
Abstract
The present study was conducted to investigate the effects of a high-lipid diet (HLD) on cyclooxygenase (Cox)-2 expression and the signalling pathways related to low-grade inflammation in the large yellow croaker (Larmichthys crocea). An isolated 2508 bp cDNA clone of cox-2 contained an open reading frame spanning 1827 bp encoding a protein with 608 amino acid residues. The over-expression of cox-2 was consistent with the activation of c-Jun N-terminal kinases (JNKs) and p38 mitogen-activated protein kinase (MAPK) in HLD-fed fish. The activation of the activator protein-1 (AP-1) and the nuclear transcription factor kappa-B (NF-κB) signalling pathways in HLD-fed fish and the significant increase of cox-2 promoter-luciferase activity in vitro indicated that AP-1 and NF-κB could combine cox-2 promoter to promote its transcription, respectively. Together, HLD-induced inflammation up-regulates cox-2 expression via JNKs and p38 MAPK-dependent NF-κB and AP-1 pathways. The present study provides important insight into the signal transduction pathways involved in HLD-induced inflammation, which is detrimental to the health and production of fish as well as to the health of fish consumers.
Similar content being viewed by others


Introduction
A high lipid diet (HLD), containing higher lipid concentration than appropriate (the control), has been increasingly used for the development of cost-effective aquaculture, mainly due to its protein sparing effect1, meaning that HLD could be more beneficial for improving protein utilization in tissue synthesis than for acting as an energy source and could maximize nitrogen retention in fish. However, an HLD often leads to abnormal lipid deposition with chronic and low-grade inflammation of farmed fish2, which is detrimental to the health and production of fish and the health of fish consumers3.
Cyclooxygenase (Cox)-2 is a prostaglandin synthesis enzyme that plays a key role in inflammation in fish4,5. Cox-2 is expressed at low levels in resting cells and can be markedly induced by lipid peroxides, as well as pro-inflammatory and mitogenic stimuli6,7,8,9. Previous reports in mammals have revealed that the increased expression of cox-2 is strongly correlated with HLD-induced inflammation10. However, no information is available regarding the regulation of cox-2 in HLD-fed fish.
In mammals, HLD-induced inflammation leads to the activation of many transcription factors such as nuclear transcription factor kappa-B (NF-κB) and transcriptional factor activator protein-1 (AP-1)11. The phosphorylation of IκB by cytoplasmic IκB-kinase (IKK) complexes liberates NF-κB, which subsequently migrates to the nucleus and binds to the cis-acting κB enhancer element of target genes12,13. The double phosphorylation of c-Jun by c-Jun N-terminal kinases (JNKs) activates c-Jun, which translocates into the nucleus and then binds Jun or Fos family members to form the AP-1 early response transcription factor14,15. In mammals, cox-2 is one of the downstream targets of both NF-κB and AP-116,17. However, no information is available on the signalling cascades of NF-κB and AP-1 or their regulatory effects on cox-2 in fish.
The mitogen-activated protein kinases (MAPKs) pathways are well conserved across vertebrates and all members of the MAPK family have been identified in fish. These members include extracellular signal-regulated kinases 1&2 (ERK1/2) that are related to cell proliferation and survival17,18, JNKs and p38 MAPK that are related to inflammation and programmed cell death19,20. Several studies in mammals have reported that MAPKs could be activated by a HLD and transduce the HLD stimulus to the nucleus by mediating downstream signalling cascades, such as NF-κB and AP-121. However, little information is available on the activation of MAPKs and their effect on downstream transcription factors in response to a HLD in fish.
Large yellow croaker (Larmichthys crocea), with its high level of production and critical role in human food or health22, has been widely cultured in southeast China. The use of a HLD has become more common in large yellow croaker commercial diets, because of its protein sparing effect. The long-term use of a HLD abnormally increases lipid accumulation in the liver23 and the level of inflammation characterized by increased immune-related gene expression8 and mitochondrial function disorder3. Furthermore, previous studies have indicated that lipid deposition and inflammatory signals of the large yellow croaker are strikingly similar to those of other fish species and mammals23. Thus, the large yellow croaker is an appropriate fish species to investigate the mechanisms of HLD-induced inflammation.
Studies on nutrition of this fish have been intensively conducted in recent years24, but no information is available on the molecular mechanism of HLD-induced inflammation. Thus, in this study, cox-2 from large yellow croaker was cloned and characterized and the effect of a HLD on cox-2 expression and the signalling transduction pathway involved were investigated.
Results
Characterization of full-length cox-2 from large yellow croaker
The full-length cDNA of cox-2 from large yellow croaker was 2508 bp (GenBank Accession No. KP259877), including a 5′ untranslated terminal region (UTR) of 106 bp, a 3′ UTR of 575 bp and an open reading frame (ORF) of 1827 bp encoding a polypeptide of 608 amino acid residues with a predicted molecular weight of 69.03 KDa and a theoretical isoelectric point of 6.74 (Fig. 1).

Nucleotide and deduced amino acid sequences of cox-2.
The deduced amino acid sequences are shown below the cDNA sequences. The initiation codon (ATG) and the stop codon were characterized in bold.
