
Abstract
Spiders store spidroins in their silk glands as high concentration aqueous solutions, spinning these dopes into fibres with outstanding mechanical properties. Aciniform (or wrapping) silk is the toughest spider silk and is devoid of the short amino acid sequence motifs characteristic of the other spidroins. Using solution-state NMR spectroscopy, we demonstrate that the 200 amino acid Argiope trifasciata AcSp1 repeat unit contrasts with previously characterized spidroins, adopting a globular 5-helix bundle flanked by intrinsically disordered N- and C-terminal tails. Split-intein-mediated segmental NMR-active isotope-enrichment allowed unambiguous demonstration of modular and malleable “beads-on-a-string” concatemeric behaviour. Concatemers form fibres upon manual drawing with silk-like morphology and mechanical properties, alongside secondary structuring and orientation consistent with native AcSp1 fibres. AcSp1 structural stability varies locally, with the fifth helix denaturing most readily. The structural transition of aciniform spidroin from a mostly α-helical dope to a mixed α-helix/β-sheet-containing fibre can be directly related to spidroin architecture and stability.
Similar content being viewed by others


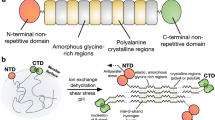
Introduction
Spiders can produce up to seven types of silk that surpass synthetic materials in ultimate tensile strength (i.e., maximum stress, or force per cross-sectional area, withstood before breaking) and toughness (i.e., energy absorbed before breaking) per unit weight1,2,3. Spider silk proteins, or spidroins, are large (250–500 kDa) and have a general architecture comprising a repetitive domain, accounting for at least 90% of the total protein sequence, flanked by non-repetitive N- and C-terminal domains. Spidroins are highly soluble in the gland and, when needed, efficiently self-assemble into insoluble fibres4. The protein secondary structure also changes during this process, typically from a soluble mixture of random-coil, polyproline-II- helices and/or α-helices to a fibre enriched in β-sheet content but still exhibiting significant disorder5,6,7.
Aciniform silk is the toughest spider silk and is composed of the protein aciniform spidroin 1 (AcSp1)8. It is the primary component of wrapping silk, which is used to wrap and immobilize prey. Present knowledge of spider silk structure and function is heavily based on dragline silk, the strongest of the spider silks9. During the transition from the soluble state to the fibre form, dragline silk converts from a disordered state10, likely exhibiting polyproline-II and transient α-helical character7,11, to a β-sheet microcrystal-rich aggregate12,13. AcSp1 from Nephila clavipes, conversely, is ~50% α-helical in the aciniform gland and ~24% α-helical and ~30% β-sheet in the solid fibre7. Retention of significant α-helical content in the insoluble form is unique to the aciniform and piriform silks, with piriform silk morphology differing in that it functions in disc form rather than as an isolated fibre7,14,15.
A typical hallmark of spidroins is the presence of small (usually ≤10 amino acid) primary structural motifs (GGX, GPGXX, An, etc.)1,16,17. These motifs have been directly linked to specific mechanical properties, particularly for dragline silk4,13,18. In contrast to this, AcSp1 is composed of concatenated ~200–400 amino acid repeat units completely lacking these short motifs8. AcSp1 primary and secondary structure as well as fibre mechanical properties therefore differ from the other spidroins and the link between these characteristics remains elusive.
To date, only three spidroin repetitive domain structures have been solved19,20 alongside several non-repetitive N- and C-terminal domain structures20,21,22,23,24,25,26. The reported repetitive domain structures are all highly similar seven-helix bundles19,20. Two of these are of tubuliform spidroin TuSp1 repeat units 1 and 220 and the third is of a putative AcSp1 repeat unit19, all recombinant proteins based upon genes annotated from Nephila antipodiana. Tubuliform (or cylindriform) spidroin is quite divergent from the other spidroin family members, with a particularly low glycine and elevated serine content. Unlike aciniform spidroin, tubuliform spidroin has also been shown to undergo a complete conversion to β-sheet/random-coil in the fibre without retention of α-helical character7. The previously reported structural similarity between AcSp1 and TuSp1 is therefore unexpected.
Here, we use solution-state NMR spectroscopy to determine the atomic-level structure and dynamics of recombinant AcSp1 based upon the Argiope trifasciata spidroin. In the native form, this AcSp1 protein is a concatemer of a 200 amino acid repeat unit (referred to as the W unit herein) iterated at least 14 times and flanked by non-repetitive C-terminal and, putatively, N-terminal domains8,27. We demonstrate the AcSp1 structure to be unlike the previously determined spidroin repeat unit structures and, in addition, validate and present the AcSp1 repeat domain in the structural context of the concatemer. Fibres may be readily drawn from our concatemer NMR samples, with morphology and secondary structure properties highly similar to native AcSp1 fibres from Argiope aurantia and mechanical properties approaching those of native silk.
Results
The Structure of W1
The soluble form of the W unit (W1: 199 amino acids, lacking the C-terminal serine of the 200 amino acid repeat) exhibits a well-folded and tightly packed ellipsoidally-shaped helical core over residues 12–149 flanked by unstructured tails (Fig. 1). Heteronuclear 1H-15N nuclear Overhauser effect (NOE) enhancement factors28 are indicative of a rigid protein core, reflected by positive enhancement factors and flexible dynamic tails, reflected by negative or near-zero enhancement factors (Figs 1b and 2c), corroborating the localization of folded vs. disordered domains29,30. The disordered tails of W1 are also evident both from a lack of inter-residue 1H-1H NOE restraints31 and according to chemical shift-derived TALOS+ dihedral angle assignments32 (Supplementary Figs. S1a and S2). The solution-state W1 structure therefore comprises a compact, predominantly helical globular core with intrinsically disordered N- and C-terminal tails.

Solution-state NMR structure of W1.
(a) Overlay of 20 lowest energy members of the NMR ensemble. Each helix in the converged domain is coloured differently, as annotated directly on the figure; the intrinsically disordered portions excluded from r.m.s.d. calculations are in green. (b) Heteronuclear 1H -15N NOE enhancement factors represented on the W1 lowest energy structure (bar graph in Fig. 2c). (c) The lowest energy structure coloured according to the Kyte-Doolittle hydrophobicity scale72 shown in ribbon/stick and surface (inset) representations.

Modularity of the repeat unit of AcSp1.
(a) Overlay of 1H-15N HSQC experiments for W1 (green) and W2 concatemers with first (blue; W2−1) or second (red; W2−2) W unit 15N-enriched. (b) Combined chemical shift difference between W2 and W1 (W2−1 blue; W2−2 red; data are presented in Supplementary Fig. S6). (c) Heteronuclear 1H-15N NOE enhancement factors for W1 (green), W2−1 (blue) and W2−2 (red). (d) Schematic of secondary structure as a function of sequence based upon DSSP63 secondary structure assessment of the 20-member ensemble in Fig. 1.
