
Abstract
Thoracic aortic aneurysm with or without dissection (TAAD) can be broadly categorized as syndromic TAAD (sTAAD) and isolated TAAD (iTAAD). sTAAD and is highly correlated with genetics. However, although the incidence of iTAAD is much higher, its monogenic contribution is not yet clear. Here, we sequenced 15 known TAAD genes for 578 iTAAD cases from four cardiac centers in China and found that 10.6% patients with a pathogenic/likely pathogenic (P/LP) variant. Other 7.27% of patients carried variants of uncertain significance in these target genes. We further investigated the correlations among genetics, clinical features, and long-term outcomes. Genetic patients showed younger onset ages (P = 1.31E-13) and larger aortic diameter (P = 1.00E-6), with the youngest age in patients with FBN1 P/LP variants. Monogenic variants were also associated with more aortic segments involved (P = 0.043) and complicated with initial dissection (P = 4.50E-5), especially for genetic patients with non-FBN1 P/LP variants. MACEs occurred in 14.9% patients during follow-up of median 55 months. Genetic status (P = 0.001) and initial dissection (P = 3.00E-6) were two major risk factors for poor prognosis. Early onset age was associated with MACEs in non-genetic cases without initial dissection (P = 0.005). Our study revealed the monogenic contribution in known TAAD genes to iTAAD patients. The genotype–phenotype correlations may complement the risk stratification of iTAAD patients and identification of higher risk subgroups, as well as assist the development of tailored precision medicine in iTAAD.
Similar content being viewed by others


Introduction
Thoracic aortic aneurysms (TAAs) are abnormal enlargements or bulging of the thoracic aortic wall. They may involve one or more aortic segments (aortic root or sinus of Valsalva (AR/SV), ascending aorta, arch, or descending aorta) and are classified accordingly [1]. Some TAA patients have aortic dissection, which is a more dangerous situation and results in high mortality despite progress in therapeutic surgical procedures. TAA with or without dissection (TAAD) can be broadly categorized as syndromic TAAD (sTAAD) and isolated TAAD (iTAAD), and most of TAAD patients are iTAAD [2].
Genetics play an important role in TAAD. Two major categories of genes involve in the pathologic process: those encoding components of the transforming growth factor beta (TGF-beta) signaling cascade (FBN1, TGFBR1, TGFBR2, TGFB2, TGFB3, SMAD2, SMAD3, and SKI), and those encoding smooth muscle contractile apparatus (ACTA2, MYH11, MYLK, and PRKG1) [3,4,5]. About 90% of sTAAD can be explained by the known TAA genes [6]. However, the monogenic contribution to iTAAD is far less clear.
In addition, iTAADs have phenotypic heterogeneity. It can occur in both young and aged people. Aortic segments involved in aneurysms and growth rate of the aneurysms are different in different patients. Some patients are associated with bicuspid aortic valve, which are frequently involves the proximal aorta, including the AR, ascending aorta, and aortic arch, but most patients have tricuspid aortic valves [7, 8]. More importantly, aortic rupture and/or dissection occur in a part of patients even when the aortas are moderately dilated, resulting in life threatening. Therefore, the onset age, the size, the location of aneurysm, acuteness, and severity vary greatly among individuals. Moreover, the outcomes also range from asymptomatic to recurrence, and sudden death. Previous studies have demonstrated that genetic heterogeneity results in different clinical presentations and affects the choice of treatment options for sTAAD [9, 10]. However, genetic basis for heterogeneous clinical features and prognosis is limited for iTAAD.
In this study, we screened TAAD causal genes in 578 iTAAD patients and a control cohort to evaluate the contribution of monogenic variants to the disease onset. Then, we systematically investigated the relationship among genetics, clinical features at diagnosis, and the occurrence of major adverse cardiovascular events (MACEs) during follow-up. These insights may facilitate more precise genetic counseling and help to improve tailored management of patients carrying monogenic variants and their families.
