
Abstract
Lysophosphatidic acid (LPA) is a pleiotropic lipid mediator that acts through G protein-coupled receptors (LPA1-6). Although several biological roles of LPA4 are becoming apparent, its role in hematopoiesis has remained unknown. Here, we show a novel regulatory role for LPA4 in hematopoiesis. Lpar4 mRNA was predominantly expressed in mouse bone marrow (BM) PDGFRα+ stromal cells, known as the components of the hematopoietic stem/progenitor cell (HSPC) niche. Compared with wild-type mice, LPA4-deficient mice had reduced HSPC numbers in the BM and spleen and were hypersusceptible to myelosuppression, most likely due to impairments in HSPC recovery and stem cell factor production in the BM. Analysis of reciprocal BM chimeras (LPA4-deficient BM into wild-type recipients and vice versa) indicated that stromal cells likely account for these phenotypes. Consistently, LPA4-deficient BM stromal cells showed downregulated mRNA expression of stem cell factor and tenascin-c in vitro. Taken together, these results suggest a critical and novel role for the LPA/LPA4 axis in regulating BM stromal cells.
Similar content being viewed by others


Introduction
Lysophosphatidic acid (LPA) is a bioactive lysophospholipid that is composed of a phosphate, a glycerol and a fatty acid1. LPA has roles in many cellular functions, including proliferation, migration, cytokine secretion and morphological changes2,3. LPA is known to be a major growth factor in serum4. Under physiological conditions, LPA in human plasma reaches approximately 80–100 nM5. A primary mechanism of LPA production involves lysophospholipase D activity of the plasma enzyme autotaxin, which converts lysophosphatidylcholine and other lysophospholipids to LPA6,7. To date, at least 6 subtypes of LPA-specific receptors have been identified8 and they are divided into two families on the basis of their amino acid sequence homology. LPA1/Edg-2/Vzg-19, LPA2/Edg-410 and LPA3/Edg-711 are members of the Edg family of LPA receptors, whereas LPA4/GPR23/p2y912, LPA5/GPR9213 and LPA6/p2y514 form a distinct family of LPA receptors, i.e., the non-Edg family of LPA receptors15,16. All LPA receptors are G protein-coupled. Each receptor couples to multiple, but specific, G proteins, including G12/13, Gi/o and Gs, that modulate a variety of intracellular signaling molecules15,17.
Following the identification of the orphan receptor GPR23/p2y9 as LPA412, our research group found that LPA4 deficiency in mice results in a partial embryonic lethal phenotype due to the abnormal development of blood and lymphatic vessels18. Additionally, an independent research group reported that LPA4-deficient mice have increased trabecular bone density compared to wild-type (WT) mice, suggesting that LPA4 has an inhibitory effect on the osteogenic differentiation of mesenchymal stem cells (MSCs)19. MSCs are multipotent and can differentiate not only into mesenchymal lineages, including osteoblasts, adipocytes and chondrocytes, but also into endothelium, neurons and skeletal muscle20. Indeed, human MSCs have been shown to express Lpar4 mRNA19. In addition, the BioGPS gene expression database (http://www.biogps.org) indicates that the mouse Lpar4 gene is expressed in mesenchymal cells such as osteoblasts and C3H10T1/2 cells. Thus, LPA4 is presumed to play pivotal roles in various cellular processes of mesenchymal cells in multiple tissues. However, this has not yet been thoroughly investigated.
The bone marrow (BM) is the main hematopoietic organ in adult mammals. In the BM, the hematopoietic stem/progenitor cells (HSPCs) give rise to all blood cell lineages. The maintenance, differentiation and proliferation of HSPCs are regulated in both cell-autonomous and non-cell-autonomous fashions21. The non-cell-autonomous regulation of HSPCs requires factors important for the proliferation, mobilization, homing and engraftment of hematopoietic stem cells (HSCs); such factors are produced by various cells surrounding HSCs22. This local microenvironment is called the HSPC niche. The HSPC niche is subdivided into two types, the osteoblastic and perivascular niches22. The cellular components of the perivascular niche have been reported to be mesenchymal stromal cells, such as CXCL12-abundant reticular cells (CAR cells)23 and nestin+ MSCs24. Although they are essential for HSPC maintenance, the molecules regulating mesenchymal stromal cells are not yet fully understood25.