Although chemical shift patterns are indicative of ~6 α-helices33, our structural refinement (Table 1) using 1H-1H NOE-based distance restraints with >20 NOE contacts per residue over most of the globular region (Supplementary Fig. S1), coupled with TALOS+32 dihedral angle restraints (Supplementary Figs. S2 and S3) and hydrogen-deuterium exchange-derived hydrogen bond restraints34 (Supplementary Figs. S1b and S3), demonstrates 5 α-helical regions. Despite the lack of a continuous helix in the putative helix 2 of the original topology33, ~65% of residues in the S40-G60 stretch adopt α-helical ϕ and ψ dihedral angles (Supplementary Fig. S4). The observation of chemical shifts implying α-helical structure is, therefore, not unreasonable. As a whole, this segment of W1 is well converged and structured, with localized helical, β- and γ-turns located in the core of the protein. Also supporting this non-canonical structuring, the N-terminal region of this segment includes several residues exhibiting chemical shifts outside the statistical norm33. Further evidence for a five-helix structural topology is clear both on the basis of regions exhibiting expected31 α-helical NOE connection patterns (Supplementary Fig. S3) and in direct analysis of the final ensemble of NMR structures. Also of note, the two phenylalanines in the central helix (F90 and F95) are mildly solvent exposed on opposing sides of W1 while the other 8 aromatic amino acids fall in the top 25% most solvent exposed residues over the NMR ensemble (Supplementary Fig. S5). Exposure of aromatic moieties in this manner may promote protein-protein association during fibre self-assembly.
Effect of Concatenation upon the W Unit
To investigate the effect of concatenation of the W unit upon protein structuring and dynamics by NMR, concatemers of two W units (W2) were studied. To unambiguously distinguish each W unit, selective enrichment of one of the two W units with NMR-active 13C- and/or 15N-isotopes was carried out using split-intein-mediated trans-splicing35,36. This was feasible by separately expressing fusion proteins of one W unit with the appropriate Ssp GyrB split-intein37 fragment in E. coli and trans-splicing the two W units together to produce a concatemer linked by a native peptide bond (Fig. 3).

Segmental labelling of W2 by split-intein-mediated trans-splicing (IN and IC refer to N- and C-terminal intein fragments, respectively).
In the illustrated splicing scheme, the W1In fusion protein is enriched with NMR active isotopes (blue), while the IcW1 fusion protein is at natural abundance (grey). In the illustrated SDS-PAGE gel, splicing products were passed through a Ni-NTA column, allowing the spliced W2 to be collected in the flow-through while all other proteins were His6-tagged and thus retained in the column.
1H-15N HSQC experiments for W1 and for each W unit in W2 demonstrate strikingly similar chemical shift patterns (Figs 2a,b and Supplementary Fig. S6). Quantitative backbone chemical shift comparison38 provides further insight, clearly demonstrating that only the residues immediately adjacent to the covalent link between W units differ between W1 and W2 (Fig. 2b and Supplementary Fig. S6a). Echoing this behaviour, W unit independence likely extends to the 3-unit concatemer (W3), as the 1D 1H-NMR spectra of W1, W2 and W3 are practically indistinguishable with exception of intensity, which increases in direct proportion to the number of W units present in the concatemer (Supplementary Fig. S6b).
The steady-state heteronuclear 1H-15N NOE enhancement factor provides a position-specific probe39 of intramolecular dynamics because it is highly sensitive to the local effective correlation time28,40. Amide H-N bonds exhibiting a heteronuclear NOE enhancement factor of 0.65 or greater are typically attributed to regions of the protein that experience minimal internal motion faster than the ~5–20 ns rotational correlation time typical of a protein41, although it should be noted that the exact value of enhancement factor used as a cut-off to identify internal motion depends upon the system in question42. Comparison of heteronuclear 1H-15N NOE enhancement factors as a function of backbone position in each W unit of W2 relative to W1 demonstrates that, in all cases, the helical domain exhibits high enhancement factors typical of tumbling as a globular protein domain while the first 11 and last 50 residues of each W unit display decreased enhancement factors typical of internal motion on the ps-ns time scale (Fig. 2c). Logically, concatenation produces some apparent damping of the dynamics in W2 at the link between units vs. at the termini. In all cases, the N- and C-terminal portions of the repeat domain retain the elevated dynamics expected of an intrinsically disordered segment30.
Hydrodynamics of AcSp1
To better characterize AcSp1 architecture, translational diffusion coefficient (DC) values for W1, W2 and W3 were determined using pulsed field gradient diffusion-ordered NMR spectroscopy (DOSY)43. For comparison, dynamic light scattering (DLS) was used to determine the hydrodynamic diameter (dH) of each W species. The observed DC of W1 was in good agreement with the NMR-derived structural ensemble (Supplementary Table S1). No component to the scattering decay curve attributable to a monomeric species was observed for filtered W1 in acetate buffer by DLS, where the predominant species giving rise to scattering appeared to be nanoparticles of ~100–200 nm size consistent with our previous DLS studies of W1 in phosphate buffer44. The source of this discrepancy between NMR (where a 100–200 nm particle would be unobservable by standard solution-state methodology) and DLS (where only the large species is observed) remains elusive. W2 and W3, conversely, exhibited clear scattering from monomeric species. The dH for W2 agrees outstandingly well with that inferred from the viscosity-corrected45 DC while the dH of W3 is inferred to be slightly more compact by DLS than by DOSY (Supplementary Table S1). As a whole, the hydrodynamic properties of W2 and W3 are highly consistent between DOSY and DLS, while those of W1 agree between DOSY and the high-resolution structural ensemble.
Concatemeric Structural Ensembles
DOSY-derived DC values were used to infer46 a radius-of gyration (Rg) for each AcSp1 species (Supplementary Table S1). Calculation of a W1 structural ensemble was carried out with addition of an Rg restraint47, using restraint weighting appropriate to ensure that other NMR-derived restraints were not violated. The incorporation of Rg as a restraint for W1 led to no significant change in the averaged hydrodynamic properties of the structural ensemble (Supplementary Table S1). Based upon the outstanding agreement between W1 and W2 backbone chemical shifts (Fig. 2), W2 and W3 structural ensembles (Fig. 4 and Supplementary Table S1) were calculated using concatenated sets of the W1 NMR restraints (Table 1). Unlike the observation with W1, the levels of agreement between the measured DC and that inferred from structural ensembles for W2 and W3 improved with addition Rg restraints47 (Supplementary Table S1) without increased violation of the other NMR-derived restraints. The W2 and W3 structural ensembles calculated incorporating Rg showed a general increase in compactness in comparison to those without.

Structural ensembles (semi-transparent colouring) underlying the representative structure (solid colouring; Rg closest to that determined by DOSY NMR) of W2 (a) and W3 (b).
Ensembles (10 members shown) were calculated using concatenated sets of W1 NMR restraints with an Rg restraint estimated from the observed DCDOSY.
Properties of W2 Fibres
The functional relevance of the NMR conditions for fibre formation was tested based on our previous demonstration that silk-like fibres can be manually drawn from solutions of concatemers comprising 2 to 4 W units48. Fibres with ~1.5 μm diameter and a surface morphology of smaller fibrils aligned parallel to the fibre long axis (Fig. 5), consistent with our previous recombinant W fibre characterization, could be readily drawn from NMR samples of W2 proteins. Interestingly, the strength and toughness of recombinant W fibres appears approximately proportional to concatemer size (Table 2), with W2 (whether produced intact or intein-spliced) exhibiting values ~half those of W4 fibres48 and ~5–10% those of native A. trifasciata aciniform silk (W14 or larger, plus non-repetitive domain(s)8).