Materials and methods
Subjects and clinical evaluation
We selected 578 sporadic iTAAD patients of Chinese origin with the following detailed inclusion criteria: all patients exhibited pathologic aneurysm in at least one segment of the thoracic aorta (AR/SV, ascending aorta, arch, or descending aorta) as shown by computed tomography angiography. Specifically, for AR/SV and ascending aorta, aneurysms were defined as permanent localized dilations with a ≥50% increase in diameter relative to the expected normal aortic diameter or with a diameter of >5.0 cm [11]. Z score was calculated as previously described [12]. For the arch and descending aorta, aneurysms were defined as a diameter of >4.5 cm. Patients were excluded if they met one of the following conditions: (1) showed a Marfan-like phenotype (the second systemic involvement besides the aorta) although had not diagnosed with Marfan syndrome (MFS) or other sTAAD; (2) had aortic lesions associated with trauma or infection; (3) were unwilling to participate in this study; (4) had any first-degree relative diagnosed with an aneurysm or dissection at any location, or any first-degree relative had a sudden or unexplained death. Onset age referred to the age at which TAAD was first detected. Left ventricular ejection fraction, left ventricular end diastolic diameter, bicuspid aortic valve, and aortic regurgitation were determined by ultrasonography. Cardiac insufficiency referred to a left ventricular ejection fraction <50%. Controls were in-house populations ethnically matched to cases, with a mean age of 32.4 years, and with no known or visible TAAD-related symptoms or history.
This study was conducted in agreement with the principles outlined in the Declaration of Helsinki. Written informed consent was obtained from all subjects according to the research protocol approved by the Ethical Review Board of Beijing Anzhen Hospital. This study was registered in ClinicalTrials.gov with ID NCT03010514 (A Registry Study on Genetics and Biomarkers of Thoracic Aortic Aneurysm/Dissection).
Targeted sequencing of 15 TAAD candidate genes
Genomic DNA was extracted from blood samples and sequenced using the Ion 318™ Chip on PGMTM instrument of Life Technologies. A custom-designed gene panel containing 15 TAAD candidate genes was used to screen 632 amplicons targeting all exons and exon–intron boundaries of the 15 genes over a total of 42 kb. Library preparation and sequencing were performed according to the manufacturer’s instructions (Ion AmpliSeqTM library kit 2.0, Life Technologies, Inc.). Sequencing quality analysis showed that 98% reads were on target, with a mean of 95% of bases for one sample having a Phred quality score (Q score) ≥ Q20. 95.8% of the samples had a >90% targeted region with a read depth of over 20× (Fig. S4). For variant calls, we chose the minimum coverage depth threshold of 20×. The sequencing data were submitted to National Population Health Data Center (NPHDC) (https://www.ncmi.cn/).
Data analysis and variant classification
Variants were called through raw paired-end sequence reads by two methods. First, raw data from sequencing runs were processed using Torrent Suite software (Version 5.04, Life Technologies, Inc.) to generate reads (BAM files), which were filtered to clean up the ones with low quality and mapped to reference genomic sequences (hg19) of target genes. Variant calling was performed with the Torrent Variant Caller Plugin of Torrent Suite software. For the second method, BAM files generated by the PGM platform were sorted and indexed, then insertion/deletion (indel) recalibration was performed to remove false positive single nucleotide polymorphisms near indel regions. Variant calling was separately performed by the Genome Analysis Tool Kit (GATK) and Samtools software, and the intersection of the results was used for the next filtering step, which was performed by the VariantFiltration module in GATK.
The union of the two results were annotated by ANNOVAR software, including genetic reference sequences, genomic and cDNA positions, amino acid changes, related information available from public databases such as dbSNP142 (2016 Jun), 1000 Genomes (2015 Aug), NHLBI Grand Opportunity Exome Sequencing Project (ESP6500siv2), ExAC03, Human Gene Mutation Database, ClinVar (2016 Mar), and multiple functional prediction databases (2015 Jan).
