angelova, M. Computational Methods for Gene Finding in Prokaryotes. ICT Innovations 1857–7288 (2010).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35, 3100–3108 (2007).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology 215, 403–410 (1990).
Powell, S. et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res 40, D284–289 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32, 1792–1797 (2004).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution 28, 2731–2739 (2011).
Loper, J. E. et al. Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLoS genetics 8, e1002784 (2012).
Li, L., Stoeckert, C. J., Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–2189 (2003).
Thompson, J. D., Gibson, T. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Current protocols in bioinformatics, 2.3. 1–2.3. 22 (2002).
Ichikawa, N. et al. DoBISCUIT: a database of secondary metabolite biosynthetic gene clusters. Nucleic acids research 41, D408–414 (2013).
Watanabe, K., Okuda, T., Yokose, K., Furumai, T. & Maruyama, H. B. Actinosynnema mirum, a new producer of nocardicin antibiotics. J Antibiot (Tokyo) 36, 321–324 (1983).
Beck, Z. Q., Burr, D. A. & Sherman, D. H. Characterization of the beta-methylaspartate-alpha-decarboxylase (CrpG) from the cryptophycin biosynthetic pathway. Chembiochem: a European journal of chemical biology 8, 1373–1375 (2007).
Carter, G. T., Nietsche, J. A., Williams, D. R. & Borders, D. B. Citreamicins, novel antibiotics from Micromonospora citrea: isolation, characterization, and structure determination. J Antibiot (Tokyo) 43, 504–512 (1990).
Coutanceau, E. et al. Modulation of the host immune response by a transient intracellular stage of Mycobacterium ulcerans: the contribution of endogenous mycolactone toxin. Cellular microbiology 7, 1187–1196 (2005).
Magarvey, N. A. et al. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from cyanobionts. ACS chemical biology 1, 766–779 (2006).
Meier, J. L., Barrows-Yano, T., Foley, T. L., Wike, C. L. & Burkart, M. D. The unusual macrocycle forming thioesterase of mycolactone. Mol Biosyst 4, 663–671 (2008).
Stinear, T. P. et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci USA 101, 1345–1349 (2004).
Oren, A. Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol 28, 56–63 (2002).
Oren, A. Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Systems 4, 2 (2008).
Brown, A. D. Microbial water stress. Bacteriol Rev 40, 803–846 (1976).
Canovas, D., Vargas, C., Calderon, M. I., Ventosa, A. & Nieto, J. J. Characterization of the genes for the biosynthesis of the compatible solute ectoine in the moderately halophilic bacterium Halomonas elongata DSM 3043. Syst Appl Microbiol 21, 487–497 (1998).
Goller, K., Ofer, A. & Galinski, E. A. Construction and characterization of an NaCl-sensitive mutant of Halomonas elongata impaired in ectoine biosynthesis. FEMS Microbiol Lett 161, 293–300 (1998).
Dijkshoorn, L., Nemec, A. & Seifert, H. An increasing threat in hospitals: multidrug-resistant Acinetobacter baumannii. Nat Rev Microbiol 5, 939–951 (2007).
Maragakis, L. L. & Perl, T. M. Acinetobacter baumannii: epidemiology, antimicrobial resistance, and treatment options. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 46, 1254–1263 (2008).
Bergogne-Berezin, E. Treatment of Acinetobacter infections. Expert opinion on investigational drugs 6, 119–127 (1997).
Edwards, D. J. & Holt, K. E. Beginner’s guide to comparative bacterial genome analysis using next-generation sequence data. Microb Inform Exp 3, 2 (2013).
Trefzer, A. et al. Biosynthetic gene cluster of simocyclinone, a natural multihybrid antibiotic. Antimicrob Agents Chemother 46, 1174–1182 (2002).
Schimana, J., Walker, M., Zeeck, A. & Fiedler, P. Simocyclinones: diversity of metabolites is dependent on fermentation conditions. J Ind Microbiol Biotechnol 27, 144–148 (2001).
Flatman, R. H., Howells, A. J., Heide, L., Fiedler, H. P. & Maxwell, A. Simocyclinone D8, an inhibitor of DNA gyrase with a novel mode of action. Antimicrob Agents Chemother 49, 1093–1100 (2005).
Salomon, A. R., Voehringer, D. W., Herzenberg, L. A. & Khosla, C. Apoptolidin, a selective cytotoxic agent, is an inhibitor of F0F1-ATPase. Chem Biol 8, 71–80 (2001).
Diogo, C. V. et al. Berberine as a promising safe anti-cancer agent - is there a role for mitochondria? Current drug targets 12, 850–859 (2011).
Wender, P. A. & Longcore, K. E. Isolation, structure determination, and anti-cancer activity of apoptolidin D. Organic letters 9, 691–694 (2007).
Zhao, C. et al. Oxazolomycin biosynthesis in Streptomyces albus JA3453 featuring an “acyltransferase-less” type I polyketide synthase that incorporates two distinct extender units. J Biol Chem 285, 20097–20108 (2010).
Yamanaka, K. et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc Natl Acad Sci USA 111, 1957–1962 (2014).
Letzel, A. C., Pidot, S. J. & Hertweck, C. Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC genomics 15, 983 (2014).
Arnison, P. G. et al. Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Natural product reports 30, 108–160 (2013).
van Heel, A. J., de Jong, A., Montalban-Lopez, M., Kok, J. & Kuipers, O. P. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res 41, W448–453 (2013).
Maksimov, M. O., Pelczer, I. & Link, A. J. Precursor-centric genome-mining approach for lasso peptide discovery. Proc Natl Acad Sci USA 109, 15223–15228 (2012).
Maksimov, M. O. & Link, A. J. Prospecting genomes for lasso peptides. J Ind Microbiol Biotechnol 41, 333–344 (2014).
Acknowledgements
We greatly thank Xu Bai from Waters Corporation for his advices in MS analysis. This study was partly supported by grants from the Science and Technology Development Fund (FDCT) of Macao SAR (Ref. No. 134/2014/A3), Research Committee of University of Macau (MYRG139(Y1-L4)-ICMS12-LMY and MYRG2015-00214-ICMS-QRCM), and the Overseas and Hong Kong, Macau Young Scholars Collaborative Research Fund by the Natural National Science Foundation of China (Grant no. 81328025). Meanwhile, this study was partly supported by grants from National Natural Sciences Foundation of China (NSFC, Grant no. 81373308 and Grant no. 81321004).
Author information
Authors and Affiliations
Contributions
S.M.Y.L. and C.H.S. conceived and designed the study. L.T. and L.G. performed the experiments. C.H. and R.K.K.L. analyzed the data. C.H. and R.K.K.L. wrote the paper. S.M.Y.L., C.H.S., M.G., Y.W.W. and I.L. reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Huang, C., Leung, RK., Guo, M. et al. Genome-guided Investigation of Antibiotic Substances produced by Allosalinactinospora lopnorensis CA15-2T from Lop Nor region, China. Sci Rep 6, 20667 (2016). https://doi.org/10.1038/srep20667
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20667
This article is cited by
-
Biocontrol potential and antifungal mechanism of a novel Streptomyces sichuanensis against Fusarium oxysporum f. sp. cubense tropical race 4 in vitro and in vivo
Applied Microbiology and Biotechnology (2022)
-
Intertidal marine sediment harbours Actinobacteria with promising bioactive and biosynthetic potential
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