W2 fibre surface observed by SEM (a) and intermittent-contact AFM (b
– colour scale for height image shown; boxed region of amplitude image shown at higher resolution to the right).
Comparison of polarized Raman spectromicroscopy of W2 fibres and of natural aciniform silk fibres from Argiope aurantia demonstrates very close agreement in amino acid composition, secondary structure and molecular orientation (Fig. 6). Curve-fitting of the amide I band of the orientation-insensitive spectra11 demonstrates a significant decrease in α-helicity and appearance of β-sheet character in W2 fibres relative to the solution-state structure, highly consistent with wrapping silk fibres produced by both A. aurantia (decomposition in Supplementary Fig. S7) and N. clavipes7 (Table 3). Analysis of relative amide I and amide III band peak height in the XX and ZZ polarized Raman spectra demonstrates that the α-helices and β-sheets are predominantly aligned along the fibre axis.

Comparative secondary structuring and orientation observed by polarized Raman spectromicroscopy for the indicated fibre type.
Local Stability Variation Within the W unit
Fibre formation by W2 is inhibited by the addition of the chaotropic reagents urea and guanidinium chloride (GdmCl) or the zwitterionic detergent dodecylphosphocholine (DPC) above its critical micelle concentration (CMC ≈ 1.1 mM49). Far-UV circular dichroism (CD) spectroscopy demonstrates complete denaturation of W1 and W2 upon titration with both chaotropes (Supplementary Figs. S8a and b), whereas DPC-induced changes are subtle at the global level reflected by CD spectroscopy (Supplementary Figs. S8c and d) but drastic at the backbone amide level observed by NMR spectroscopy once the CMC is exceeded (Supplementary Figs. S9 and S10). To rule out effects of ionic strength vs. the chaotrope activity of GdmCl, a control titration was carried out for W1 using NaCl (Supplementary Fig. S11). Ionic strength-dependent chemical shift perturbation is clearly apparent, but the typical W1 HSQC spectral pattern is maintained with NaCl, unlike with GdmCl, where a contraction in cross-peak dispersion consistent with denaturation is observed.
In each of the chaotrope and DPC titrations, helix 5 (residues 135–149, yellow in Fig. 1a) and the residues located directly underneath it are the most readily perturbed portions of the W unit (Fig. 7 and Supplementary Fig. S9). Correspondingly, of all helical segments in W1, helix 5 was the least protected from H/D exchange in buffer (Supplementary Fig. S1b). In converse to the perturbation at helix 5, even at the DPC endpoint, the W2 linker region remained unperturbed (Supplementary Fig. S10). The propensity of the fifth helix towards unfolding may be rooted in its primary structure, given that a variety of sequence-based analysis algorithms predict the entire stretch of W1 over residues 125–199, including helix 5, to be intrinsically disordered (Supplementary Fig. S2).

Site-specific perturbation of W1 upon denaturation or detergent treatment.
(a) Normalized combined chemical shift displacement (CSD)38 as a function of amino acid position in W1 for titrations with the detergent dodecylphosphocholine (DPC; 20 mM endpoint) or the indicated chaotropic denaturant (2.5 M urea or 0.8 M GdmCl titration points). (b) Representation by thickness and colour of average CSD caused by urea, GdmCl and DPC on a cartoon representation of the lowest-energy member of the W1 structural ensemble.
Discussion
The mechanical properties of spider silks are predominantly linked to the spidroin repetitive domain. Comparison of repetitive domain structuring before and after fibre formation is fundamental to understanding both the exceptional mechanical properties of spider silks and their self-assembly. We present the structure of the repeat unit of the toughest spider silk, aciniform silk, from A. trifasciata. The observed globular 5-helix W1 architecture has no structural resemblance (Supplementary Fig. S12) to the 7-helix bundle previously reported for tubuliform spidroin repetitive unit and for a truncated, putative aciniform spidroin repetitive unit. These latter two spidroin repetitive units were both identified from an expressed sequence tag library from N. antipodiana and share only ~27% and 22% pairwise sequence identity with W1, respectively19. W unit structuring generally appears insensitive to conditions with robust refolding following thermal denaturation44; therefore, the observed difference in fold between W unit and the other spidroin repetitive units seems unlikely to be caused by differences in experimental conditions. Rather, this appears to be a fundamental architectural difference with as yet unknown source.
In each of the previous studies, only isolated repeats were employed and the impact of repeat unit concatenation was not investigated at the atomic level or discussed. Chemical shift and backbone amide dynamics comparison, alongside hydrodynamics characterization, allowed us to demonstrate that the repeat units in W1 and W2 are structurally indistinguishable and that there are no detectable persistent interactions between the two repeat units in W2. Modularity of behaviour also extends to W3, implying that the W1 structure is representative of the repeat unit structure in the context of a large, multiple-repeat protein. For the first time, we demonstrate the modularity of a spidroin repetitive domain at the atomic level. As a whole, the W1 unit is a highly tractable module for study, providing direct insight into much larger proteins otherwise infeasible to study at atomic resolution.
Notably, the conditions under which we have characterized W2 are clearly directly relevant to fibre formation given that fibres formed by manual pulling, including directly from NMR buffer, have architecture and orientation of structural units consistent with natural wrapping silk from A. aurantia. Fibres, conversely, cannot be pulled from solutions of W1 alone48. Structural study of the functional W2 fibre precursor, consisting of identical W1 19 kDa subunits (with ~50% of content being Gly, Ala and Ser) concatenated into a repetitive 38 kDa protein was possible only through intein-mediated protein trans-splicing. As highlighted in recent literature reviews50,51,52, such a strategy has strong potential for direct characterization of other comparably challenging and interesting systems. The similarity in both structure and dynamics of W1 and W2 implies that W1 is directly representative of the functional AcSp1 repeat unit in the state where it is primed for fibre formation.
Concatemers larger than W2 tend to oligomerize in solution, with faster rates observed for larger concatemers, precluding extended solution-state NMR studies. W4, for example, oligomerized into a visible precipitate during DOSY experiments, rendering NMR-based hydrodynamics characterization infeasible. Structural studies of tubuliform spidroin TuSp1 domains were performed in DPC micelles, as these conditions stabilized the monomeric forms of a variety of TuSp1 constructs20. We had, therefore, hoped that DPC would act to stabilize large W unit concatemers sufficiently for hydrodynamics characterization. Unexpectedly, however, we observed that DPC specifically inhibited fibre formation of W2 despite minimal structural perturbation evident by CD spectroscopy. This observation, in light of similar inhibition of fibre-formation behaviour upon addition of urea and GdmCl, spurred our investigation of the effects of DPC titration upon W unit structuring. Fortuitously, the NMR spectroscopic behaviour of the W unit in the presence of DPC allowed for much clearer tracking of individual HSQC-based N-H correlations than in chaotrope solutions, allowing unambiguous demonstration of localized perturbation in both W1 and W2.
