
Abstract
Genetic etiology in majority of patients with sporadic thoracic aortic aneurysm and dissections (STAAD) remains unknown. Recent GWAS study suggested common variant(s) in FBN1 is associated with STAAD. The present study aims to test this hypothesis and to identify mutation spectrum by targeted exome sequencing of the FBN1 gene in 146 unrelated patients with STAAD. Totally, 15.75% of FBN1 variants in STAAD were identified, including 5 disruptive and 18 missense mutations. Most of the variants were novel. Genotype-phenotype correlation analysis suggested that the maximum aortic diameter in the disruptive mutation group was significantly larger than that in the non-Cys missense mutation group. Interestingly, the variant Ala27Thr at −1 position, which is predicted to change the cleavage site of the signal peptidase of fibrillin-1, was detected in two unrelated patients. Furthermore, genotyping analysis of this variant detected 10 heterozygous Ala27Thr from additional 666 unrelated patients (1.50%), versus 7 from 1500 controls (0.47%), indicating a significant association of this variant with STAAD. Collectively, the identification of the variant Ala27Thr may represent a relatively common genetic predisposition and a novel pathogenetic mechanism for STAAD. Also, expansion of the mutation spectrum in FBN1 will be helpful in genetic counselling for Chinese patients with STAAD.
Similar content being viewed by others


Introduction
Thoracic aortic aneurysm and dissection (TAAD) is a frequent cause of morbidity and mortality in patients with cardiovascular disorders. Thoracic aortic aneurysms are often asymptomatic and underdiagnosed and may lead to an acute aortic dissection with life-threatening complications. Many presentations of TAAD show classic Mendelian inheritance with high or complete penetrance, suggesting the major contribution of a single gene1. The most common genes mutated in familial TAAD encode proteins involved in transforming growth factor-beta (TGF-β) signaling (TGFBR1/22,3, TGFB24, SKI5 and SMAD36) or proteins that comprise the contractile apparatus of aortic smooth muscle cells (MYH117, ACTA28 and MYLK9). Mutations of FBN1 were more frequently detected in patients with Marfan syndrome (MFS) including many Chinese patients10,11 compared to that in patients with STAAD12,13,14.
However, in the absence of familial history and features of syndromic forms, the proportion of patients with sporadic TAAD (STAAD) resulting from a genetic predisposition remains unknown. A recent GWAS study15 has mapped a single susceptibility locus of STAAD to 15q21.1, suggesting that one or more common variants within the FBN1 gene are associated with STAAD. This hypothesis whether FBN1 common variants that may affect the gene’s function and expression can be identified in affected individuals with STAAD needs to be tested by large-sample sequencing studies.
With the progress in next-generation sequencing (NGS), targeted NGS of disease-specific genes can be reliably implemented as a stand-alone diagnostic test. Recent studies by us and others16,17 have demonstrated this technology has higher efficiency but lower cost compared with traditional methods. Here, we report the results from 146 STAAD patients by target NGS analysis and additional 666 STAAD patients by mutation association study.
Results
Patient cohort
A total of 146 patients (115 males and 31 females) with a mean age of 50.31 years (16–81 years) were consecutively recruited in our hospital. Half of the patients were younger than 50 years old when the diagnosis was first given. Only 10 of the individuals were under the age of 31 years. Aortic dissections were found in 92 (63.01%) cases, while thoracic aortic aneurysms (TAA) were diagnosed in 54 (36.99%). Additional 8 individuals were classified into MFS18,19, because of accompanying skeletal and/or ocular complications (such as arachnodactyly, scoliosis, pectus carinatum and severe myopia) and thus were excluded from this study. No patient was found with Shprintzen-Goldberg syndrome5 or a newly defined aortic aneurysm syndrome due to TGFβ receptor mutations3.