Multiple sequence alignment analysis and phylogenetic analysis of Cox-2
The BLAST analysis revealed that Cox-2 of the large yellow croaker shared high identity with the known Cox-2 of teleosts including Atlantic croaker (Micropogonias undulatus), 95%; rock bream (Oplegnathus fasciatus), 92%; red seabream (Pagrus major), 91%; bicolour damselfish (Stegastes partitus), 91%; Antarctic rock cod (Notothenia coriiceps), 89%; and giant killifish (Fundulus heteroclitus), 88%. All marine fish species and freshwater fish species clustered together and formed a sister group to the branch of mammal species such as human and mouse (Fig. 2 and 3).

Multiple sequence alignment of Cox-2 and other invertebrate Cox-2.
Alignment was performed using ClustalW2. Identical residues are indicated in black and similar residues in light gray. Dashes indicate gaps. Identities are shown as black boxes and shaded boxes represent similar amino acids.

Phylogenetic tree of Cox-2.
The phylogenetic tree was constructed using MEGA 4.0 software by the Neighbor-joining method based on sequence alignment using ClustalW2 and 1000 replications of bootstrap. The scale bar indicated a branch length of 0.2. Amino acid sequences of Cox-2 are obtained from invertebrate and vertebrate animals.
Tissue distribution of cox-2 in the large yellow croaker
The tissue-specific expression of cox-2 was examined in multi-tissues, including gill, intestine, kidney, liver, adipose, muscle, stomach and eye. The cox-2 transcripts were broadly expressed in all of the detected tissues. The highest expression was found in gill, followed by intestine, kidney, liver, adipose, muscle and stomach, whereas the lowest expression level was observed in eye (Fig. 4).

Tissue distribution of cox-2 in large yellow croaker.
Relative cox-2 mRNA expression was determined by quantitative realtime PCR (qRT-PCR) and expressed relative to β-actin levels. Results are expressed as means ± S.E.M. (n = 3). Different letters above the bars denote significant differences among tissues at the P < 0.05 level (P = 0.000) as determined by one-way ANOVA followed by Tukey’s test (SPSS).
Cox-2 and pro-inflammatory cytokine expression in response to dietary fatty levels
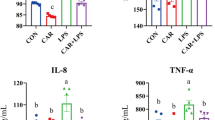
Compared with that in the control group, cox-2 transcript levels were up-regulated in the liver of HLD fish by approximately 2.78-fold (P < 0.05) (Fig. 5). The levels of cox-2 translation paralleled its transcript levels (Fig. 6). The transcript expression levels of the pro-inflammatory cytokine TNFα and IL-1β were also up-regulated in HLD fish compared with the control group (P < 0.05) (Fig. 5). These data indicate that cox-2 might be induced by an HLD and may play a critical role in inflammation.

Relative cox-2, TNFα and IL-1β mRNA levels.
Relative cox-2, TNFα and IL-1β mRNA levels were evaluated by quantitative realtime PCR (qRT-PCR) and expressed relative to β-actin levels in the liver of experimental fish. Results are expressed as means ± S.E.M. (n = 3). *P < 0.05, **P < 0.01.

The Cox-2 protein expression levels.
The Cox-2 protein expression levels were evaluated by Western blot and expressed relative to β-actin levels in the liver of experimental fish. Data are expressed as A.U. of the Western blots and are depicted as a ratio of Cox-2 to β-actin, (n = 3 in each group). All data are presented as mean ± S.E.M. *P < 0.05, **P < 0.01.
MAPK activity, AP-1 and NF-κB signal pathways involved in dietary lipid level-induced cox-2 expression
The HLD increased the phosphorylation of JNK and p38 MAPK in HLD fish compared to the control group, whereas no significant effect on the phosphorylation status of ERK1/2 was observed among the different lipid levels (Fig. 7). No significant difference in ERK1/2, JNK and p38 total protein was observed among the different dietary lipid levels. Therefore, it is likely that JNK and p38 MAPK but not ERK 1/2 up-regulated cox-2 expression. The phosphorylation of c-Jun was increased by the HLD, indicating the nuclear translocation level of c-Jun. Similarly, the HLD increased IKKα/β and IκBα protein phosphorylation, which induced the degradation of cytoplasmic IκBα and subsequently increased the nuclear level of NF-κB subunits p65. These data indicate that the HLD stimulated the expression of cox-2 depending on the AP-1 and NF-κB pathways via the activation of the JNK and p38 MAPK pathways (Fig. 8).

HLD activates the phosphorylation of JNK1/2 and p38 MAPK but not ERK1/2.
Quantitative results of p-ERK1/2, p-JNK1/2 and p-p38 protein levels, which were adjusted with the total ERK1/2, p38 and JNK1/2 protein levels in the liver of experimental fish were analysed using Western blot. Data are expressed as A.U. of the Western blots and are depicted as a ratio of p-ERK1/2 (pThr202/Tyr204) to total ERK1/2, p-JNK1/2 (pThr183/Thr185) to total JNK1/2 and p-p38 (pThr180/Thr182) to total p38 (n = 3 in each group). All data are presented as mean ± S.E.M. *P < 0.05, **P < 0.01.