To identify clinically significant variants, variants were first excluded for both cases and controls with minor allele frequencies >0.1% in one of the following databases: 1000 Genomes (2015 Aug)_All, 1000 Genomes (2015 Aug)_EAS, ESP6500siv2_All, ExAC03_All, or ExAC03_EAS (Fig. S1). Second, disruptive variants such as frameshift indels, stop-gain variants, and splice acceptor/donor changes with dPSI in SPANR [13] >4 or <–4 were selected as suspected functional variants. Next, the potential functions of all missense variants were predicted by in silico-based computational analysis. Those meeting the following three criteria were also selected as suspected functional variants: (1) M-CAP score >0.025 [14]; (2) REVEL score >0.5 [15]; and (3) meeting at least five of the following six requirements, SIFT < 0.1, Polyphen2 (HVAR) > 0.9, CADD score > 20, GERP > 4, MutationTaster = “A” or “D”, and LRT = “D”. Finally, the variants that also existed in controls were deleted, and the remaining suspected functional variants were all confirmed using Sanger sequencing. A team of clinicians and geneticists evaluated these variants and classified them as pathogenic/likely pathogenic (P/LP) variants or variants of uncertain significance (VUS) using the American College of Medical Genetics (ACMG) variant classification scheme [16]. The P/LP variants identified in this study cause disease under dominant genetic model. The variants identified in this study were submitted to ClinVar (https://www.clinicalgenome.org/data-sharing/clinvar/). The accession numbers of all variants can be found in Supplemental material.
Follow-up and outcomes
The primary outcome was cardiovascular events related death. Secondary outcome included aneurysm or dissection recurrence and other cardiovascular adverse events. When patients appeared at least one outcome described above, they were classified as suffering from MACEs. The outcomes were obtained by contacting each patient or their relatives individually.
Statistical analyses
Characteristics of different groups were compared using the χ2 test for categorical variables if appropriate; otherwise, Fisher’s exact tests were used. For all continuous variables, Shapiro–Wilk test was applied to determine whether data were normally distributed. Independent sample t-tests combined with Levene’s tests were used to compare normally distributed variables between two groups, and Mann–Whitney U tests were used for non-parametric variables. One-way analysis of variance (ANOVA) plus post-hoc Fisher’s least significant difference test or the non-parametric Kruskal–Wallis test were used to compare among three or more groups. All statistical analyses were performed using SPSS software (Version 23).
Results
Study design and population characteristics
Study design was shown in Fig. S1. Five hundred seventy-eight unrelated iTAAD patients were included in this study. The median onset age was 51 years for men (interquartile range [IQR]: 43 to 59 years) (n = 456, 78.89%) and 54.5 years for women (IQR: 44–61 years). We identified distinct phenotypic profiles in different genders. The proportion of AR/SV aneurysms was greater in males than females (53.3% vs. 28.7%, 1.00E-6), but the proportion of ascending aortic aneurysms was lower (44.8% vs. 63.1%, 3.34E-4). Cardiac dysfunction and aortic regurgitation were also more common in men. Detailed population information is presented in Table S1. The 1454 controls were a self-reported non-TAAD population ethnically matched with cases.
Variant classification and diagnostic yield
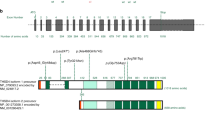
To identify clinically significant variants, we established a set of criteria (See “Materials and methods”) based on previous publications [17,18,19] and selected 99 suspected functional variants (Table S2, P = 4.57E-25 when compared with control). Of these, 93 (93.94%) were unique, and six (6.06%) were carried by two patients. FBN1, MYH11, and TGFBR2 variants were the most common (79.8%) (Fig. 1a). Sixty-seven suspected functional variants identifying in FBN1 were widespread across the gene, with the most common variation being the substitution of cysteine (23/67, 34.3%, Fig. S2). For MYH11 and TGFBR2, the suspected functional variations mainly resided in the myosin tail domain and protein kinase domain, respectively (Fig. S2). Variants on other genes were shown in Fig. S3. The numbers of total variants, variants in functional regions, and non-synonymous variants in samples with or without suspected functional variations are similar (Fig. 1b), suggesting similar genomic variation rates. The prevalence of nonsense variants in females was higher than in males (males: 10.6% vs. females: 33.3%; P = 0.033, Fig. 1c).

a Variants of individual genes (rows) are shown for 103 TAAD patients (columns). Percentages represent the fractions of variants in the specified gene (right) and in the specified gene per kb (left). Different variant types are shown in different colors. b Variation rate of target genome for samples with/without suspected functional variants. c Proportion of different types of variants in males and females.