Identification of FBN1 variants
In order to identify the mutation spectrum of FBN1 in Chinese STAAD, we sequenced each of the 146 DNA samples using the NGS technology. An average sequencing depth of these samples in the targeted regions reached 53.07× (ranging from 43.61× to 74.21×). Each sample had more than 81.1% of the targeted regions being covered. An average coverage of targeted exons (>10 reads) was 75.0% and also the coverage (>20 reads) reached 74.0%.
A total of 23 FBN1 variants in 23 unrelated patients (15.75%) were identified from the cohort of 146 individuals (Fig. 1a). Average sequencing depth in these 23 patients achieved 52-fold in 80.55% of the target regions (Table S1), which is similar to the sequencing depth reached in the 146 subjects. All detected variants were bidirectionally confirmed by Sanger sequencing (Fig. S1). All variants were found to be present in highly conserved nucleotides of the coding region (Fig. S2-S3) and were mostly predicted to be damaging by SIFT, PolyPhen-2 and MutationTaster. Notably, 14/23 of variants were first identified, not shown in the commonly used databases (ESP6500, 1000G, dbSNP132 and 1500 Chinese Han in-house database). The rest 9/23 variants (Table 1) were rare with low MAF (0.0003–0.0077).

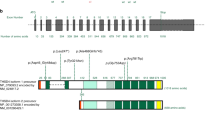
Research steps and identification of FBN1 variants.
(a) The scheme illustrates the main steps of patient classification and targeted sequencing results. (b) Schematic representation of locations of 23 FBN1 variants. Previously reported variants are labeled by stars*. (c) Symbol keys: Selected exons (numbered in each box) encoding for signal peptide are in yellow, for hybrid motifs in red, for epidermal growth factor (EGF)-like domains in blue, for calcium-binding epidermal growth factor (cb EGF)-like domains in green and for transforming growth factor beta (TGF-β)-like domains in purple.
Among the 23 variant carriers detected from the 146 STAAD samples (15.75%), further analysis stratified by phenotype divided 14.81% (8/54) variants to the TAA group and 16.30% (15/92) to the aortic dissections group (12 type A, 3 type B). Only 2 of them were younger than 31 years.
Recurrent FBN1 variants
Of the nine rare SNP variants, p.Ala27Thr was detected in two unrelated patients (Table 1). Ala27 is at the −1 position of the signal peptidase cleavage site (Fig. 2a), as predicted by SignalP 4.1 Server. The substitution of Ala27 with tyrosine (Fig. 2b) was predicted to change the cleavage site of fibirillin-1 (Fig. S4). This substitution was confirmed by Sanger sequencing (Fig. 2c). Ala27 is evolutionarily conserved in most species (Fig. 2d), but not in few others like sheep. And yet, this variant is present in 1000G database (rs25397) with low MAF (=0.0018, given a rare variant is defined by fewer than one in 1,000).

Effect of Ala27Thr on Signal peptide.
(a) Analysis of signal peptide structure and its amino acid properties. The signal peptide in secreted proteins is comprised of a charged region, a central hydrophobic region that is usually flanked by charged residue and a C-terminal region containing signal peptidase cleavage site(s). The cleavage site (arrow) between Ala27 and Asn28 is predicted by SignalP 4.1. These features are critical for the translocation of secretory proteins and their cleavage by signal peptidase. (b) Effect of Ala27Thr on the cleavage site (arrow) in two patients with STAAD. (c) The c.79G>A variant is confirmed in Patient 1 and 2 by bidirectional Sanger sequencing. (d) Evolutionary analysis of p.Ala27Thr in selected species by multiple sequence alignment.
In order to investigate if the c.79G>A (p.Ala27Thr) variant is associated with the disease in a larger cohort of STAAD patients, we performed genotyping analysis using a TaqMan assay in additional 666 unrelated subjects with STAAD. We identified the heterozygous Ala27Thr substitute in 10 of them (1.50%). These substitutions were all confirmed by Sanger sequencing. On the contrary, only 7 were found from 1500 Chinese Han individuals from our in-house database (0.47%), indicating that the c.79G>A is significantly associated with STAAD (χ2 test, P = 0.012, Table 2).