HLD activates the NF-κB and AP-1 pathway.
Quantitative results of p-c-Jun, p-IKKα/β, p-IκBα protein levels, which were adjusted with the total c-Jun, IKKα/β and IκBα protein levels, were analysed using Western blot. Nuclear NF-κB p65 protein levels which were adjusted with the total NF-κB p65 protein levels, were also analysed using Western blot in the liver of experimental fish. Data are expressed as A.U. of the Western blots and are depicted as a ratio of p-c-Jun (pSer73) to total c-Jun, p-IKKα/β (pSer176/Ser180) to total IKKα/β, p-IκBα (pSer32/Ser36) to total IκBα and nuclear-NF-κB p65 to total NF-κB (n = 3 in each group). All data are presented as mean ± S.E.M. *P < 0.05, **P < 0.01.
Dual-luciferase reporter assays
Compared with the empty vector pCS2+, the expression of recombinant pCS-NF-κB and pCS-AP-1 resulted in 2.91- and 2.22-fold (P < 0.05) increases in pGL3-Cox-2 expression, respectively and the combination of pCS-NF-κB and pCS-AP-1 resulted in a 4.15-fold (P < 0.05) increase in pGL3-Cox-2 expression. Compared to the combination of pCS-NF-κB and pCS-AP-1 without an inhibitor, the combination of pCS-NF-κB and pCS-AP-1 with a NF-κB or AP-1 inhibitor resulted in a significant decrease in pGL3-Cox-2 expression. Compared to pCS-AP-1 or pCS-NF-κB independent treatments, no significant differences were observed in pGL3-Cox-2 expression of the combination of pCS-NF-κB and pCS-AP-1 with a NF-κB or AP-1 inhibitor treatment. These results indicate that both NF-κB and AP-1 activate the expression of luciferase reporter genes, suggesting that they could promote the transcription of cox-2 and that their regulation could be regarded as independent actions (Fig. 9).

Relative dual-luciferase activity analysis of NF-κB and AP-1 in the cox-2 promoter in HEK293 cells.
The bars indicated relative luciferase activity (n = 3). PRL-CMV and pGL3-Basic used as control. The amount relative to the internal control is expressed as mean ± S.E.M. (n = 3). Significant differences across control were indicated.
Discussion
The ORF of cox-2 encodes a 608 amino acid polypeptide, which showed a high similarity to Cox-2 from other fish and mammals. The evolutionary relationship of Cox-2 is consistent with traditional taxonomy. Additionally, the highest cox-2 expression was found in gills, a primary defence against external pathogens, thus indicating that cox-2 may play a significant role in innate immunity.
Previous reports on fish species have indicated that cox-2 is expressed at low levels in resting cells and is significantly induced by various fatty acids, such as saturated fatty acids8 and n-6 polyunsaturated fatty acids (PUFAs)9. Saturated fatty acids are positively associated with inflammation, mainly due to the significant effects of palmitic and stearic acids. Many studies have demonstrated that saturated fatty acids increase the saturation of membrane phospholipids30, initiate the unfolded protein response31, lead to endoplasmic reticulum stress32, affect mitochondrial metabolism33 and promote ROS accumulation34. In general, n-6 PUFAs have pro-inflammatory activity and can play important roles in immune function. Human inflammatory cells typically contain high proportions of the n-6 PUFA arachidonic acid (20:4n-6) and low proportions of n-3 PUFA, mainly because arachidonic acid is the precursor of two-series prostaglandins and four-series leukotrienes, which are highly-active mediators of inflammation35. Furthermore, some negative effects of obesity are associated with excessive levels of n-6 PUFAs36,37,38,39. In the present study, HLD-induced inflammation may be due to the high levels of saturated fatty acid and n-6 PUFAs in the HLD. Although many studies involving fatty acids induced inflammation have been reported in mammals, limited information is available in fish.
The over-expression of cox-2 plays a crucial role in the development and progression of inflammation40,41. In the present study, high levels of cox-2 expression were found in the liver of HLD-fed fish and the increased cox-2 expression was associated with increased pro-inflammatory cytokine TNFα and IL-1β transcripts (Fig. 5). In mammals, the up-regulated cox-2 expression in liver is associated with pathological conditions, such as acute liver failure, hepatic fibrosis and cirrhosis and hepatocarcinogenesis41,42. Given the conservation of cox-2 function, cox-2 over-expression in the present study supports its important role in HLD-induced hepatic inflammation.
In fish, MAPKs are associated with cox-2 expression through several stimuli, including metals43,44 and parasites45, but the effects of an HLD on MAPKs remain unknown. A number of reports in mammals have demonstrated that several signal transduction pathways are simultaneously stimulated by an HLD and these signals appear to converge at the common MAPK pathway46,47. In the present study, the HLD caused a marked increase in the level of phosphorylated JNK and p38 MAPK, whereas the phosphorylation of ERK was not affected (Fig. 7). These findings suggest that two types of MAPKs, JNK and p38 MAPK, but not ERK, are involved in the HLD-induced up-regulation of cox-2.
