
Abstract
De novo mutations that contribute to rare Mendelian diseases, including neurological disorders, have been recently identified. Whole-exome sequencing (WES) has become a powerful tool for the identification of inherited and de novo mutations in Mendelian diseases. Two important guidelines were recently published regarding the investigation of causality of sequence variant in human disease and the interpretation of novel variants identified in human genome sequences. In this study, a family with supposed movement disorders was sequenced via WES (including the proband and her unaffected parents) and a standard investigation and interpretation of the identified variants was performed according to the published guidelines. We identified a novel de novo mutation (c.2327C > T, p.P776L) in DYNC1H1 gene and confirmed that it was the causal variant. The phenotype of the affected twins included delayed motor milestones, pes cavus, lower limb weakness and atrophy and a waddling gait. Electromyographic (EMG) recordings revealed typical signs of chronic denervation. Our study demonstrates the power of WES to discover the de novo mutations associated with a neurological disease on the whole exome scale and guidelines to conduct WES studies and interpret of identified variants are a preferable option for the exploration of the pathogenesis of rare neurological disorders.
Similar content being viewed by others

Introduction
Recent advances in sequencing technologies and bioinformatics offer the opportunity to more easily identify the disease-causing mutations in rare Mendelian diseases. However, the contribution of genetic variants to sporadic disease remains largely unknown1,2. It is difficult for researchers to identify the causal gene in apparently sporadic conditions using traditional sequencing approaches, especially in small families3. Whole exome sequencing (WES), which enables researchers to interrogate all known protein-coding genes in a single experiment, facilitates the discovery of causal variants in rare diseases4. In addition, MacArthur et al.5 developed guidelines for the investigation causality of sequence variants causality in human disease. The American College of Medical Genetics and Genomics (ACMG) have also developed guidelines for the interpretation of sequence variants6.
Recently, many neurological diseases such as alternating childhood hemiplegia7, Kabuki syndrome8 and amyotrophic lateral sclerosis (ALS)9, have been identified that are caused by de novo mutations. In this study, we searched for the disease-causing variants by simultaneously sequencing the exome of the proband and her unaffected parents. In our study design and sequencing data analysis, we utilized the two recently published guidelines to investigate the causality of the sequence variants and to interpret the novel variants that were identified via the human genome sequencing. Consequently, we determined that a novel de novo missense mutation (c.2327C > T, p.P776L) in the DYNC1H1 gene was the disease-causing mutation. Our study demonstrated that WES is a preferable strategy for the detection of de novo mutations. The objective investigation and interpretation of sequence variants is an accessible means to molecularly diagnose Mendelian diseases.
Results
Clinical features
The proband and her twin sister, aged 24 years old, were born from an uneventful pregnancy. The twin sisters had mild foot deformities (pes cavus) at birth. Late infantile motor milestones were apparently delayed. The twins could walk independently at approximately 7 years of age but a waddling gait persisted thereafter. Then, the twins slowly developed progressive weakness and wasting of the lower extremity muscles from 9 to 14 years of age. According to the parents, no motor decline was apparent during the last years. A physical examination in our hospital revealed pes cavus (Fig. 1A), mild distal lower limb-dominant muscle atrophy and absent deep tendon reflexes. Muscle strength and tendon reflexes were preserved in the upper limbs. No fasciculations or other neurological deficits were observed. No sensory disturbances or ataxia were recognized. The twins demonstrated normal intellectual development according to measurements performed by the Mental Health Center of Xiangya Hospital.

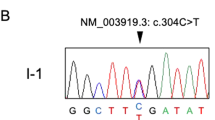
Clinical features of the patients and the pedigree with the c.2327C> T, p.P776L mutation in DYNC1H1.
(A) Foot deformities (pes cavus) of the two patients. (B) Pedigree structure of the studied family. In the family, WES was performed in I:1, I:2 and II:3. (C) Electropherograms of the Sanger sequences of the DYNC1H1 c.2327C > T, p.P776L variant; II: 2 and II: 3 are heterozygous mutations. (D) The conservation of this variant among different species.
Clinical laboratory evaluations revealed normal blood counts and creatine kinase levels and slightly increased lactic acid levels (3.2 mmol/L before exercise and 3.5 mmol/L after exercise; normal range 1.42–1.90 mmol/L). Non-contrast brain magnetic resonance imaging scans were also normal. Sensory and motor nerve conduction studies revealed normal conduction velocities and amplitudes for the upper and lower extremities. Electromyographic (EMG) recordings revealed a small amount of denervated potentials, which indicated anterior horn involvement in the lower limbs. In addition, large-amplitude and long-duration motor unit potentials were observed in the lower limbs. Neurogenic recruitment patterns were present in all leg muscles. All features were consistent with typical chronic denervation.