The observation of a locally destabilized region of the W unit folded domain implies potential for localized structural modulation during fibre formation. Structural transition could be initiated by application of shear forces, for example, during the manual fibre pulling process. Helix 5, which is positioned on the surface of the helical bundle without extensive inter-helical interactions, normally positions the globular bead-like domain proximally to the linker (Fig. 1). Its denaturation and concomitant extension would therefore allow for structural decompaction through decreased constraint upon the linker. Protein-protein interaction would, in turn, be facilitated through an overall increase in surface area and introduction of a longer and more flexible string-like linker with potential to entangle with proximal molecules. Future testing of both the effects of stabilizing this helix (e.g. through replacement with a stabilized helix, as employed probing β2-microglobulin amyloid formation53) and of holding the helix in place (e.g., with a disulfide, as employed to trap the SH3 domain transition state54) would be ideal to provide a more detailed characterization of the role of localized unfolding in aciniform silk fibre formation.
Notably, the W2 fibre demonstrates a secondary structural transformation from its soluble precursor very similar to that of native wrapping silk of N. clavipes. Although the dope has not been characterized in the A. aurantia aciniform gland, the W2 fibre also exhibits outstanding agreement with A. aurantia wrapping silk fibre structural properties. In short, partial α-helix to β-sheet transition is observed in all cases upon conversion from the soluble spidroin to fibrous silk state. Chemical shift-based secondary structure propensity (SSP) analysis55 implies that the only regions of W2 with nascent β-strand propensity in the soluble state are found flanking helix 5 and proximal to the junction between W units (Supplementary Fig. S10c). It is therefore probable that β-Sheet formation is seeded on the helix 5 face of the W unit and within the linker between neighbouring W units. A significant portion of the linker, however, likely retains its disordered state given that the final proportion of disorder in the fibre is ~40%. The fact that 6 of 8 prolines are found in a 20-residue stretch of the linker (between residue numbers 172 and 191 of the W unit) provides a further, significant constraint against complete conversion of the disordered linker to β-sheet.
Based upon these findings, fibrillogenesis may be hypothesized to take place as follows. First, localized unfolding of helix 5, encouraged by shear forces, decompacts a given W unit, inducing increased intermolecular interactions. Intermolecular β-sheet formation subsequently occurs, perhaps following alleviation of some mechanical force. The resulting fibre consists of helical domains composed of the stable helix 1–4 core of the bead-like globular portion of AcSp1 alongside β-sheet-rich domains. A significant proportion of disorder, or non-canonical secondary structuring, would arise in the linker and, potentially, the structured but non-helical segment between helices 1 and 2 (residues 40–60 of W1), providing elasticity to the fibre. The end-result would be a fibre composed of a mixture of discrete α-helical and β-sheet domains embedded in a disordered and elastic protein milieu.
In summary, the soluble form of the repetitive domain of the spider wrapping silk protein AcSp1 is composed of globular domains (“beads”) containing 5 helical segments held together by compact but intrinsically disordered linkers (“strings”). This beads-on-a-string architecture was shown unambiguously by independently and selectively enriching each W unit in the two-unit concatemer (W2) with NMR-active isotopes using split-intein-mediated trans-splicing. Comparison of Raman spectra of fibres drawn directly from recombinantly produced W2 protein samples and of natural A. aurantia aciniform silk fibres demonstrate strong similarity in amino acid composition, secondary structure and molecular orientation. A fibrous wrapping silk architecture composed of discrete oriented α-helix and β-sheet rich modules in a setting of intrinsically disordered protein would logically arise from the observed locally variable helical stability in the soluble form of the AcSp1 repeat unit. The mixture of α-helical, β-sheet and non-canonical secondary structures making up the outstandingly tough spider wrapping silk fibre thus can be related directly to the modular architecture and properties of its soluble precursor protein.
Methods
Protein Expression and Labelling
SUMO-W1 and -W2 fusion proteins were constructed as contiguous genes in a modified pET32 plasmid, expressed, labelled with NMR active isotopes, cleaved using SUMO protease and reverse purified as previously described33,48. For segmental-labelling, two constructs containing the N-precursor (W1IN: W1 + intein N-fragment (IN) + His6 tag) and C-precursor (ICW1: His6 tag + intein C-fragment (IC) + W1; this fusion protein required addition of urea for solubilisation at all stages from lysis (4 M) through to purification (2 M)) were constructed for use with the split-intein Ssp GyrB37. N- and C-precursors were purified by Ni-NTA affinity chromatography. To segmentally-label W2 proteins through intein trans-splicing, excess of the unlabelled N- or C-precursor was mixed with the corresponding isotope-enriched C- or N- precursor, respectively, to make efficient use of isotope enrichment. The splicing reaction was carried out in purification elution buffer (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, pH 8.0) with 1 mM DTT at 4 °C for > 6 hours. The mixture was then dialyzed against 50 mM potassium phosphate, pH 7.5 at 4 °C for > 2 hours and reverse purified by passing through Ni-NTA Sepharose. Any remaining unreacted precursors and all intein fragments had His6 tags and were thus trapped, leaving the tag free W2 protein to flow through the column. Splicing and purification efficiencies were analysed by SDS-PAGE and visualized by staining with Coomassie Brilliant Blue R-250.
NMR Spectroscopy
W1 (~0.2 mM) NMR experiments in NMR buffer (20 mM sodium acetate, 1 mM 2,2-dimethyl-2-sila-pentane-5-sulfonic acid (DSS), 1 mM NaN3; pH 5) in H2O:D2O at 9:1 (v:v) were acquired, processed and assigned as previously described33 with the addition of an aromatic 13C-edited NOESY-HSQC (mixing time: 85 ms). Backbone NMR experiments were acquired for 13C/15N enriched segmentally-labelled W2 proteins (~0.2–0.3 mM in NMR buffer) using a 16.4 T Avance III spectrometer equipped with a 5 mm TCI cryoprobe (Bruker, Milton, ON, Canada) at 303.15 K in the same manner as for W1. 1H-15N HSQC experiments were used to monitor H/D exchange at 0 h (control), 6 h and 40 h time point at 16.4 T with 24 scans, 2048 and 192 points in the 1H and 15N dimensions respectively and a recovery delay of 1.5 s. H/D exchange was performed by replacement of the original NMR buffer in 90% H2O and 10% D2O with NMR buffer in 100% D2O using centrifugal 15 mL spin dialysis filters (EMD Millipore, Billerica, MA). Heteronuclear NOE enhancement factors were measured using the standard Bruker sensitivity enhanced 1H-15N HSQC experiment with saturation during the recycle delay performed in an interleaved manner for recording relative NOE enhanced vs. unenhanced signal intensity (hscqnoef3gpsi3d; d1 of 5 s). Enhancement factors are reported as I(sat)/I(ref), where I(sat) is a given peak height under saturation and I(ref) the corresponding height without. Experimental data were processed with NMRPipe56 and assigned via CcpNmr Analysis57. Combined chemical shift displacements38 for each residue in each W unit in W2 relative to W1 were calculated using the backbone H, N, CA and CO chemical shifts weighted by gyromagnetic ratio.