Mutation spectrum and genotype–phenotype correlation
Further analysis disclosed that 17 of these variants were novel and only 6 of them were previously reported in MFS, including, p.Arg861×20,21, p.Cys2307Arg (FBN1 mutations database), p.Asp2329Glu22, p.Met2347delAT23, p.Cys2448Arg24 and p.Gly2536Arg25. No different clinical manifestations were found between the reported patients and our cases under the same mutations (Table S2).
Remarkably, we identified a total of 5 variants (c.2581C>T, c.4160_4161insA, c.5249_5258del GTCAAAGGCC, c.5913T>A and c.7039_7040delAT) that lead to nonsense or frameshift-indel mutations, which were predicted to disrupt the function of fibrillin-1 (Fig. 3a). Three of the disruptive mutations (in bold, Table 1), to the best of our knowledge, were identified for the first time. Based upon available three-dimensional structural data of fibrillin-1, we were able to predict the effect of a premature stop mutation thereof on the function of the protein. Clearly, the variant p.Arg861X was located in the hybrid motif 2 and could result in the loss of calcium-binding activity and disrupt the protein’s function (Fig. 3b).

Disruptive mutations and protein structure.
(a) Sanger sequencing chromatograms of five disruptive mutations identified in five patients with STAAD. (b) Structural analysis of fibrillin-1 with a premature stop mutation in the cb EGF-like domain. Locations of residues R861 are shown in yellow. Two calcium binding sites are shown in cyan. 3D structural model is established using PyMOL Molecular Graphics System (Schrödinger L. version 1.3r1, 2010). This structure contains 147 amino acids (residues 807–951) (ID# 2W86, PDB Bank, http://www.pdb.org).
Furthermore, we evaluated potential correlations between different types of mutations and clinical conditions. No clinical difference was found between the group with missense mutations involving Cys residues in the calcium-binding epidermal growth factor (cb EGF)-like modules and the group with missense in the non-Cys residues (Table 3). However, we found that the maximum aortic diameter (70.8 ± 18.5 mm) in the disruptive mutation group was significantly larger than that in the groups with non-Cys missense mutation (53.9 ± 12.6 mm, P = 0.044). Likewise, the maximum aortic diameter in the disruptive mutation group was also larger than that in the groups with Cys missense. However, this difference did not reach statistical significance.
Clinical analysis in patients with or without FBN1 mutations
To find whether the patients with FBN1 mutations had different clinical manifestations from the patients without, we analyzed their clinical characteristics. No differences were found between these two groups in following six cardiovascular system-related parameters, including maximum aortic diameter, incidence of aortic dissection, aortic regurgitation, mitral valve regurgitation, tricuspid valve regurgitation and bicuspid aortic valve (Table 4). However, the patients with FBN1 mutations were 6.7 years younger than the patients without FBN1 mutation (P = 0.028) when the first diagnosis of TAAD was given. In addition, 30.4% of the patients with FBN1 mutation had a clinical history of hypertension vs. 62.6% of the patients without (P = 0.006).
Discussion
In the present study, we have reported the results of FBN1 mutations in 146 patients with STAAD as well as a rare SNP association study with 666 additional patients. This screening, the largest to date in Chinese cohort, has identified a total of 23 variants, including 17 novel variants, 5 disruptive mutations and 2 recurrent variants. Majority variants are rare and lead to missense, nonsense and frameshift-indel mutations. Interestingly, the variant (p.Ala27Thr) of FBN1 is a relatively frequent SNP, which is significantly associated with the disease.