Previous studies have suggested that MAPKs activate separate effector pathways, demonstrating crosstalk between receptor systems that converged further downstream in the cell nucleus in mammals. The AP-1 and NF-κB activation can be selectively triggered by all MAPKs in a stress-dependent manner48,49. The qRT-PCR results indicated that an HLD up-regulated cox-2 expression at the transcriptional level (Fig. 5). The activation of transcription factor AP-1 and NF-κB pathways was thoroughly studied to investigate the downstream molecular mechanism of HLD action during cox-2 induction. In the present study, a high level of c-Jun phosphorylation was observed in HLD-fed fish, which revealed increased c-Jun nuclear translocation and AP-1 activation (Fig. 8). The HLD also activated NF-κB signalling pathways by the rapid phosphorylation of IKKα/β and the ubiquitination and proteolytic degradation of IκBα, which then resulted in the translocation of NF-κB to the nucleus (Fig. 8). In support of the findings in the present study, Dembinska et al. have demonstrated that the human obesity-induced inflammatory process depends on the AP-1 and NF-κB signalling pathways50. Therefore, the HLD-induced over-expression of cox-2 is regulated by JNK and p38 MAPK through the AP-1 and NF-κB pathways.
The human cox-2 promoter contains multiple potential cis-activating elements, including NF-κB and AP-1 binding sites51. The transcription factors AP-1 and NF-κB act as environmental sensors, detecting changes in the extracellular milieu through multiple signalling cascades52. Previous studies have demonstrated that NF-κB is not required for lipopolysaccharide (LPS)-induced cox-2 expression in murine macrophages by the dominant negative inhibition of NF-κB and cox-2 reporter gene activity53, whereas AP-1 binding activity is increased by LPS in macrophages54. Kim et al. have demonstrated that the application of a JNK inhibitor has no significant effect on papillomavirus E5-induced cox-2 expression compared with NF-κB inhibitor and have suggested that NF-κB is a major modulator of papillomavirus E5-induced cox-2 expression with AP-1 playing a reduced role55. These studies indicate that the regulation of AP-1 and NF-κB in cox-2 transcription is not necessary through different stimuli. In the present study, dual-luciferase reporter assays showed that AP-1 and NF-κB significantly increased cox-2 promoter-luciferase activity. As confirmed by pathway inhibitors, the regulation of cox-2 expression by AP-1 and NF-κB occurred independently, in agreement with previous studies (Fig. 9). These findings indicate that AP-1 and NF-κB interact with cognate cis-acting elements within the cox-2 promoter to promote cox-2 transcription independently.
In conclusion, the full-length cDNA encoding Cox-2 from the large yellow croaker was cloned and characterized. The transcription factor AP-1 and NF-κB specifically bind to the cox-2 promoter and independently promote its expression. A HLD increases cox-2 transcription levels, which is mediated by JNKs and p38 MAPK-dependent NF-κB and AP-1 activation. The present study provides important insight into the signal transduction pathways, which may potentially be applied to the discovery of disease mechanisms, thereby offering great benefits to the aquaculture industry and to human health.
Materials and Method
Ethics statement
The present study was performed in strict accordance with the Standard Operation Procedures (SOPs) of the Guide for the Use of Experimental Animals of Ocean University of China. All animal care and use procedures were approved by the Institutional Animal Care and Use Committee of Ocean University of China. Fish were anesthetized with eugenol (1:10,000) (Shanghai Reagent Corp., Shanghai, China) to minimize suffering before being assigned to cages and sampling.
Experimental diets and feeding process
The experimental diets design and the feeding process were given in Yan et al.56. The ingredient and nutrient composition of the experimental diets are showed in Tables 1 and 2. In brief, whitefish meal and soybean meal were chosen as the main protein sources. Fish oil and soybean lecithin were chosen as the main lipid sources, which provided essential fatty acids and phosphatides, respectively. Three isoproteic (43% crude protein) diets were formulated to contain graded levels of lipid (12% and 18% on a dry basis) (Table 1). The diet with 12% crude lipid was used as the control because this dietary lipid level is optimal for the growth of large yellow croaker57.
At the start of the experiment, the fish were fasted for 24 h. The fish (average body weight 150.0 g) were randomly distributed into 6 cages (1.5 × 1.5 × 2.0 m) with 40 fish in each cage. Each diet was randomly allocated to triplicate cages and the fish were hand-fed twice daily for 10 weeks.
Sample collection
At the termination of the experiment, fish were fasted for 24 h and anesthetized with eugenol (1:10,000) (purity 99%, Shanghai Reagent, China) before sampling. Tissues including kidney, intestine, spleen, heart, liver, brain and muscle from experimental fish were collected, frozen in liquid nitrogen and then stored at −80 °C for the analysis of immune related gene expression.