Ninety-nine suspected functional variants were further classified as P/LP variants (Table S3) and VUS (Table S4) according to ACMG guidelines. 10.6% of patients (61 out of 578 patients) had P/LP variants, and other 7.27% (42 out of 578 patients) carried VUS. We compared the known P/LP variants with our newly identified ones (Table S5). Except for onset age was younger in newly identified P/LP variants, other features showed no significant difference (Table S6), suggesting the clinical significance of newly identified P/LP variants and the efficiency of our filtering criteria.
Genotype–phenotype correlations
The 578 iTAAD patients were then classified into three categories based on their genetic status: (1) genetic, with P/LP variants; (2) possibly genetic, with VUS; and (3) non-genetic. The onset age was much younger in genetic cases (38.13 vs. 47.62 (possibly genetic) vs. 51.20 (non-genetic), P for trend = 1.31E-13) (Table 1). Figure 2 shows that the onset age distribution of genetic cases was significantly different from that in the other two groups. Furthermore, the onset ages of FBN1 P/LP variants carriers were even younger than others genetic cases (36.5 (27.0–45.0) vs. 44.0 (38.0–56.0), P = 0.061).

Chance of having a mutation decreased with age (odds ratio: 0.950 per year; 95% confidence interval: 0.935–0.966; P = 1.08E-9).
Genetic cases were more likely to suffer from AR/SV aneurysms (P = 5.88E-14), but those without monogenic variants were more likely to suffer from ascending aortic aneurysms (P = 0.053) (Table 1). Monogenic factor was associated with aneurysms involving multiple segments (P = 0.043), and the incidence of which was much higher in non-FBN1 genetic cases compared with FBN1 genetic cases (57.1% vs. 7.4%, P = 0.004). In addition, genetic cases are more likely to have larger aneurysm. Z score of AR/SV (in patient with AR/SV aneurysm), Z score of ascending aorta (in patient with ascending aortic aneurysm), and maximum diameter of aorta were larger in genetic cases, with a P value of 4.44E-4, 0.005, and 1.00E-6, respectively (Table 1). Conversely, the proportion of risk factors such as hypertension and bicuspid aortic valves was lower in genetic cases (18.6% vs. 40.5% vs. 46.8% and 6.8% vs. 14.3% vs. 24.6%, P for trend = 5.50E-5 and 0.001, respectively).
The risk of dissection in individuals with aorta dilation is clinically important. 4.7% iTAAD patients had dissections at the time of first diagnosis (initial dissection, dissection occurred during follow-up were not included). The risk of dissection was significantly higher in genetic cases compared with the other groups (16.4% vs. 2.4% (possibly genetic) vs. 3.4% (non-genetic group), P = 4.50E−5). On univariable logistic regression analyses, the higher risk of dissection was associated with genetic cases (OR: 2.35; 95% CI: 1.57 to 3.51; P = 3.40E-5). When adjusted by onset age, hypertension, and maximal aortic diameter in multivariable logistic regression analyses, genetic status still showed a significant association (P = 0.002), suggesting it was an independent predictor of initial dissection. In addition, the incidence of initial dissection is lower in FBN1 P/LP variants carriers (FBN1 vs. non-FBN1: 13.0% vs. 42.9%, P = 0.08).
Effect of monogenic variants on prognosis
Four sixty-one cases with more than 3 years follow-up were used to assess the association between monogenic variants and poor prognosis. Follow-up was complete in 85.9% (396 of 461) of individuals, with a median time of 55 months (IQR: 45 to 64 months). Except with a less proportion of men (69.2% vs. 82.1%; P = 0.016) and lower left ventricular ejection fraction (P = 0.023), there were no other significant differences in the general situation or clinical phenotype for those with incomplete follow-up when compared with those with complete follow-up (Table S7). The proportion of genetic cases, possibly genetic cases, and non-genetic cases was also similar in two groups (10.1%/7.3%/82.6% vs. 7.7%/6.2%/86.2%, P = 0.769).