Bioinformatics analyses have shown that most, if not all, of these variants are pathogenic mutations. We have shown that the FBN1-mutation rate is approximately 15.75% in 146 Chinese patients with STAAD, compared to 62.5% in the 8 patients with MFS (Fig. 1; Table S3). The rate of FBN1-variant was much higher in patients with MFS, which is in agreement with previous studies26. However, the incidence of FBN1-mutation may show substantial differences in different subjects and different sample sizes. Also, current Ghent nosology for the clinical diagnosis of MFS, given no family history of the disease, include aortic root enlargement plus one of either ectopia lentis or pathogenic FBN1 mutation or a systemic core greater than or equal to seven19. Therefore, all of these 28 patients could be classified into MFS given the pathological effect of each FBN1 variant is ascertained. By this definition, the overall molecular diagnostic yield for MFS achieved 18.18% (28/154 patients) in our cohort. This mutation rate is similar to previous report (18.42%, 14/76) in patients who fulfill the clinical criteria for diagnosis making of MFS27.
Although these 23 FBN1 variants seemed not to be clustered as a “hot-spot”, most variants either affect highly conserved sequences and/or are predicted to be deleterious. Although more than 1,000 different FBN1 mutations have been documented28,29 since the first mutation of the gene was discovered in 199130, most mutations identified here are novel, suggesting highly diversified genetic resources in Chinese patients. A majority of the mutations (15/23) occurred within the cb EGF-like domains of the protein fibrillin-1, which are required for protecting fibrillin-1 from the activity of proteases31,32. On the other hand, mutations that directly disrupt the calcium-binding consensus sequence or result in perturbation of protein conformation (e.g., cysteine substitutions) could give rise to a disordered interbedded region and thus increase the susceptibility of the monomer to proteolysis33.
A comparison between the type or position of each mutation and the patient´s phenotype revealed a potential relation. It appeared that patients with disruptive mutations were diagnosed at younger age. And yet, patients with FBN1 mutation were 6.7 years younger than patients without FBN1 mutation (Table 4). Indeed, synthesis of normal-sized fibrillin-1 protein in the disruptive mutation samples was only about 50% of the control’s level20. Intriguingly, a history of hypertension was significantly more prevalent in patients without FBN1 mutation. Herein, it is inferred that hypertension is not a primary element involving the primary pathogenesis of STAAD with FBN1 mutations.
The p.Ala27Thr variant is predicted to eliminate the cleavage site at Ala27/Asn28 of the signal peptide and thus to produce a shorter signal peptide (1–24) rather than normally 27 residues. This might affect maturation and cellular function of fibrillin-1, suggesting a novel causative mechanism of the disease. In the N’-fibrillin overexpressed SF9 and CHO-K1 cells, two cleavage sites actually were observed at Gly24/Ala25 and Ala27/Asn28 by amino-terminal sequencing of secreted fibrillin-134. However, cleavage site(s) of the endogenous protein as well as the mutant p.Ala27Thr in human aortic artery cells remains to be determined.
Sequence variations/mutations of a signal peptide have been identified in multiple human disorders35,36,37. Most signal peptide mutations were found in the hydrophobic regions that lead to impaired protein-protein interaction and defective intracellular translocation38,39. In relatively rare cases, mutations can be found to affect the −1 or −3 residues at the signal peptidase cleavage site. For instance, p.Ala25Thr mutation at the −1 position of the ADAMTS10 signal peptide significantly diminished secretion of the protein into extracellular region and thus caused Weill-Marchesani syndrome36, a rare inherited disorder with generalized mesodermal dysplasia. Likewise, an Ala to Thr substitution at −1 position of the signal peptide in preprovasopressin led to failure of signal peptide processing and reduced secretion and resulted in familial central diabetes insipidus40.