Genetic findings
The average sequencing depth and depth distribution on target of the family are shown in Supplementary Fig. S1. To date, there have been 19 genes (AARS, ATP7A, BICD2, BSCL2, DCTN1, DYNC1H1, GARS, HINT1, HSJ1, HSPB1, HSPB3, HSPB8, IGHMBP2, MYH14, PLEKHG5, REEP1, SLC5A7, SETX and TRPV4) have been associated with lower limb weakness and wasting and foot deformity10,11. Using the Integrative Genomics Viewer v.2.3 (IGV, Supplementary Fig. S2), we checked the coverage of these genes manually. No exons or exon-intron regions were located in these genes with <10× coverage.
With the data analysis and variants filtering strategy described above, 5 genes withhomozygous variants (HSD17B7, MYH6, PCLO, DIXDC1 and BPTF), 1 gene with compound heterozygous variants (MUC19) and 4 genes (DYNC1H1, PHOX2B, AKAP12 and NEK5) with de novo variants were identified. Among them DYNC1H1 is a known causal gene of SMA-LED. Sanger sequencing was conducted to exclude the false positives in the WES. Finally, the de novo mutation (c.2327C > T, p.P776L in DYNC1H1) in the proband was confirmed by Sanger sequencing (Fig. 1C). This mutation was a true de novo mutation as it was absent in her parents. Interestingly, the mutation was present in her twin sister (another patient), but did not exist in her two unaffected siblings (Fig. 1C, Supplementary Table S1). It was not found in more than 60,000 people (including the Exome Aggregation Consortium database, 1000 genomes, dbSNP 141 and the NHLBI Exome Sequencing Project). According to ACMG guidelines, the de novo mutation (c.2327C > T, p.P776L in DYNC1H1) is categorized to be the disease “pathogenic variant” because it belongs to both PS2 (de novo in a patient with the disease and no family history) and PS4 (the prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls) in ACMG6. This variant is located in a highly conserved domain (Fig. 1D) and is “probably damaging” as predicted by PolyPhen2, Mutation Taster, SIFT and CADD (Table 12).
We also conducted Sanger sequencing of the other genes (HSD17B7, MYH6, PCLO, DIXDC1, BPTF, MUC19, PHOX2B, AKAP12 and NEK5). Another variant passed the co-segregation analysis, which was a splicing variant (Table 1; Supplementary Table S1) in HSD17B7, with a MAF score 0.0018 in dbSNP. This gene encodes an enzyme that functions both as a 17-beta-hydroxysteroid dehydrogenase (EC 1.1.1.62) during the biosynthesis of sex steroids and as a 3-ketosteroid reductase (EC 1.1.1.270) during the biosynthesis of cholesterol ( http://omim.org/entry/606756). These functions have no overlap with the pathogenesis of SMA-LED. Thus, we suggest that HSD17B7 is not a candidate gene for our phenotype of interest. The Sanger sequencing results indicated that all the other candidate variants mentioned above did not segregate appropriately in the family or were false positives from WES and could not be the causative mutations (Supplementary Table S1). Our exome data revealed only one de novo mutation and this result is consistent with previous reports (approximately 1 de novo mutation/generation/exome)12,13.
Discussion
DYNC1H1 is located on chromosome 14q32 and encodes the heavy chain of cytoplasmic dynein 1, which is a component of a multi-subunit motor complex that is essential for retrograde axonal transport and other intracellular functions14. In humans, DYNC1H1 mutations are present in autosomal dominant pedigrees of CMT15, cHSP (complex hereditary spastic paraplegia)16, severe cognitive disability, microcephaly, MCD17 and SMA-LED18 (Supplementary Table S2). Additionally, de novo mutations in DYNC1H1 are present in patients with sporadic mental retardation and hypotonia19, severe intellectual disability and peripheral neuropathy20, SMA-LED21,22, MCD17, or a combination phenotype23 (Supplementary Table S2).
Here, we suggest the molecular diagnosis to be SMA-LED according to WES and we determined the causal variant to be a de novo SNV (c.2327C > T, p.P776L) located in exon 8 of DYNC1H1. The process of our study design, genomic sequence data analysis and the interpretation of the sequencing variants are consistent with the guidelines published by MacArthur et al.5 and the ACMG6. In our study design phase, we identified the disease as a rare Mendelian disorder. The family reported no history of movement disorders. We inferred the mutation type to be a homozygous mutation, a compound heterozygous mutation or a de novo mutation. Thus, we utilized WES to sequence the proband and her unaffected parents to explore the potential disease causal variants5. Quality controls (QC) were conducted during the whole process, including QC for the DNA library, testing raw sequence data and testing base calling.