During follow-up, MACEs occurred in 59 patients, including aneurysm or other cardiovascular events related death (24, 40.7%), aneurysm or dissection recurrence (31, 52.5%), other cardiovascular adverse events (4, 6.8%). The follow-up time was similar for patients with and without MACEs (Table 2). We then compared general situations and clinical features of cases with and without MACEs, and found that there were differences in genetic status, onset age, and proportion of multi-segments involved and initial dissection (Table 2). On univariable regression, all the four variables showed the significant association with MACEs. However, only genetic status (P = 0.001) and initial dissection (P = 3.00E-6) remained to be the risk factors for poor prognosis on multivariable regression analysis (Table 3). As shown in Fig. 3a, the incidence of MACEs was significant higher in genetic and possibly genetic cases compared with non-genetic cases, and patients with initial dissection had an even much higher risk of poor prognosis no matter whether they had monogenic factor.

a Proportions of patients with/without MACE in different groups divided by genetic status and initial dissection. D with initial dissection; ND without initial dissection. b Density plots of onset age for (possibly) genetic cases and non-genetic cases without initial dissection. c Probability of MACE during follow-up of median 55 months based on onset age estimated by logistic regression models. Gray bands indicate 95% confidence intervals.
In subgroup analysis, we found that early onset age was associated with MACEs in non-genetic cases without initial dissection but not in genetic and possibly genetic cases (Fig. 3b and Table 3). Risk of MACEs varied markedly from 16.7% in the early onset age group to 3.7% in the late onset age group (Table 4). From 60 years old, the risk of MACE increased with the decrease of onset age. For every 10 years of onset age reduction, the risk of MACE increases by 58%. The estimated risk of MACEs was 16.55%, 10.45%, and 6.42% for onset age of 30, 50, and 70 years respectively in non-genetic cases without initial dissection (Fig. 3c).
Discussion
Our comprehensive analyses revealed that the burden of P/LP variants in the TAAD causal genes was 10.6% among our iTAAD cohort. Genetic cases showed younger onset ages, larger aortic diameter, and higher risk of multiple segments involved and initial dissection. MACEs occurred in 14.9% patients during follow-up of median 55 months. Monogenic factor, dissection at presentation, and early onset age were the main risk factors for poor long-term outcomes in iTAAD.
In our study, we classified the variants as VUS using the ACMG variant classification scheme. These VUS may have potential functions, but their existing evidence is not enough to classify them into “pathogenic” or “likely pathogenic” variants. With the increase of the co-segregation and family studies, de novo occurrence and genotype–phenotype correlation studies, the evidence of the causal effect of these variants will be further accumulated. When the criteria for “pathogenic” or “likely pathogenic” are met, these variants will be reclassified.
Our study may help international guidelines to complement the detailed instructions regarding genetic evaluation and family screening of iTAAD patients. First, we provide evidence for criteria to identify iTAAD patients who most likely to have a genetic predisposition. It has been suggested that patients with family history should undergo genetic testing [20]. However, even after excluding patients with a clear family history, P/LP variants could be detected in more than 10% of apparently sporadic patients in our study. Due to that most iTAAD remain asymptomatic for a long time, as well as that sometimes the causal variants are de novo, a negative family history does not exclude a genetic predisposition. Therefore, out results suggest that patients with young onset age, large aortic size, multiple segments involved, little risk factors and initial dissection should be advised to have genetic testing whether they have family history or not. Second, for individuals with a genetic predisposition, genotype–phenotype correlations serve more precise genetic counseling, prediction of the potential clinical phenotypes, and guidance of early clinical surveillance and prevention. For example, for individual with a genetic predisposition to aneurysms involving multiple segments, imaging should be more comprehensive to identify latent lesions. If a patient is predicted to be at a high risk of dissection, an improved control of risk factors such as hypertension, and regular examinations should be implemented to delay and monitor the occurrence of dissection. Third, family members of patients with a genetic predisposition can benefit from family screening. Although gene testing of family members cannot diagnosis the disease, it can promote the regular medical monitoring and even early preventive measures for family members with positive results. More importantly, genetic testing is the only way for prenatal diagnosis or pre-implantation diagnosis opportunities.