RNA extraction and cDNA synthesis
RNA extraction and cDNA synthesis has been previously described in Yan et al.56 with slight modification. In brief, total RNA was extracted from kidney, intestine, spleen, heart, stomach, liver, brain and muscle samples with Trizol Reagent (Invitrogen, USA) according to the manufacturer’s instructions and electrophoresed on a 1.2% denaturing agarose gel to test the integrity. The quantity and quality of the total RNA were assessed using the Nano Drop® ND-1000 spectrophotometer (Nano-Drop Technologies, Wilmington, DE, USA). The 260/280 nm absorbance ratios of all samples ranged from 1.90 to 2.07, indicating a satisfactory purity of the RNA samples. First-strand cDNA was reverse transcribed from the DNase-treated RNA using PrimeScript TM RT reagent Kit (Takara, Japan).
The cloning and characterization of full-length cox-2 cDNA
The cloning has been previously described in Dong et al. with slight modification58. Degenerate primers were designed based on highly conserved regions from the genes of other fish to amplify internal fragments and gene-specific primers were designed based on the known sequences of the internal fragments cDNA to clone the 3′- and 5′-end by rapid amplification of cDNA ends (RACE) through a two-round PCR using the SMARTerTM RACE cDNA Amplification Kit (Clontech, California, USA) (Table 3). PCR amplifications using the primers (Table 3) and Taq DNA Polymerase (Takara, alian, China) were performed with an initial denaturation at 95 °C for 3 min and 35 cycles of “95 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min”, followed by a final extension at 72 °C for 10 min. All PCR products were run on a 1.5% agarose gel and then purified by SanPrep PCR urification Kit (Sangon Biotech, Shanghai, China). PCR products were cloned into pEASY-T1 simple cloning vector (TransGen, Beijing, China) and sequenced in BioSune (Shanghai, China).
The nucleotide and deduced amino acid sequence of cox-2 from large yellow croaker were analyzed using BioEdit 7.0.1 and Expasysearch program (http://au.expasy.org/tools/). The sequences of cox-2 from different species were compared by the NCBI BLAST search program. A multiple sequence alignment was performed using ClustalW (http://www.ebi.ac.uk/clustalw/) and a phylogenetic tree of Cox-2 was made by MEGA 4.0 (http://www.megasoftware.net).
Quantitative realtime PCR (qRT-PCR) analysis
The first-strand cDNA synthesis was the same as described above. First-strand cDNA was diluted by 4 times using sterilized double-distilled water. Quantitative realtime PCR (qRT-PCR) was carried out in a quantitative thermal cycler (Mastercyclerep realplex, Eppendorf, Germany). The amplification was performed in a total volume of 25 μl, containing 12.5 μl of 2× SYBR® Premix Ex TaqTMII (Takara, Japan), 9.5 μl of sterilized double-distilled water, 1 μl of each primer (10 μM) (Table 3) and 1 μl of the diluted first strand cDNA product. The real-time qPCR amplification began with 2 min at 95 °C, followed by 40 cycles of 10 s at 95 °C, 10 s at 60 °C and 20 s at 72 °C. Melting curve (1.85 °C increment/min from 58 °C to 95 °C) was performed after the amplification phase for confirmation. Each sample was run in triplicate. Reference Beta-actin gene was used as internal control59. A four-fold serial dilution of the cDNA samples quantified 5 concentrations in triplicate was used to assess PCR efficiencies for each assay. The primer amplification efficiency was analyzed according to the following equation E = 10(−1/Slope)−1. To calculate the expression of cox-2, the comparative CT method (2−ΔΔctmethod) was used as described by Livak et al.60.
Western blot
Total protein was extracted from the liver of experimental fish with Total Protein Extraction Kit (Applygen Technologies Inc, Beijing, China) and nuclear and cytosolic fractions were collected using a nuclear protein extraction kit (Pierce, Nashville, TN, USA), according to the instructions of the manufacturers. Protein concentration was measured using BCA kit (Pierce). Fifty micrograms of protein was loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (Pall Corporation, Port Washington, NY, USA). The membranes were incubated with 5% skimmed milk for 1 h at room temperature. Immunoblots were obtained using antibodies against the following proteins: ERK1/2, ERK1/2 (pThr202/Tyr204), JNK1/2, JNK1/2(pThr183/Thr185), p38, p38 (pThr180/Thr182), IKKα/β, IKKα/β (pSer176/Ser180), IκBα, IκBα (pSer32/Ser36), c-Jun, c-Jun (pSer73), NF-κB p65 and beta-actin (Cell Signaling Technology, Danvers, MA, USA). Western blots were exposed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific, Waltham, MA, USA) and film images were scanned by Epson Perfection V33 (China).
Plasmid constructs
For expression plasmids, NF-κB p65 ORF (GenBank Accession No. XM_010731731.1) and c-Jun ORF (GenBank Accession No. XM_010739773.1) fragments were amplified with the primers (Table 3) and subcloned into the EcoRI/Xhol site of pCS2+ vectors (Invitrogen, USA) respectively to construct pCS- NF-κB and pCS-Ap-1. For reporter plasmids, the cox-2 promoter (a 2000bp fragment, GenBank Accession No. NW_011322590.1) was inserted into the KpnI/Xhol site of the promoterless pGL3-basic vector (Promega, USA) to construct pGL3-Cox-2. Plasmids for transfection were prepared using the EndoFree Plasmid Mini Kit (OMEGA, USA) according to the manufacturer’s instruction.