After choosing the study strategy and ensuring strict QC during WES, we interpreted the sequence variants in an objective way, as suggested by MacArthur et al.5 and the ACMG6. The ACMG states “investigators should begin by examining sequence variations in genes known to be associated with that phenotype and assessing sequence coverage of the coding sequences and splice junctions for these genes before exploring the possibility of new candidate genes in the affected individuals”6. As mentioned above, 19 genes are currently associated with lower limb weakness and wasting and foot deformity10,11. However, there were no other known causal genes identified in the WES results except for DYNC1H1. DYNC1H1 is a known causal gene of SMA-LED18,24,25. Then, the question was whether the identified variant was the disease causing mutation or not. The ACMG guideline offers us an objective way to answer this question6. Both parental samples were obtained from the biological parents of the patients and our patient had a family history of disease that was consistent with de novo inheritance (e.g., unaffected parents for a dominant disorder). The phenotype (foot deformity at birth, delayed motor milestones and lower limb weakness and atrophy) matched the DYNC1H1’s disease association with reasonable specificity. Thus, the de novo c.2327C > T, p.P776L mutation in DYNC1H1 has strong pathogenic criterion (PS2) according to ACMG6. Additionally, this mutation was not found in a large control population (including the Exome Aggregation Consortium database, 1000 genomes, dbSNP 141 and the NHLBI Exome Sequencing Project) indicating its strong pathogenic criterion (PS4) according to ACMG6. Literature and database sources as well as computational (in silico) predictive programs were also considered. Sanger sequencing and co-segregation analyses of all the identified candidate novel variants further confirmed our molecular diagnosis of the patient5. We also summarized our results as a supplement (Table 2, Supplementary Section1) according to the ACMG recommendations in order to facilitate the consultation of the proband and her affected twin sister and refer them for prenatal counseling when they become pregnant.
In conclusion, using WES and the related guidelines, we identified a novel de novo mutation (c.2327C > T, p.P776L) in the DYNC1H1 gene and confirmed it as the causal variant of SMA-LED. This paradigm facilitates the molecular diagnosis of rare Mendelian diseases. WES, combined with the guidelines for investigating the causality of sequence variants in human diseases and the interpretation of the sequence variants, as recommended by MacArthur et al.5 and the ACMG6, has become a preferable option to explore the pathogenesis of many rare neurological disorders and to make a molecularly diagnose the diseases.
Method and Materials
Subjects
A non-consanguineous Chinese family including healthy parents, two affected twins and two unaffected siblings were enrolled in our study (Fig. 1B). The family presented with no history of movement disorders. Genomic DNA was extracted from peripheral blood leukocytes via standard phenol-chloroform extraction methods. This study was approved by the ethics committee of Xiangya Hospital affiliated to Central South University. The methods in this study were performed in accordance with the approved guidelines. Written informed consent was obtained from all subjects.
Whole exome sequencing and data analysis
We conducted WES of a proband-parent trio to identify the causal gene. The SureSelect Human All ExonV5 Kit (Agilent) was used for exome capture. The IlluminaHiseq 2500 platform (San Diego, CA) was utilized for genomic DNA sequencing of the proband and her parents in Novogene (Beijing, China).
Raw image analyses and base calling were performed using Illumina’s Pipeline (version 1.3.4) with default parameters. Sequence data were aligned to the reference human genome (hg19) using the Burrows-Wheeler Aligner (BWA)26 and duplicate reads were removed using Picard tools. We used the Genome Analysis ToolKit (GATK)27 to perform the re-alignment and variation (SNP and Indel) detection. Annovar was utilized to catalogue the detected variations. Then, we filtered variations with a homopolymer length >6 (and synonymous substitutions) or that were common (>0.5%) in dbSNP ( http://www.ncbi.nlm.nih.gov/projects/SNP/), HapMap and the 1000 Genomes Project ( http://www.1000genomes.org). Variants that were not present in any of the above databases were considered novel.
Given to the characteristics of the pedigree, homozygous, compound heterozygous or de novo variations were considered to be candidate causal variations28.
Polymerase chain reaction (PCR) and Sanger sequencing
We confirmed the candidate causal variations identified via WES and conducted co-segregation analyses among the family. The primers of the candidate variations were designed using Primer 3 ( http://primer3.ut.ee/). The genomic DNA was PCR-amplified using Roche Fast Start PCR Master Mix polymerase (Roche Diagnostics Corp, USA). PCR products were sequenced with Applied BiosystemsBigDye terminator sequencing chemistry and then run on an ABI3730xl genetic analyzer according to the manufacturer’s instructions (Applied Biosystems,CA, USA). Sequence analysis was performed with Lasergene software (DNASTAR, Madison, WI, USA).
Additional Information
How to cite this article: Ding, D. et al. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Sci. Rep. 6, 20423; doi: 10.1038/srep20423 (2016).
References
Steinberg, K. M. et al. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep 5, 9124 (2015).
Feng, L. et al. Exome sequencing identifies a de novo mutation in HDAC8 associated with Cornelia de Lange syndrome. J Hum Genet 60(3), 165 (2015).