The Ala27Thr is a recurrent variant (1.37% or 2/146 patients). Although Ala27Thr is also present in 1000G database (low MAF = 0.0018), a large sample screening in 666 unrelated patients confirmed that this variant is significantly associated with the disease, arguing that this variant is a susceptibility allele for STAAD. This assumption is also supported by the GWAS results, i.e., the FBN1-containing region was the only locus linked to STAAD15. The significantly linked SNPs, such as rs2118181 and rs1036477 in the FBN1 region, may be served as biomarkers for identifying patients at risk for STAAD. However, whether these biomarkers are functionally related to the disease is difficult to be verified. Therefore, targeted-exome sequencing of the linked region in large samples is an ideal approach to quickly identify potential variants that may affect the gene’s function.
Alternatively, the Ala27Thr variant might be a recessive allele with reduced expression of FBN1 in STAAD, suggesting a threshold model that requires a combination of mild genetic defects to trigger clinical manifestation. Although this hypothesis has not been experimentally demonstrated, there is a convincing precedent in a familiar TAAD with recessive mutations in the first aspartic acid of a cb EGF-like domain of FBN-141.
Furthermore, similar cases with mutations in single peptide regions in several other diseases have been previously reported. For example, a common polymorphism, either Ala17 or Thr17 in the signal peptide of CTLA-4 T-cell receptor, resulted in decreased cell surface presentation, which was associated with a higher risk of autoimmune disorders42. Therefore, our findings support a model in which the subtle effects on signal peptidase cleavage by Ala27Thr may predispose individuals to STAAD.
It is worthy to mention that we also identified a disruptive nonsense mutation p.Arg861X in the hybrid motif 2 domain. Only a few other missense mutations in the hybrid motif 1 or 2 domain were previously described27,43. Interactions between the residues within the hybrid motif 1 and LTBP-1 and LTBP-4 were previously observed44, indicating the hybrid motif involves TGF pathway. However, the underlying pathogenic mechanism remains unclear.
These findings collectively highlight the importance of these correlations in anticipating the clinical consequences of specific FBN1 mutations. Furthermore, it is worthy to apply whole-exome sequence to identify potential genetic defects in the rest 126 patients who do not carry FBN1 mutations.
Taken together, we have determined that mutations in FBN1 are responsible for approximately 15.75% (23/146) of Chinese patients with STAAD who did not fulfil the clinical criteria of MFS. A total of 17 novel mutations, particularly those located in the signal peptide or the hybrid motif regions, may involve different pathogenetic mechanisms. To the best of our knowledge, the Ala27Thr may represent a relatively frequent variant that predisposes individuals to STAAD. Our findings will be helpful in genetic counselling for patients with STADD or MFS as well as their at-risk family members.
Methods
Patient population
A total of 146 unrelated Chinese Han patients with STAAD, who were evaluated at Beijing Anzhen Hospital and referred for surgery repair, were recruited from the year 2011 to 2012. Eight patients who showed skeletal and ocular complications were not accounted as STAAD but were included for sequencing analysis. Additional 666 unrelated patient samples recently recruited through Beijing Anzhen hospital were used for SNP genotyping association study. No patients had a family history of TAAD or evidence of a syndromic form of TAAD. Patients with aortic lesions associated with trauma, infection, or aortitis were excluded. Written informed consent was obtained from each of the subjects according to the research protocol approved by the Ethical Review Board of Beijing Anzhen Hospital. The methods were carried out in accordance with the approved guidelines.
Echocardiographic examination was performed for assessment of the aortic aneurysm and dissection. Aortic computed tomography scan was also applied as deemed necessary by treating physicians. Individuals were diagnosed as TAAD if they had a thoracic aneurysm (diameter ≥ 3.6 cm) or dissection of the thoracic aorta. Diagnosis and conditions of type A or B aortic dissections were assessed according to the classification of International Registry of Aortic Dissection (IRAD)45,46. Aortic, tricuspid valve and mitral valve regurgitations, as well as associated congenital disorders (bicuspid aortic valve, atrial septal defect and patent ductus arteriosus) were systematically screened. All patients were also examined for musculoskeletal, pulmonary, dermatologic and ophthalmic phenotypes in MFS based on recent Ghent criteria19.