Cell culture and transfection
HEK293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Gibco, USA) supplemented with 10% foetal bovine serum (FBS, Invitrogen) at 37 °C in a humidified incubator under 5% CO2. For the over-expression experiments, cells were seeded in 24-well plates. Then, 24 h after seeding, 300 ng expression plasmid, 100 ng reporter gene plasmid, 10 ng pRL-CMV Renilla luciferase plasmid and 1 μl LipofectamineTM 2000 were co-transfected into cells in each well in a 24-well plate. All assays were performed with three independent transfections. The BAY 11-7082 (1 μM, Sigma) and JNK-IN-8 (1 μM, Sigma) were used as NF-κB pathway and AP-1 pathway inhibitors, respectively.
Dual-luciferase reporter assays
Firefly and renilla luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacturer’s instruction. Briefly, at 24 h post-transfection, HEK293 cells in 24-well plates were washed twice with 100 μl PBS and then lysed with 2 μl 1 × passive lysis buffer at room temperature for 15 min. The cell lysate (20 μl) was transferred to a plate and 50 μl luciferase assay reagent II and 1 × Stop & Glo reagent were added in sequence. Then, firefly and Renilla luciferase activities were measured.
Statistical analysis
Software SPSS 17.0 (SPSS Inc.) was used for all statistical evaluations. All data were subjected to a one-way analysis of variance (ANOVA) and followed by Tukey’s multiple-range test. The level of significance was chosen at P < 0.05 and the results were presented as means ± standard error of the mean.
Additional Information
How to cite this article: Wang, T. et al. Characterization of Cyclooxygenase-2 and its induction pathways in response to high lipid diet-induced inflammation in Larmichthys crocea. Sci. Rep. 6, 19921; doi: 10.1038/srep19921 (2016).
References
Mohanta, K. N., Mohanty, S. N., Jena, J. K. & Sahu, N. P. Optimal dietary lipid level of silver barb, Puntius gonionotus fingerlings in relation to growth, nutrient retention and digestibility, muscle nucleic acid content and digestive enzyme activity. Aquac. Nutr. 14, 350–359 (2008).
Refstie, S., Storebakken, T., Baeverfjord, G. & Roem, A. J. Long-term protein and lipid growth of Atlantic salmon (Salmo salar) fed diets with partial replacement of fish meal by soy protein products at medium or high lipid level. Aquaculture, 193, 91–106 (2001).
Cabello, F. C. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ. Microbiol. 8, 1137–1144 (2006).
Ishikawa, T., Griffin, K. J. P., Banerjec, U. & Herschman, H. R. The zebrafish genome contains two inducible, functional cyclooxygenase-2 genes. Biochem. Biophys. Res. Commun. 352, 181–187 (2007).
Ishikawa, T. & Herschman, H. R. Two inducible, functional cyclooxygenase-2 genes are present in the rainbow trout genome. J. Cell Biochem. 102, 1486–1492 (2007).
Chettri, J. K., Raida, M. K., Holten-Andersen, L., Kania, P. W. & Buchmann, K. PAMP induced expression of immune relevant genes in head kidney leukocytes of rainbow trout (Oncorhynchus mykiss). Dev. Comp. Immunol. 35, 476–482 (2011).
Leite, C. E. et al. Involvement of purinergic system in inflammation and toxicity induced by copper in zebrafish larvae. Toxicol. Appl. Pharm. 272, 681–689 (2013).
Kim, H. et al. Oleic acid ameliorates Aβ-induced inflammation by downregulation of COX-2 and iNOS via NFκB signaling pathway. J. Funct. Foods. 14, 1–11 (2015).
Calder, P. C. n-3 Polyunsaturated fatty acids and inflammation: from molecular biology to the clinic. Lipids 38, 343–352 (2003).
Delage, B., Bairras, C., Buaud, B., Pallet, V. & Cassand, P. A high-fat diet generates alterations in nuclear receptor expression: Prevention by vitamin A and links with cyclooxygenase-2 and β-catenin. Int. J. Cancer 116, 839–846 (2005).
Luedd, T. & Schwabe, R. F. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma, Nat. Rev. Gastro. Hepat. 8, 108–118 (2011).
Blonska, M. & Lin, X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 21, 55–70 (2011).
Schmid, J. A. & Birbach, A. IκB kinase β (IKKβ/IKK2/IKBKB)-A key molecule in signaling to the transcription factor NF-κB. Cytokine Growth F. R. 19, 157–165 (2008).
Hess, J., Angel, P. & Schorpp-Kistner, M. AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 117, 5965–5973 (2004).
D’Acquisto, F. et al. Involvement of NF-κB in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. Febs Lett. 418, 175–178 (1997).
Surh, Y. J. et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat. Res-Fund. Mol. M. 480, 243–268 (2001).
MacDougal, K. G., Merickob, P. A. & Burnett, K. G. Antigen receptor-mediated activation of extracellular related kinase (ERK) in B lymphocytes of teleost fishes. Dev. Comp. Immunol. 23, 221–230 (1999).
Hashimoto, H. et al. Existence of two isoforms of extracellular signal regulated kinase in fish. J. Biochem. (Tokyo) 123, 1031–1035 (1998).