W1 Restraint Refinement and Structure Calculation
NOE-derived distance restraints for W1 were assigned in CcpNmr Analysis57 for the following spectra: a 15N-edited NOESY-HSQC, a 1H-13C HSQC-NOESY-1H-15N HSQC, a 13C-edited NOESY-HSQC and an aromatic 13C-edited NOESY-HSQC. Dihedral angle restraints were produced using TALOS+32. H-bond restraints were generated on the basis of assigned 1H-15N HSQC peaks remaining following 6 h of H/D exchange based on the premise that backbone amide peaks resistant to exchange are located in helical regions34. Highly ambiguous NOEs were filtered with ARIA 2.158, using NOE distances and TALOS+ dihedral restraints. In total, 8 iterations were performed in ARIA, folding the structure in torsional space and employing network anchoring for the first 3 iterations. 40 structures were calculated, saving the 15 lowest energy structures for the next round, with the exception of the final iteration where 100 structures were calculated and the 20 lowest energy structures were imported into Analysis. Automatically assigned ambiguous NOEs were manually checked and ambiguity was reintroduced based on a 20 Å distance cut-off.
Following ARIA refinement, an NOE restraint list was generated for structure calculation with Xplor-NIH 2.32 employing the RAMA multi-dimensional torsion angle database potential term59. Dihedral and H-bond restraints were also employed (final restraints summarized in Table 1). During iterative Xplor-NIH restraint refinement, NOE restraints were heavily weighted until the structure converged; the weight of dihedral angle restraints was then increased, followed by H-bonds, until the energies began to increase for each class of restraint. Following ensemble calculation with the final set of restraints, water refinement was performed using Xplor-NIH56. The 20 lowest energy structures out of 100 calculated in this manner were retained for the final ensemble and visualized using Chimera60 and VMD61. PROCHECK-NMR62, in-house tcl/tk scripts (freely available upon request) and DSSP63 were used to assess structure quality, restraint violations and structural features, respectively. The average backbone and all-heavy atom r.m.s.d. values were calculated relative to the lowest energy structure using VMD59.
DOSY-Based Hydrodynamics Measurements
Translational diffusion coefficient (DC) values for W1, W2 and W3 proteins (0.2 mM in NMR buffer, 1 mM DSS and 1 mM NaN3 at pH 5 (H2O:D2O = 9:1); 0.06% dioxane as an internal viscosity control56) were determined at 303.15 K from 1H diffusion ordered spectroscopy (DOSY) experiments employing pulsed field gradient (PFG) NMR43 using an 11.7 T Avance NMR spectrometer equipped with a z-axis gradient and a BBFO SmartProbe (Bruker Canada). DOSY (64 scans, sweep width 12 ppm, relaxation delay incorporating presaturation 2 s) employed stimulated echo and longitudinal eddy current delay (LED) with bipolar gradient pulses and two spoil gradients64. The envelope of 1H signals was attenuated by increasing the gradient strength from 2% to 95% in 16 steps. The observed signal intensity as a function of gradient strength was fit using a single component exponential fit of the signal decay and the DC was determined from the fit using the simfit program within the T1/T2 Relaxation module of Bruker Topspin 3.1 using the Stejskal-Tanner formula65:

where I is the observed signal intensity, I(0) is the back-calculated unattenuated signal intensity, γ is the gyromagnetic ratio of 1H (4257.7 Hz/G), g is the gradient strength (based on maximum amplitude 53.5 G/cm at 100%), δ is the gradient pulse length (8 ms) and Δ is the diffusion time (100 ms). The radius-of-gyration (Rg, in Å) is calculated as46:

where T is the temperature (in K), η is the viscosity (in cP) estimated using the experimentally observed DC of a dioxane internal standard45 and DC is the observed diffusion coefficient (in cm2/s). Protein hydrodynamic diameter (dH) was inferred from the DOSY-based DC (DCprotein) and corrected for viscosity using the relationship:

where DCdioxane is the measured DOSY-based DC of the dioxane internal standard in a given sample and dHdioxane is the known hydrodynamic diameter of dioxane (0.424 nm)45.
DLS-Based Hydrodynamics Measurements
DLS was carried out at an angle of 173° using a 633 nm He-Ne laser using a Zetasizer nano ZS (Malvern, Worcestershire, UK) as described previously44. Scattering of filtered (0.45 μm, EMD Millipore) W1, W2 or W3 at both equimolar (~20 μM) and at ~equivalent W-unit concentrations (56 μM W1, 28 μM W2 and 20 μM W3) in NMR buffer was measured (in duplicate) in disposable, 10-mm path length, polystyrene cuvettes (Sarstedt, Montréal, QC, Canada) at 30 °C. Duplicate measurements were made using an automated attenuator 4.65 mm away from the cuvette wall and the resulting autocorrelation function obtained was analysed using Zetasizer software ver. 7.10 under the Protein Analysis Model to determine the average dH of the primary species in each W sample.
W2 and W3 Structure Calculation
The W1 NOE, H-bond and dihedral angle restraints from the final round of structure calculations (summarized in Table 1) were propagated over residues 200–400 (W2 and W3) and 400–600 (W3) to generate restraint files for W2 and W3. 100-member structural ensembles were calculated in the same manner as for W1 without water refinement using Xplor-NIH 2.3259 under the final simulating annealing energy weights used for W1 with visualization in Chimera60. Direct comparison of the lowest energy 20 ensemble members produced either in the absence of or with DOSY-derived Rg restraints (the scaling of Rg in the simulated annealing potential59, determined iteratively, was the maximum which did not induce a major increase in overall energy) was performed. An in-house python script was used to test for restraint violations. HYDROPRO66 was used to calculate the DC of each ensemble member based upon its coordinate file, allowing direct comparison of DOSY derived DC to the ensemble-average behaviour.
W2 Fibre Formation and Mechanical Properties
W2 fibres were pulled from 20–200 μM W2 protein dissolved either in 50 mM potassium phosphate (pH 7.5) or in NMR buffer, as previously described48. Before tensile strength testing, the diameter of each fibre was measured using light microscopy at 400× magnification. Three micrographs were employed for each fibre, two taken near each end and one in the ~middle of the fibre. From each micrograph, three locations were analysed using ImageJ 1.47 v67 to determine the diameter of the fibre and the resulting nine diameter estimates for each fibre were averaged. The cross sectional area of each fibre was calculated, assuming that the fibres were circular in cross-section. Tensile strengths of fibres were measured at 22 ± 2 °C and ~40% humidity, using an Agilent T150 UTM, following previously reported procedures48.
Electron and Atomic Force Microscopy
Scanning electron microscopy (SEM) (S-4700, Hitachi, Tokyo, Japan) was used to observe fibre surfaces at 3 kV. Fibres were fixed on conductive adhesive tape glued onto an SEM stub and then coated with gold particles by an SC7620 mini sputter coater (Quorum Technologies, East Sussex, UK) before SEM imaging. For atomic force microscopy (AFM), a ~5 μL drop of a given protein solution in phosphate buffer ([W2] ~20 μM) or in NMR buffer ([W2] ~200 μM) was deposited onto a clean glass microscope slide. A ~1 cm long fibre was pulled from the solution and placed back onto the slide, then allowed to air-dry at ambient temperature and pressure. Dry fibres were imaged in intermittent contact mode (22 ± 2 °C, at 47 ± 5% relative humidity) using an atomic force microscope (NanoWizard II Ultra, JPK, Berlin, Germany) mounted on an inverted optical microscope (Axio Observer A1, Carl Zeiss Canada, Toronto, Canada). Cantilevers with ~300 kHz resonance frequency and force constant of 40 N/m with a tip height of 17 μm and nominal radius of curvature of <10 nm at the tip were employed (Tap 300-G, Budget Sensors, Sofia, Bulgaria). AFM image files were processed using v3.3.32 of the NanoWizard IP software (JPK).