iTAAD shares some disease causal genes with sTAAD, such as FBN1 for MFS and TGFBR1/2 for Loeys-Dietz syndrome, implying a shared genetic predisposition to isolated and sTAAD. Moreover, most reported TGFBR2 variants in sTAAD were predicted to disrupt the kinase domain [21,22,23,24], similar with our results in iTAAD. On the other hand, we found the youngest onset age in patients with FBN1 variants. Consistently, the overall onset age of previously reported FBN1 variants carriers was younger than others (Table S8). We found that non-FBN1 variants were associated with aneurysm involving multiple segments and initial dissection, in consistent with which, a much higher incidence of aneurysms of descending aorta or other vessels were evident in TGFBR2 variants carriers, but rare in MFS caused by FBN1 variants [25, 26], and the proportion of dissection was also lower for previously reported MFS with FBN1 variants (Table S8). However, there are also differences between the genetic predisposition of isolated and sTAAD. Unlike most of the sTAADs that have a clear family history and causal mutation, this situation accounts for only about 20% of the iTAADs. This implies that the genetic landscapes of iTAAD are more complex and heterogeneous than that of sTAAD.
Besides clinical features, we also found that monogenic factor was the independent risk factors for poor prognosis. It has been reported that genotype affects the risk of cardiovascular death in MFS patients [17], while our study first time links the genetic status with the prognosis of non-sTAAD. Hence, genetic status may guide the modality and frequency of follow-up. In addition, unlike genetic cases, early onset age was found to be association with MACEs in non-genetic case. Previous studies have found that the early onset age of some diseases is significantly related to the poor prognosis, such as breast cancer [27, 28]. However, it remains unclear why early onset age is associated with poor prognosis of non-genetic TAADs, and future work is needed.
With methodological advances in genetics, increasing number of novel iTAAD genes have been revealed in recent years. For example, SMAD2, encoding proteins involved in the TGF-beta signaling pathway, were found to be associated with arterial aneurysms and dissections [29]. FOXE3 mutations lead to a reduced number of aortic SMCs during development and increased SMC apoptosis in the ascending aorta, thus lead to familial thoracic aortic disease [30]. Deficiency of LOX in mice or inhibition of lysyl oxidases in turkeys and rats causes aortic dissections [31]. More recently, we identified focal adhesion scaffold genes as a novel catalog of TAAD causal genes [32]. Although these novel genes were not included in our panel, they should not be ignored and will have the opportunity to be added to the TAAD gene testing panel in the future.
Limitations
Our study has a number of limitations. First, family members of our cases did not undergo genetic testing, so we cannot determine whether the variants are de novo or inherited. Second, the proportion of genetic patients might be underestimated because VUS variant could be reclassified in the future as P/LP variant, and moreover, beyond the 15 genes in this study, there must be other genes contributing to the disease. Third, our subgrouping resulted in a limited number of cases in individual group, and some analyses were based on a relatively small number of patients. More cases for validation and refining are therefore needed in the future.
Conclusions
Taken together, this study represents the most comprehensive investigation into phenotype, genotype, and the genotype–phenotype correlations for iTAAD, which is a heterogeneous condition. Our study optimizes the risk stratification of iTAAD patients, and assists the development of tailored precision medicine in iTAAD.
References
Cury M, Zeidan F, Lobato AC. Aortic disease in the young: genetic aneurysm syndromes, connective tissue disorders, and familial aortic aneurysms and dissections. Int J Vasc Med. 2013;2013:267215.
Quintana RA, Taylor WR. Cellular mechanisms of aortic aneurysm formation. Circ Res. 2019;124:607–18.
Isselbacher EM, Lino Cardenas CL, Lindsay ME. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation. 2016;133:2516–28.
Prakash SK, LeMaire SA, Guo DC, Russell L, Regalado ES, Golabbakhsh H, et al. Rare copy number variants disrupt genes regulating vascular smooth muscle cell adhesion and contractility in sporadic thoracic aortic aneurysms and dissections. Am J Hum Genet. 2010;87:743–56.
LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC 3rd, Johnson RJ, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000.
Pyeritz RE. Heritable thoracic aortic disorders. Curr Opin Cardiol. 2014;29:97–102.
Blunder S, Messner B, Aschacher T, Zeller I, Turkcan A, Wiedemann D, et al. Characteristics of TAV- and BAV-associated thoracic aortic aneurysms-smooth muscle cell biology, expression profiling, and histological analyses. Atherosclerosis. 2012;220:355–61.