Disease-related gene enrichment and sequencing
DNA was extracted from whole blood with Blood DNA Purification Kit (Promega, Beijing branch, China). Purity of the DNA samples was assessed with a NanoDrop2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). FBN1 was screened by a GenCap custom enrichment kit (MyGenostics, Beijing, China) as described previously47,48. Biotinylated single-strand capture probes were designed to tile along the exonic non-repeated regions and intron-exon boundaries of the gene (Table S4). Capture experiment was conducted according to the manufacturer’s protocol. The FBN1 gene enriched libraries were sequenced by Illumina HiSeq 2000 sequencer, which produced paired reads around 90-bp for further bioinformatics analysis.
Bioinformatics analysis
Bioinformatics analysis began with the raw sequencing data generated from the Illumina pipeline. After the adapter and low-quality reads were discarded, clean reads were aligned to reference FBN1 sequence (Hg19) by Burrows-Wheeler Aligner (BWA)49. Final BAM files were used for variant calling. Single nucleotide polymorphisms (SNPs) and small insertion/deletions (InDels) were detected by SOAPsnp50 and SAMtools51, respectively. Variants were annotated by ANNOVAR52. Quality Control (QC) was performed at each step of the analysis pipeline.
Validation and evaluation of variants
All identified rare variants in FBN1 were bidirectionally sequenced by ABI 3130XL sequencer (Applied Biosystems, Foster City, CA). Variant frequency was compared with 4 major SNP databases: ESP6500 (http://evs.gs.washington.edu/EVS/), 1000G (http://www.1000genomes.org/), dbSNP132 (http://www.ncbi.nlm.nih.gov/projects/SNP/) and 1500 Chinese Han in-house database. Deleterious effect of each variant was assessed by various algorithms, such as SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/). Signal peptide analysis was evaluated by SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP/).
SNP Genotyping
Genotyping of 666 patients was performed using TaqMan platform in 384-well plates and read with the Sequence Detection Software on ABI Prism 7900 instrument (Applied Biosystems, Foster City, CA). Primers and probes specific for the variant c.79G>A were supplied by Applied Biosystems. PCR conditions were: 95 °C for 5 min, followed by 8 cycles (0.5 °C touchdown) for 10 s at 95 °C, 25 s at 64 °C and 20 s at 72 °C and then followed by 32 cycles of 10 s at 95 °C, 25 s at 60 °C and 20 s at 72 °C. 1500 controls’ data were obtained from 1500 Chinese Han in-house database.
Statistical analysis
Most data were presented as mean ± SD. Qualitative variables were compared by χ2 test and Fisher test. Quantitative variables were analyzed by Students t test and one-way ANOVA with Bonferroni post-test. All statistical tests were two-sided and a P value of ≤0.05 was considered significant. SPSS (version 19, IBM, Armonk, NY, USA) was used for all statistical analyses.
Additional Information
How to cite this article: Guo, J. et al. Wide mutation spectrum and frequent variant Ala27Thr of FBN1 identified in a large cohort of Chinese patients with sporadic TAAD. Sci. Rep. 5, 13115; doi: 10.1038/srep13115 (2015).
References
Lindsay, M. E. & Dietz, H. C. Lessons on the pathogenesis of aneurysm from heritable conditions. Nature 473, 308–316 (2011).
Mizuguchi, T. et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet 36, 855–860 (2004).
Loeys, B. L. et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37, 275–281 (2005).
Boileau, C. et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 44, 916–921 (2012).
Doyle, A. J. et al. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet 44, 1249–1254 (2012).
Regalado, E. S. et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res 109, 680–686 (2011).
Zhu, L. et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet 38, 343–349 (2006).
Guo, D. C. et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39, 1488–1493 (2007).
Wang, L. et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87, 701–707 (2010).