Hashimoto, H., Matsuo, Y., Yokoyama, Y., Toyohara, H. & Sakaguchi, M. Structure and expression of carp mitogen-activated protein kinases homologous to mammalian JNK/SAPK. J. Biochem. (Tokyo) 122, 381–386 (1997).
Fujii, R., Yamashita, S., Hibi, M. & Hirano, T. Asymmetric p38 activation in zebrafish: its possible role in symmetric and synchronous cleavage. J. Cell Biol. 150, 1335–1348 (2000).
Chen, H. J. et al. Canna indica L. attenuates high-glucose-and lipopolysaccharide-induced inflammatory mediators in monocyte/macrophage. J Ethnopharmacol. 148, 317–321. (2013).
Yearbook, C. F. S. China Fishery Statistics Yearbook. Bureau of Fisheries, Ministry of Agriculture, Beijing, China. (2014).
Wang, X. X., Li, Y. J., Hou, C. L., Gao, Y. & Wang, Y. Z. Influence of different dietary lipid sources on the growth, tissue fatty acid composition, histological changes and peroxisome proliferator-activated receptor γ gene expression in large yellow croaker (Pseudosciaena crocea). Aquac Res. 43, 281–91 (2012).
Ai, Q. H. et al. Effects of dietary vitamin C on survival, growth and immunity of large yellow croaker, Pseudosciaena crocea. Aquaculture 261, 327–336 (2006).
Ai, Q. H. et al. Effects of dietary β-1, 3 glucan on innate immune response of large yellow croaker, Pseudosciaena crocea. Fish Shellfish Immunol. 22, 394–402 (2007).
Ai, Q. H. et al. Optimal dietary lipid level for large yellow croaker (Pseudosciaena crocea) larvae. Aquacult. Nutr. 14, 515–522 (2008).
Wang, J. et al. Effects of dietary ethoxyquin on growth performance and body composition of large yellow croaker Pseudosciaena crocea. Aquaculture 306, 80–84 (2010).
Zuo, R. T. et al. Effects of dietary n-3 highly unsaturated fatty acids on growth, nonspecific immunity, expression of some immune related genes and disease resistance of large yellow croaker (Larimichthys crocea) following natural infestation of parasites (Cryptocaryon irritans). Fish Shellfish Immunol. 32, 249–258 (2012).
Cohen, B. L. et al. Cyclooxygenase-2 (COX-2) expression is an independent predictor of prostate cancer recurrence. Int. J. Cancer 119, 1082–1087 (2006).
Hardy, S. et al. Saturated fatty acid-induced apoptosis in MDA-MB-231 breast cancer cells - A role for cardiolipin. J. Biol. Chem. 278, 31861–70 (2003).
Turpin, S., Lancaster, G., Darby, I., Febbraio, M. & Watt, M. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am. J. Physiol-Endocrinol. Metab. 291, E1341–50 (2006).
Pagliassotti, M., Wei, Y. & Wang, D. Saturated fatty acids induce cytotoxicity in hepatocytes via effects on the endoplasmic reticulum. Obesity Res. 13, A31 (2005).
Srivastava, S. & Chan, C. Application of metabolic flux analysis to identify the mechanisms of free fatty acid toxicity to human hepatoma cell line. Biotechnol. Bioeng. 99, 399–410 (2008).
Okere, I. et al. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution and serum leptin. Am. J. Physiol-Heart Circulatory Physiol. 291, H38–44 (2006).
Calder, P. C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 142, 592S–599S (2012).
Ailhaud, G., Guesnet, P. & Cunnane, S. C. An emerging risk factor for obesity: does disequilibrium of polyunsaturated fatty acid metabolism contribute to excessive adipose tissue development? Br. J. Nutr. 100, 461–470 (2008).
Anderson, A. K., McDougald, D. M. & Steiner-Asiedu, M. Dietary trans fatty acid intake and maternal and infant adiposity. Eur. J. Clin. Nutr. 64, 1308–1315 (2010).
Massiera, F. et al. Arachidonic acid and prostacyclin signaling promote adipose tissue development: a human health concern? J. Lipid Res. 44, 271–279 (2003).
Muhlhausler, B. S. & Ailhaud, G. P. Omega-6 polyunsaturated fatty acids and the early origins of obesity. Curr. Opin. Endocrinol. 20, 56–61 (2013).
Richardsen, E., Uglehus R. D., Due J., Busch, C. & Busund, L. T. COX-2 is overexpressed in primary prostate cancer with metastatic potential and may predict survival. A comparison study between COX-2, TGF-beta, IL-10 and Ki67. Cancer Epidemiol. 34, 316–322 (2010).
Demirel, Ö. et al. The lysosomal polypeptide transporter TAPL is stabilized by interaction with LAMP-1 and LAMP-2. J. Cell Sci. 125, 4230–4240. (2012).
Giannitrapani, L. et al. Cyclooxygenase-2 Expression in Chronic Liver Diseases and Hepatocellular Carcinoma. Ann. Ny. Acad. Sci. 1155, 293–299 (2009).