Norton E, Yang B. Managing thoracic aortic aneurysm in patients with bicuspid aortic valve based on aortic root-involvement. Front Physiol. 2017;8:397.
Teixido-Tura G, Franken R, Galuppo V, Gutierrez Garcia-Moreno L, Borregan M, Mulder BJ, et al. Heterogeneity of aortic disease severity in patients with Loeys-Dietz syndrome. Heart. 2016;102:626–32.
Franken R, den Hartog AW, Radonic T, Micha D, Maugeri A, van Dijk FS, et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circ Cardiovasc Genet. 2015;8:383–8.
Johnston KW, Rutherford RB, Tilson MD, Shah DM, Hollier L, Stanley JC. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J Vasc Surg. 1991;13:452–8.
Campens L, Demulier L, De Groote K, Vandekerckhove K, De Wolf D, Roman MJ, et al. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories. Am J Cardiol. 2014;114:914–20.
Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science. 2015;347:1254806.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Franken R, Groenink M, de Waard V, Feenstra HM, Scholte AJ, van den Berg MP, et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;37:3285–90.
Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36:2327–37.
Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–35a.
Verhagen JMA, Kempers M, Cozijnsen L, Bouma BJ, Duijnhouwer AL, Post JG, et al. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int J Cardiol. 2018;258:243–8.
Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, et al. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–60.
Ki CS, Jin DK, Chang SH, Kim JE, Kim JW, Park BK, et al. Identification of a novel TGFBR2 gene mutation in a Korean patient with Loeys-Dietz aortic aneurysm syndrome; no mutation in TGFBR2 gene in 30 patients with classic Marfan’s syndrome. Clin Genet. 2005;68:561–3.
Yetman AT, Beroukhim RS, Ivy DD, Manchester D. Importance of the clinical recognition of Loeys-Dietz syndrome in the neonatal period. Pediatrics. 2007;119:e1199–202.
Watanabe Y, Sakai H, Nishimura A, Miyake N, Saitsu H, Mizuguchi T, et al. Paternal somatic mosaicism of a TGFBR2 mutation transmitting to an affected son with Loeys-Dietz syndrome. Am J Med Genet A. 2008;146A:3070–4.
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81.
Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–20.
Colzani E, Liljegren A, Johansson AL, Adolfsson J, Hellborg H, Hall PF, et al. Prognosis of patients with breast cancer: causes of death and effects of time since diagnosis, age, and tumor characteristics. J Clin Oncol. 2011;29:4014–21.
Anders CK, Hsu DS, Broadwater G, Acharya CR, Foekens JA, Zhang Y, et al. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. J Clin Oncol. 2008;26:3324–30.
Micha D, Guo DC, Hilhorst-Hofstee Y, van Kooten F, Atmaja D, Overwater E, et al. SMAD2 mutations are associated with arterial aneurysms and dissections. Hum Mutat. 2015;36:1145–9.
Kuang SQ, Medina-Martinez O, Guo DC, Gong L, Regalado ES, Reynolds CL, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Investig. 2016;126:948–61.
Lee VS, Halabi CM, Hoffman EP, Carmichael N, Leshchiner I, Lian CG, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci USA. 2016;113:8759–64.
Li Y, Gao S, Han Y, Song L, Kong Y, Jiao Y, et al: Variants of focal adhesion scaffold genes cause thoracic aortic aneurysm. Circ Res. 2021;128:8–23.
Acknowledgements
We thank all the patients for their participation in this study.
Funding
This study was supported by grants from the National Key Research and Development Program of China (2016YFC0903000), the National Science Foundation of China (81800218 and 81930014), the Key Laboratory of Remodeling-Related Cardiovascular Diseases, Ministry of Education of China (PXM2014-014226-000012), International Cooperation Project from the Ministry of Science and Technology of China (2015DFA31070), and the Beijing Collaborative Innovative Research Center for Cardiovascular Diseases.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Li, Y., Kong, Y., Duan, W. et al. Evaluating the monogenic contribution and genotype–phenotype correlation in patients with isolated thoracic aortic aneurysm. Eur J Hum Genet 29, 1129–1138 (2021). https://doi.org/10.1038/s41431-021-00857-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00857-2