Zhao, F., Pan, X., Zhao, K. & Zhao, C. Two novel mutations of fibrillin-1 gene correlate with different phenotypes of Marfan syndrome in Chinese families. Mol Vis 19, 751–758 (2013).
Wang, F., Li, B., Lan, L. & Li, L. C596G mutation in FBN1 causes Marfan syndrome with exotropia in a Chinese family. Mol Vis 21, 194–200 (2015).
Milewicz, D. M. et al. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation 94, 2708–2711 (1996).
Brautbar, A. et al. FBN1 mutations in patients with descending thoracic aortic dissections. Am J Med Genet A 152A, 413–416 (2010).
Pepe, G. et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Med Genet 15, 23 (2014).
Lemaire, S. A. et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet 43, 996–1000 (2011).
Carvill, G. L. et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45, 825–830 (2013).
Sikkema-Raddatz, B. et al. Targeted next-generation sequencing can replace Sanger sequencing in clinical diagnostics. Hum Mutat 34, 1035–1042 (2013).
De Paepe, A., Devereux, R. B., Dietz, H. C., Hennekam, R. C. & Pyeritz, R. E. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 62, 417–426 (1996).
Loeys, B. L. et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 47, 476–485 (2010).
Schrijver, I. et al. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet 71, 223–237 (2002).
Katzke, S. et al. TGGE screening of the entire FBN1 coding sequence in 126 individuals with marfan syndrome and related fibrillinopathies. Hum Mutat 20, 197–208 (2002).
Liu, W. O., Oefner, P. J., Qian, C., Odom, R. S. & Francke, U. Denaturing HPLC-identified novel FBN1 mutations, polymorphisms and sequence variants in Marfan syndrome and related connective tissue disorders. Genet Test 1, 237–242 (1997).
Korkko, J., Kaitila, I., Lonnqvist, L., Peltonen, L. & Ala-Kokko, L. Sensitivity of conformation sensitive gel electrophoresis in detecting mutations in Marfan syndrome and related conditions. J Med Genet 39, 34–41 (2002).
Sakai, H. et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet A 140, 1719–1725 (2006).
Comeglio, P., Evans, A. L., Brice, G. W. & Child, A. H. Detection of six novel FBN1 mutations in British patients affected by Marfan syndrome. Hum Mutat 18, 251 (2001).
Wang, W. J. et al. Exon 47 skipping of fibrillin-1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. J Mol Med (Berl) 91, 37–47 (2013).
Rommel, K., Karck, M., Haverich, A., Schmidtke, J. & Arslan-Kirchner, M. Mutation screening of the fibrillin-1 (FBN1) gene in 76 unrelated patients with Marfan syndrome or Marfanoid features leads to the identification of 11 novel and three previously reported mutations. Hum Mutat 20, 406–407 (2002).
Faivre, L. et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet 81, 454–466 (2007).
Keane, M. G. & Pyeritz, R. E. Medical management of Marfan syndrome. Circulation 117, 2802–2813 (2008).
Dietz, H. C. et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 352, 337–339 (1991).
Reinhardt, D. P., Ono, R. N. & Sakai, L. Y. Calcium stabilizes fibrillin-1 against proteolytic degradation. J Biol Chem 272, 1231–1236 (1997).
Reinhardt, D. P., Gambee, J. E., Ono, R. N., Bachinger, H. P. & Sakai, L. Y. Initial steps in assembly of microfibrils. Formation of disulfide-cross-linked multimers containing fibrillin-1. J Biol Chem 275, 2205–2210 (2000).
Handford, P. et al. The calcium binding properties and molecular organization of epidermal growth factor-like domains in human fibrillin-1. J Biol Chem 270, 6751–6756 (1995).
Ritty, T. M., Broekelmann, T., Tisdale, C., Milewicz, D. M. & Mecham, R. P. Processing of the fibrillin-1 carboxyl-terminal domain. J Biol Chem 274, 8933–8940 (1999).