Mok, K. et al. Homozygosity analysis in amyotrophic lateral sclerosis. Eur J Hum Genet 21(12), 1429 (2013).
Johar, A. S. et al. Candidate gene discovery in autoimmunity by using extreme phenotypes, next generation sequencing and whole exome capture. Autoimmun Rev 14(3), 204 (2015).
MacArthur, D. G. et al. Guidelines for investigating causality of sequence variants in human disease. Nature 508(7497), 469 (2014).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5), 405 (2015).
Rosewich, H. et al. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: a whole-exome sequencing gene-identification study. Lancet Neurol 11(9), 764 (2012).
Ng, S. B. et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet 42(9), 790 (2010).
Chesi, A. et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 16(7), 851 (2013).
Gess, B. et al. HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology 83(19), 1726 (2014).
Rossor, A. M., Kalmar, B., Greensmith, L. & Reilly, M. M. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 83(1), 6 (2012).
Ku, C. S., Tan, E. K. & Cooper, D. N. From the periphery to centre stage: de novo single nucleotide variants play a key role in human genetic disease. J Med Genet 50(4), 203 (2013).
Neale, B. M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485(7397), 242 (2012).
Garrett, C. A. et al. DYNC1H1 mutation alters transport kinetics and ERK1/2-cFos signalling in a mouse model of distal spinal muscular atrophy. Brain 137(Pt 7), 1883 (2014).
Weedon, M. N. et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 89(2), 308 (2011).
Strickland, A. V. et al. Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J Neurol 262(9), 2124 (2015).
Poirier, K. et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet 45(6), 639 (2013).
Harms, M. B. et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78(22), 1714 (2012).
Vissers, L. E. et al. A de novo paradigm for mental retardation. Nat Genet 42(12), 1109 (2010).
Willemsen, M. H. et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet 49(3), 179 (2012).
Peeters, K. et al. Novel mutations in the DYNC1H1 tail domain refine the genetic and clinical spectrum of dyneinopathies. Hum Mutat 36(3), 287 (2015).
Punetha, J. et al. Exome Sequencing Identifies DYNC1H1 Variant Associated With Vertebral Abnormality and Spinal Muscular Atrophy With Lower Extremity Predominance. Pediatr Neurol 52(2), 239 (2015).
Fiorillo, C. et al. Novel dynein DYNC1H1 neck and motor domain mutations link distal spinal muscular atrophy and abnormal cortical development. Hum Mutat 35(3), 298 (2014).
Tsurusaki, Y. et al. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics 13(4), 327 (2012).
Scoto, M. et al. Novel mutations expand the clinical spectrum of DYNC1H1-associated spinal muscular atrophy. Neurology 84(7), 668(2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14), 1754 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5), 491 (2011).
Fromer, M. et al. de novo mutations in schizophrenia implicate synaptic networks. Nature 506(7487), 179 (2014).
Acknowledgements
The authors are grateful to all subjects for their participation in our study. This study was supported by the National Basic Research Program (973 Program) (Nos 2012CB944601and 2012CB517902 Hong Jiang), the National Natural Science Foundation of China (Nos 81471156, 81271260 to Hong Jiang; No. 31401135 to RongQiu), Hunan Funds for Distinguished Young Scientists (No. 14JJ1008 to Hong Jiang), Fundamental Research Funds for the Central Universities of Central South University (No. 2015zzts110 to Dongxue Ding) and High-level medical personnel of Hunan province “225”Project.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: D.X.D. and H.J. Performed the experiments: D.X.D. Analyzed the data: D.X.D., Z.C. and K.L. Contributed reagents/materials/analysis tools: D.X.D., Z.C., K.L., Z.L., W.Y., Z.L.T., B.S.T., R.Q., K.X. and H.J. Contributed to the writing of the manuscript: D.X.D. Reviewed and critiqued the first manuscript: Z.C., K.L. and H.J.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ding, D., Chen, Z., Li, K. et al. Identification of a de novo DYNC1H1 mutation via WES according to published guidelines. Sci Rep 6, 20423 (2016). https://doi.org/10.1038/srep20423
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20423
This article is cited by
-
The regulatory function of the AAA4 ATPase domain of cytoplasmic dynein
Nature Communications (2020)
-
Identification of novel mutations in TSC1 and TSC2 for tuberous sclerosis complex by targeted next-generation sequencing and ACMG guidelines
Child's Nervous System (2020)
-
Molecular mechanism of cytoplasmic dynein tension sensing
Nature Communications (2019)
-
The genotypic and phenotypic spectrum of PARS2-related infantile-onset encephalopathy
Journal of Human Genetics (2018)
-
Novel mutations in ADSL for Adenylosuccinate Lyase Deficiency identified by the combination of Trio-WES and constantly updated guidelines
Scientific Reports (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