Raman Spectromicroscopy of Fibres
W2 fibres produced and prepared as described above were fixed onto glass slides by taping two ends and middle section of the fibres for Raman spectromicroscopy. A. aurantia spiders (collected in Florida, USA) were farmed in 20 × 50 × 60 cm cages at 58 ± 5% relative humidity (RH) and 24 ± 2 °C, fed four times weekly with small crickets and weekly with 3 droplets of 10% w/v glucose solution. A. aurantia wrapping silk fibres were directly reeled by the spiders around small plastic caps for spectromicroscopy. Spectra were obtained at 22.0 ± 0.5 °C and 20 ± 5% RH using a LABRAM 800HR Raman spectrometer (Horiba Jobin Yvon, Villeneuve d’Ascq, France) coupled to a BX30 (Olympus, Richmond Hill, ON, Canada) fixed stage microscope. An Ar+ laser (514 nm; 50 mW) was focused through a 100× objective lens onto a given fibre. To obtain information about molecular orientation, four polarized spectra, labelled XX, XZ, ZX and ZZ, were recorded68. The first and second letters indicate the polarization of the incident and scattered radiation, respectively, where Z corresponds to the fibre long-axis and X to the perpendicular direction. 2 × 15 sec acquisitions were collected 3–5 times at 5 positions on 3 W2 fibres. Reported spectral data are the average of these acquisitions. A similar procedure was applied to natural fibres, with the polarized spectra resulting from the average of ~12 acquisitions. Spectral manipulations were performed using GRAMS/AI 7.0 (ThermoGalactic, Salem, NH). The spectra were baseline-corrected using a cubic function, 5-point smoothed and averaged for each fibre. The orientation-insensitive spectrum was calculated and the amide I band decomposed to estimate the content of α-helices and β-sheets, as described previously11,69.
Titration-Induced Protein Unfolding
To monitor the global folded state, far-UV CD spectra of W1 and W2 protein samples (9–20 μM in 20 mM sodium acetate buffer) were recorded at 100 nm/min in 0.1 nm intervals from 260 to 195 nm using a J-810 spectropolarimeter (Jasco, Easton, MD, USA) at 22 ± 2 °C in 0.1 cm quartz cuvettes (Hellma, Müllheim, Germany). Protein concentration in a given sample was determined by the absorbance at 210 nm (calculated ε-values: εW1 = 270858 M−1cm−1, εW2 = 543596 M−1cm−1). Samples were analysed in duplicate, blank corrected, averaged and converted to mean residue ellipticity [θ]. DPC was titrated using a relative molar ratios to W1 or W2 (Wn:DPC 1:0, 1:1, 1:10; 1:50; 1:100 and 1:500). Guanidinium chloride (GdmCl) and urea were titrated as a function of concentration from 0 to 5 M for urea and 0 to 4 M for GdmCl. The fraction folded as a function of denaturant concentration (C), F(C), was calculated (similarly to thermal denaturation70) as:

where θ(C), θN and θD are the ellipticities observed at concentration C, in the native (C = 0) and denatured states (in 5 M urea or 4 M guanidine chloride), respectively.
To monitor localized detergent-induced unfolding by NMR spectroscopy, DPC was added to 15N-enriched W1 (0.34 mM in NMR buffer) at an increasing molar ratio of W1:DPC (10:1, 5:1, 1:1, 1:5, 1:10, 1:50), with 1H-15N HSQC experiments (8 scans, 2048 × 128 points, recovery delay of 1.5 s) acquired at each titration point using a 16.4 T Bruker Avance III spectrometer equipped with a 5 mm indirect detection TCI cryoprobe at 303.15 K. The change in W1 concentration due to volume increase was negligible. 3D backbone NMR experiments (HNCO, HNcaCO, HNCA, HNcoCA, as previously acquired for W133) were acquired at the DPC endpoint (0.34 mM W1, 20 mM DPC) to facilitate assignment. Perturbation of W2 was monitored using an intein-spliced variant with the first W-unit uniformly 13C- and 15N-enriched and the second W-unit only 15N-enriched (0.17 mM in NMR buffer) via the isotopically discriminated (IDIS) 1H-15N HSQC experiment71 at 0 mM and 20 mM DPC (48 scans, 2048 × 128 × 2 points, recovery delay of 1.5 s), allowing simultaneous acquisition of data for both W domains in a single sample. To monitor effects of chaotropes, W1 samples (0.34 mM in NMR buffer) were titrated with GdmCl (0, 0.4, 0.6, 0.8, 1, 1.2 and 2 M) and urea (0, 0.5, 1, 1.2, 1.4, 2.0, 2.3, 2.5 M) and monitored using 1H-15N HSQC experiments (24–32 scans, 2048 × 196 points, recovery delay of 1.5 s) acquired at 11.7 T and 303.15 K on a Bruker Avance spectrometer equipped with a 5 mm BBFO SmartProbe. Concentration variation due to volume change was accounted for to accurately set denaturant concentration at each titration point. W1 was diluted over the course of each titration, leading to a final concentration of ~0.25 mM at the GdmCl and urea endpoints. All acquired spectra were processed with NMRPipe56 and assigned in CcpNmr Analysis57. Combined chemical shift displacement (CSD), weighed by gyromagnetic ratio for 1H and 15N nuclei38, was calculated as a function of amino acid residue between the samples containing no titrant and those containing 20 mM DPC, 0.8 M GdmCl (0.30 mM W1), or 2.5 M urea. The spectra chosen for analysis of GdmCl and urea perturbation were not the titration endpoints; rather, these were chosen as the most assignable spectra before denaturation made backbone amide chemical shifts indiscriminable. A control titration using NaCl as the titrant was carried out using a series of 40 μL samples (0, 0.4, 0.6, 0.8, 1 and 1.2 M) though 1H-15N HSQC experiments (24 scans, 2048 × 96 points, recovery delay of 1.5 s) acquired at 303.15 K on a 16.4 T Bruker Avance III spectrometer equipped with a 1.7 mm TCI probe.
Additional Information
Accession codes. Protein Data Bank (PDB): the atomic coordinates for the 20 lowest energy structures for W1 described in this manuscript has been deposited with the following accession code: 2MU3 and appended to previously published NMR assignments (BMRB 17899)33. NMR assignments for W2 were deposited to the Biological Magnetic Resonance Data Bank (BMRB) by combining chemical shifts of W2-1 and W2-2 with accession code BMRB 25197. http://www.nature.com/srep
How to cite this article: Tremblay, M.-L. et al. Spider wrapping silk fibre architecture arising from its modular soluble protein precursor. Sci. Rep. 5, 11502; doi: 10.1038/srep11502 (2015).
References
Lewis, R. V. Spider silk: ancient ideas for new biomaterials. Chem. Rev. 106, 3762−3774 (2006).
Vollrath, F. & Knight, D. P. Liquid crystalline spinning of spider silk. Nature 410, 541–548 (2001).
Omenetto, F. G. & Kaplan, D. L. New opportunities for an ancient material. Science 329, 528–531 (2010).