Urushibara, N. et al. JNK and p38 MAPK are independently involved in tributyltin-mediated cell death in rainbow trout (Oncorhynchus mykiss) RTG-2 cells. Comp. Biochem. Phys. C 149, 468–475 (2009).
Zheng, G. H., Liu, C. M., Sun, J. M., Feng, Z. J. & Cheng, C. Nickel-induced oxidative stress and apoptosis in Carassius auratus liver by JNK pathway. Aquat. Toxicol. 147, 105–111 (2014).
Sheng, H., Shao, J., Morrow, J. D., Beauchamp, R. D. & DuBois, R. N. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 58, 362–6 (1998).
Wan, Z., Durrer, C. & Mah, D. Reduction of AMPK activity and altered MAPKs signalling in peripheral blood mononuclear cells in response to acute glucose ingestion following a short-term high fat diet in young healthy men. Metabolism, 63, 1209–1216 (2014).
Chen, C., Zhang, W. & Shi, H. A novel benzenediamine derivative FC98 reduces insulin resistance in high fat diet-induced obese mice by suppression of metaflammation. Eur. J. Pharmacol. 761, 298–308 (2015).
Ulivi, V., Giannoni, P., Gentili, C., Cancedda, R. & Descalzi, F. p38/NF-kB-dependent expression of Cox-2 during differentiation and inflammatory response of chondrocytes. J. Cell Biochem. 104, 1393–1406 (2008).
Kim, A. R. et al. Phlorofucofuroeckol A inhibits the LPS-stimulated iNOS and Cox-2 expressions in macrophages via inhibition of NF-κB, Akt and p38 MAPK. Toxicol. in Vitro. 25, 1789–1795 (2011).
Dembinska-Kiec, A., Mykkänen, O., Kiec-Wilk, B. & Mykkänen, H. Antioxidant phytochemicals against type 2 diabetes. Brit. J. Nutr. 99, ES109–ES117 (2008).
Guo, Y. S., Hellmich, M. R., Wen, X. D. & Townsend Jr., C. M. Activator protein-1 transcription factor mediates bombesin-stimulated cyclooxygenase-2 expression in intestinal epithelial cells. J. Biol. Chem. 276, 22941–22947 (2001).
Chuang, S. M., Wang, I. C. & Yang, J. L. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis 21, 1423–1432 (2000).
Wadleigh, D. J., Reddy, S. T., Kopp, E., Ghosh, S. & Herschman, H. R. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated RAW 264.7 macrophages. J. Biol. Chem. 275, 6259–6266 (2000).
Lee, A. K., Sung, S. H., Kim, Y. C. & Kim, S. G. Inhibition of lipopolysaccharide-inducible nitric oxide synthase, TNF-α and COX-2 expression by sauchinone effects on I-κBα phosphorylation, C/EBP and AP-1 activation. Brit. J. Pharmacol. 139, 11–20 (2003).
Kim, M. K. et al. Human papillomavirus type 16 E5 oncoprotein as a new target for cervical cancer treatment. Biochem. Pharmacol. 80, 1930–1935 (2010).
Yan, J., Liao, K. & Wang, T. Dietary Lipid Levels Influence Lipid Deposition in the Liver of Large Yellow Croaker (Larimichthys crocea) by Regulating Lipoprotein Receptors, Fatty Acid Uptake and Triacylglycerol Synthesis and Catabolism at the Transcriptional Level. PloS one 10, e0129937 (2015).
Duan, Q., Mai, K., Zhong, H., Si, L. & Wang, X. Studies on the nutrition of the large yellow croaker, Pseudosciaena crocea R. I: growth response to graded levels of dietary protein and lipid. Aquaculture Research, 32, 46–52 (2001).
Dong, X. et al. Cloning and characterization of SREBP-1 and PPAR-α in Japanese seabass Lateolabrax japonicus and their gene expressions in response to different dietary fatty acid profiles. Comp. Biochem. Phys. B 180, 48–56 (2015).
Yao, C. L. et al. Molecular cloning and expression of MyD88 in large yellow croaker, Pseudosciaena crocea. Fish Shellfish Immun. 26, 249–255 (2009).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Acknowledgements
This study was financially supported by National Science Fund for Distinguished Young Scholars of China (31525024) and National Natural Science Foundation of China grants (31372541, 31172425). We are grateful to K.L., S.L.L. and X.J.D. for their experimental assistance.
Author information
Authors and Affiliations
Contributions
T.J.W., Q.H.A., K.S.M. and W.X. designed the research; T.J.W. and J.Y. conducted the research; T.J.W. analyzed the data and wrote the paper. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, T., Yan, J., Xu, W. et al. Characterization of Cyclooxygenase-2 and its induction pathways in response to high lipid diet-induced inflammation in Larmichthys crocea. Sci Rep 6, 19921 (2016). https://doi.org/10.1038/srep19921
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep19921
This article is cited by
-
Shrimp lectin–conjugated copper sulfide nanoparticles enhance immune response and gene expression in Etroplus suratensis infected with Aeromonas hydrophila
Aquaculture International (2021)
-
Gene expression analysis of Atlantic salmon gills reveals mucin 5 and interleukin 4/13 as key molecules during amoebic gill disease
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