Godi, M. et al. A recurrent signal peptide mutation in the growth hormone releasing hormone receptor with defective translocation to the cell surface and isolated growth hormone deficiency. J Clin Endocrinol Metab 94, 3939–3947 (2009).
Kutz, W. E. et al. Functional analysis of an ADAMTS10 signal peptide mutation in Weill-Marchesani syndrome demonstrates a long-range effect on secretion of the full-length enzyme. Hum Mutat 29, 1425–1434 (2008).
Datta, R., Waheed, A., Shah, G. N. & Sly, W. S. Signal sequence mutation in autosomal dominant form of hypoparathyroidism induces apoptosis that is corrected by a chemical chaperone. Proc Natl Acad Sci USA 104, 19989–19994 (2007).
Hughes, A. E. et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet 24, 45–48 (2000).
Pidasheva, S., Canaff, L., Simonds, W. F., Marx, S. J. & Hendy, G. N. Impaired cotranslational processing of the calcium-sensing receptor due to signal peptide missense mutations in familial hypocalciuric hypercalcemia. Hum Mol Genet 14, 1679–1690 (2005).
Ito, M. et al. Possible involvement of inefficient cleavage of preprovasopressin by signal peptidase as a cause for familial central diabetes insipidus. J Clin Invest 91, 2565–2571 (1993).
Hilhorst-Hofstee, Y. et al. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat 31, E1915–1927 (2010).
Anjos, S., Nguyen, A., Ounissi-Benkalha, H., Tessier, M. C. & Polychronakos, C. A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem 277, 46478–46486 (2002).
Arbustini, E. et al. Identification of sixty-two novel and twelve known FBN1 mutations in eighty-one unrelated probands with Marfan syndrome and other fibrillinopathies. Hum Mutat 26, 494 (2005).
Ono, R. N. et al. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem 284, 16872–16881 (2009).
Nienaber, C. A. & Eagle, K. A. Aortic dissection: new frontiers in diagnosis and management: Part I: from etiology to diagnostic strategies. Circulation 108, 628–635 (2003).
Rogers, A. M. et al. Sensitivity of the aortic dissection detection risk score, a novel guideline-based tool for identification of acute aortic dissection at initial presentation: results from the international registry of acute aortic dissection. Circulation 123, 2213–2218 (2011).
He, J. et al. IgH gene rearrangements as plasma biomarkers in Non- Hodgkin’s lymphoma patients. Oncotarget 2, 178–185 (2011).
Wu, J. et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 3, 92ra66 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, R. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res 19, 1124–1132 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164 (2010).
Acknowledgements
This study was supported financially by National Natural Science Foundation of China (81200074), International Science & Technology Cooperation Program of China (2010DFB30040) and Program for Changjiang Scholars and Innovative Research Team in University (IRT1074). We thank Dr. Jinyu Wu’s assistance on bioinformatics analysis of FBN1 mutations.
Author information
Authors and Affiliations
Contributions
J.G., T.C. and J.D. conceived and designed the experiments. J.G., L.C. and L.J. performed the experiment. J.G., X.L. and Z.S. analyzed the data. X.X., S.Z., X.L., C.P. and T.L. prepared the samples. J.G. and T.C. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Guo, J., Cai, L., Jia, L. et al. Wide mutation spectrum and frequent variant Ala27Thr of FBN1 identified in a large cohort of Chinese patients with sporadic TAAD. Sci Rep 5, 13115 (2015). https://doi.org/10.1038/srep13115
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep13115
This article is cited by
-
Multi-omics in thoracic aortic aneurysm: the complex road to the simplification
Cell & Bioscience (2023)
-
Alpha-actin-2 mutations in Chinese patients with a non-syndromatic thoracic aortic aneurysm
BMC Medical Genetics (2016)
-
Targeted next-generation sequencing reveals multiple deleterious variants in OPLL-associated genes
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