Heim, M., Keerl, D. & Scheibel, T. Spider silk: from soluble protein to extraordinary fiber. Angew. Chem. Int. Ed. 48, 3584–3596 (2009).
Hijirida, D. H. et al. 13C NMR of Nephila clavipes major ampullate silk gland. Biophys. J. 71, 3442–3447 (1996).
Asakura, T., Suzuki, Y., Nakazawa, Y., Holland, G. P. & Yarger, J. L. Elucidating silk structure using solid-state NMR. Soft Matter 9, 11440–11450 (2013).
Lefèvre, T., Boudreault, S., Cloutier, C. & Pézolet, M. Diversity of molecular transformations involved in the formation of spider silks. J. Mol. Biol. 405, 238–253 (2011).
Hayashi, C. Y., Blackledge, T. A. & Lewis, R. V. Molecular and mechanical characterization of aciniform silk: uniformity of iterated sequence modules in a novel member of the spider silk fibroin gene family. Mol. Biol. Evol. 21, 1950–1959 (2004).
Tokareva, O., Jacobsen, M., Buehler, M., Wong, J. & Kaplan, D. L. Structure-function-property-design interplay in biopolymers: spider silk. Acta Biomater 10, 1612–1626 (2014).
Hronska, M., van Beek, J. D., Williamson, P. T. F., Vollrath, F. & Meier, B. H. NMR characterization of native liquid spider dragline silk from Nephila edulis. Biomacromolecules 5, 834–839 (2004).
Lefèvre, T., Rousseau, M.-E. & Pézolet, M. Protein secondary structure and orientation in silk as revealed by Raman spectromicroscopy. Biophys. J. 92, 2885–2895 (2007).
Parkhe, A. D., Seeley, S. K., Gardner, K., Thompson, L. & Lewis, R. V. Structural studies of spider silk proteins in the fiber. J. Mol. Recognit. 10, 1–6 (1997).
Gosline, J. M., Guerette, P. A., Ortlepp, C. S. & Savage, K. N. The mechanical design of spider silks: from fibroin sequence to mechanical function. J. Exp. Biol. 202, 3295–3303 (1999).
Geurts, P. et al. Synthetic spider silk fibers spun from Pyriform Spidroin 2, a glue silk protein discovered in orb-weaving spider attachment discs. Biomacromolecules 11, 3495–3503 (2010).
Blasingame, E. et al. Pyriform spidroin 1, a novel member of the silk gene family that anchors dragline silk fibers in attachment discs of the black widow spider, Latrodectus hesperus. J. Biol. Chem. 284, 29097–29108 (2009).
Heidebrecht, A. & Scheibel, T. Recombinant production of spider silk proteins. Adv. Appl. Microbiol. 82, 115–153 (2013).
Kluge, J. A., Rabotyagova, O., Leisk, G. G. & Kaplan, D. L. Spider silks and their applications. Trends Biotech 26, 244–251 (2008).
Keten, S., Xu, Z., Ihle, B. & Buehler, M. J. Nanoconfinement controls stiffness, strength and mechanical toughness of β-sheet crystals in silk. Nat. Mater. 9, 359–367 (2010).
Wang, S., Huang, W. & Yang, D. NMR structure note: repetitive domain of aciniform spidroin 1 from Nephila antipodiana. J. Biomol. NMR. 54, 415–420 (2012).
Lin, Z., Huang, W., Zhang, J., Fan, J.-S. & Yang, D. Solution structure of eggcase silk protein and its implications for silk fiber formation. Proc. Natl. Acad. Sci. U.S.A. 106, 8906–8911 (2009).
Askarieh, G. et al. Self-assembly of spider silk proteins is controlled by a pH-sensitive relay. Nature 465, 236–238 (2010).
Gao, Z. et al. Structural characterization of minor ampullate spidroin domains and their distinct roles in fibroin solubility and fiber formation. PLoS ONE 8, e56142 (2013).
Hagn, F. et al. A conserved spider silk domain acts as a molecular switch that controls fibre assembly. Nature 465, 239–242 (2010).
Andersson, M. et al. Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. PLoS Biol. 12, e1001921 (2014).
Jaudzems, K. et al. pH-dependent dimerization of spider silk N-terminal domain requires relocation of a wedged tryptophan side chain. J. Mol. Biol. 422, 477–487 (2012).
Wang, S., Huang, W. & Yang, D. Structure and function of C-terminal domain of aciniform spidroin. Biomacromolecules 15, 468–477 (2014).
Chaw, R. C. et al. Intragenic homogenization and multiple copies of prey-wrapping silk genes in Argiope garden spiders. BMC Evol. Biol. 14, 31 (2014).
Gust, D., Moon, R. B. & Roberts, J. D. Applications of natural-abundance nitrogen-15 nuclear magnetic resonance to large biochemically important molecules. Proc. Natl. Acad. Sci. U.S.A. 72, 4696–4700 (1975).
Barbato, G., Ikura, M., Kay, L. E., Pastor, R. W. & Bax, A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry 31, 5269–5278 (1992).
Eliezer, D., Yao, J., Dyson, H. J. & Wright, P. E. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat Struct Mol Biol 5, 148–155 (1998).
Wüthrich, K. (1986) NMR of Proteins and Nucleic Acids. John Wiley & Sons, NewYork, NY.
Shen, Y., Delaglio, F., Cornilescu, G. & Bax, A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR. 44, 213–223 (2009).
Xu, L., Tremblay, M.-L., Meng, Q., Liu, X.-Q. & Rainey, J. K. 1H, 13C and 15N NMR assignments of the aciniform spidroin (AcSp1) repetitive domain of Argiope trifasciata wrapping silk. Biomol NMR Assign 6, 147–151 (2012).
Wüthrich, K. & Wagner, G. Nuclear magnetic resonance of labile protons in the basic pancreatic trypsin inhibitor. J. Mol. Biol. 130, 1–18 (1979).
Wu, H., Hu, Z. & Liu, X. Q. Protein trans-splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc. Natl. Acad. Sci. U.S.A. 95, 9226–9231 (1998).
Southworth, M. W. et al. Control of protein splicing by intein fragment reassembly. EMBO J. 17, 918–926 (1998).
Appleby, J. H., Zhou, K., Volkmann, G. & Liu, X.-Q. Novel split intein for trans-splicing synthetic peptide onto C terminus of protein. J. Biol. Chem. 284, 6194–6199 (2009).
Schumann, F. H. et al. Combined chemical shift changes and amino acid specific chemical shift mapping of protein-protein interactions. J. Biomol. NMR. 39, 275–289 (2007).
Kay, L. E., Torchia, D. A. & Bax, A. Backbone dynamics of proteins as studied by nitrogen-15 inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979 (1989).
Doddrell, D., Glushko, V. & Allerhand, A. Theory of nuclear overhauser enhancement and 13C–1H dipolar relaxation in proton‐decoupled carbon‐13 NMR spectra of macromolecules. J. Chem. Phys. 56, 3683–3689 (1972).
Tjandra, N., Feller, S. E., Pastor, R. W. & Bax, A. Rotational diffusion anisotropy of human ubiquitin from 15N NMR relaxation. J. Am. Chem. Soc. 117, 12562–12566 (1995).
Pawley, N. H., Wang, C., Koide, S. & Nicholson, L. K. An improved method for distinguishing between anisotropic tumbling and chemical exchange in analysis of 15N relaxation parameters. J. Biomol. NMR. 20, 149–165 (2001).
Morris, K. F. & Johnson, C. S., Jr. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 114, 3139–3141 (1992).
Xu, L. et al. Nanoparticle self-assembly by a highly stable recombinant spider wrapping silk protein subunit. FEBS Lett 587, 3273–3280 (2013).
Jones, J. A., Wilkins, D. K., Smith, L. J. & Dobson, C. M. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR. 10, 199–203 (1997).
Tyn, M. T. & Gusek, T. W. Prediction of diffusion coefficients of proteins. Biotechnol. Bioeng. 35, 327–338 (1990).
Kuszewski, J., Gronenborn, A. M. & Clore, G. M. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J. Am. Chem. Soc. 121, 2337–2338 (1999).
Xu, L., Rainey, J. K., Meng, Q. & Liu, X.-Q. Recombinant minimalist spider wrapping silk proteins capable of native-like fiber formation. PLoS ONE 7, e50227 (2012).
Palladino, P., Rossi, F. & Ragone, R. Effective critical micellar concentration of a zwitterionic detergent: a fluorimetric study on n-dodecyl phosphocholine. J. Fluoresc. 20, 191–196 (2010).
Liu, D., Xu, R. & Cowburn, D. Segmental isotopic labeling of proteins for nuclear magnetic resonance. Meth. Enzymol. 462, 151–175 (2009).
Skrisovska, L., Schubert, M. & Allain, F. H.-T. Recent advances in segmental isotope labeling of proteins: NMR applications to large proteins and glycoproteins. J. Biomol. NMR. 46, 51–65 (2010).
Volkmann, G. & Iwaï, H. Protein trans-splicing and its use in structural biology: opportunities and limitations. Mol Biosyst 6, 2110–2121 (2010).
Feige, M. J. et al. The structure of a folding intermediate provides insight into differences in immunoglobulin amyloidogenicity. Proc. Natl. Acad. Sci. U.S.A. 105, 13373–13378 (2008).
Grantcharova, V. P., Riddle, D. S. & Baker, D. Long-range order in the src SH3 folding transition state. Proc. Natl. Acad. Sci. U.S.A. 97, 7084–7089 (2000).
Marsh, J. A., Singh, V. K., Jia, Z. & Forman-Kay, J. D. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804 (2006).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 (1995).
Vranken, W. F. et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 (2005).
Rieping, W. et al. ARIA2: automated NOE assignment and data integration in NMR structure calculation. Bioinformatics 23, 381–382 (2007).
Schwieters, C. D., Kuszewski, J. J. & Clore, G. M. Using Xplor–NIH for NMR molecular structure determination. Prog. Nucl. Mag. Res. Sp. 48, 47–62 (2006).
Pettersen, E. F. et al. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1612 (2004 ).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–8– 27–8 (1996).
Laskowski, R. A., Rullmann, J. A., MacArthur, M. W., Kaptein, R. & Thornton, J. M. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 8, 486 (1996).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577 (1983).
Wu, D. H., Chen, A. D. & Johnson, C. S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Mag. Res. Ser. A 115, 260–264 (1995).
Stejskal, E. O. & Tanner, J. E. Spin diffusion measurements: spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 42, 288 (1965).
Ortega, A., Amorós, D. & García de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 (2011).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Rousseau, M.-E., Lefèvre, T., Beaulieu, L., Asakura, T. & Pézolet, M. Study of protein conformation and orientation in silkworm and spider silk fibers using Raman microspectroscopy. Biomacromolecules 5, 2247–2257 (2004).
Rousseau, M.-E. et al. Characterization by Raman microspectroscopy of the strain-induced conformational transition in fibroin fibers from the silkworm Samia cynthia ricini. Biomacromolecules 7, 2512–2521 (2006).
Persikov, A. V., Xu, Y. & Brodsky, B. Equilibrium thermal transitions of collagen model peptides. Protein Sci. 13, 893–902 (2004).
Bermel, W., Tkach, E. N., Sobol, A. G. & Golovanov, A. P. Simultaneous measurement of residual dipolar couplings for proteins in complex using the isotopically discriminated NMR approach. J. Am. Chem. Soc. 131, 8564–8570 (2009).
Kyte, J. & Doolittle, R. F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 (1982).
Acknowledgements
Thanks to Drs. Stephen Bearne and David Waisman for CD spectropolarimeter access; Bruce Stewart for technical assistance; Xudong Dai for fibre mechanical properties testing; Dr. Mike Lumsden for 11.7 T NMR spectrometer support at the Nuclear Magnetic Resonance Research Resource Facility (NMR3, Dalhosie University); Ian Burton and Drs. Nadine Merkley and Ray Syvitski for 16.4 T NMR spectrometer support at the National Research Council Biological Magnetic Resonance Facility (NRC-BMRF, Halifax, NS); Dr. Tara Sprules for 11.7 T NMR data acquisition for initial chemical shift assignment at the Québec Eastern Canada High Field NMR Facility (QANUC), supported by the Canada Foundation for Innovation (CFI), the Groupe de Recherche Axé sur la Structure des Protéines (GRASP), McGill University Faculty of Science and Department of Chemistry and PROTEO; and Dr. John Archibald for comments on the manuscript. This work was supported by Discovery and Research Tools and Instruments Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC; to JKR, XQL, MP and MA); a Leaders Opportunity Fund award from the CFI (to JKR); a Dalhousie Medical Research Foundation Capital Equipment Grant (to JKR and XQL); research grants (to QM) from the National High Technology Research and Development Program 863 (NO 2006AA03Z451), the National Natural Science Foundation of China (NO 31070698), the Ph.D. Programs Foundation of Ministry of Education of China (No. 20120075110007); and, funds (to MP and MA) from PROTEO and CERMA. The TCI probes for the 16.4 T NMR spectrometer at the NRC-BMRF were provided by Dalhousie University through an Atlantic Canada Opportunities Agency Grant. JKR is supported by a Canadian Institutes for Health Research New Investigator Award; MLT by studentships from the Nova Scotia Health Research Foundation and an NSERC Postgraduate Scholarship; and, KEO by NSERC Undergraduate Student Research Awards.
Author information
Authors and Affiliations
Contributions
X-Q.L., Q.M and J.K.R. conceived the research; M-L.T., L.X., T.L., M.S., K.E.O., J.L. and J.K.R. acquired experimental data; M-L.T., L.X., T.L., M.S., M.A., M.P., X-Q.L. and J.K.R. analysed experimental data; J.K.R., M-L.T. and L.X. wrote the manuscript; all authors edited and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Tremblay, ML., Xu, L., Lefèvre, T. et al. Spider wrapping silk fibre architecture arising from its modular soluble protein precursor. Sci Rep 5, 11502 (2015). https://doi.org/10.1038/srep11502
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11502
This article is cited by
-
Digestive enzymes and sphingomyelinase D in spiders without venom (Uloboridae)
Scientific Reports (2023)
-
Aerodynamics and the role of the earth’s electric field in the spiders’ ballooning flight
Journal of Comparative Physiology A (2021)
-
Novel Amino Acid Assembly in the Silk Tubes of Arid-Adapted Segestriid Spiders
Journal of Chemical Ecology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