In this report, we observed that LPA4-deficient mice were highly sensitive to myelosuppression and showed a delay in the recovery of HSPC numbers. LPA4 was predominantly expressed in the BM mesenchymal stromal cells. In addition, LPA4 in the BM mesenchymal stromal cells was shown to regulate the production of factors involved in HSPC proliferation both in vivo and in vitro. Our present study consistently demonstrates a significant role for LPA4 in maintaining the HSPC niche.
Methods
Mice
LPA4-deficient mice on a C57BL/6 genetic background have been described previously18. C57BL/6 mice congenic for the Ly5 locus (B6-Ly5.1) were purchased from Sankyo Labo Service (Tokyo, Japan) by permission of Prof. Hiromitsu Nakauchi (Institute of Medical Science, The University of Tokyo). Male mice of 9–12 weeks old were used for these studies. Mice were housed under specific pathogen-free conditions in an air-conditioned room and fed standard laboratory chow ad libitum (CE-2; CLEA Japan, Tokyo, Japan), in accordance with institutional guidelines. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Akita University.
Myelosuppression models
Mice were intravenously injected with 5-fluorouracil (5-FU; Kyowa Hakko, Tokyo, Japan) at a dose of 250 mg/kg body weight and their survival was monitored for 24 days. Some mice were euthanized for analysis of the BM cells and peripheral blood (PB) on days 0, 2, 4, 7 and 10. For experiments with the BM chimeric mice (shown below), mice were intraperitoneally injected with 5-FU (150 mg/kg body weight) twice, at a 1-week interval and their survival was monitored for 24 days after initial injection. In another model, mice were irradiated with 8 Gy in two split doses with a 3-hr interval using a cabinet X-ray system CP-160 (Faxitron X-ray Corporation, Wheeling, IL). Their survival was then monitored for 24 days.
PB collection
Under isoflurane anesthesia, PB was collected from the retro-orbital sinus using capillaries and analyzed using a hematological analyzer (Celltac MEK-5258, Nihon Kohden, Tokyo, Japan).
Stromal and hematopoietic cell isolation
Hematopoietic BM cells were obtained from femurs and tibias by flushing the bones with PBS containing 0.5% BSA (Sigma-Aldrich). For isolation of stromal cells, the flushed bones were minced with scissors. Then, the bone fragments were incubated with DMEM (Sigma-Aldrich) containing 10% FBS (Gibco) and 3 mg/ml type I collagenase (Worthington Biochemical, Lakewood, NJ) for 60 min at 37 °C. The cell suspensions were filtered with a 100-μm cell strainer. Red blood cells were lysed using BD Pharm Lyse Buffer (BD Biosciences).
Flow cytometry and cell cycle analysis
The cell suspension was preincubated with Fc block (BD Biosciences) to avoid nonspecific binding of antibodies. The following primary antibodies were used: anti-CD3 (145-2C11), anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-Gr-1 (RB6-8C5), anti-Ter119 (TER-119), anti-CD45 (30-F11), anti-FcRγII-III (93), anti-Sca-1 (E13-161.7), anti-c-Kit (2B8), anti-CD41 (MWReg30), anti-CD48 (HM-48-1), anti-CD150 (TC15-12F12.2), anti-IL7Rα (A7R34), anti-Flt3 (A2F10), anti-PDGFRα (APA5) (all from BioLegend, San Diego, CA) and anti-CD34 (RAM34; eBioscience, San Diego, CA). A mixture of CD4, CD8, CD11b, B220, Ter-119 and Gr-1 antibodies was used as the lineage (Lin) mixture. 7-AAD was used to identify and exclude dead cells. For cell cycle analysis, the cells were fixed and permeabilized using the Fixation/Permeabilization Solution kit (BD Biosciences) and were stained with PE-conjugated anti-Ki-67 antibody (BD Biosciences) and DAPI (BioLegend). The stained cells were analyzed and sorted using a FACSAria and an Accuri C6 flow cytometer (BD Biosciences), respectively. The flow cytometry data were analyzed using FlowJo ver. 10.0.5 (Treestar, Ashland, OH).
Colony-forming units assay
The nucleated 2 × 104 BM cells were plated in methylcellulose media supplemented with a cocktail of recombinant cytokines (Methocult 3434; StemCell Technologies, Vancouver, Canada). Cultures were plated in duplicate and placed in a humidified chamber with 5% CO2 at 37 °C. Colonies containing at least 30 cells were counted at day 12 of culture.
Bone marrow transplantation
Lethally irradiated mice were transplanted with 2 × 106 BM cells. Three months later, their peripheral blood was collected from retro-orbital venous plexus. The percentages of donor-derived cells were analyzed with an Accuri C6 flow cytometer (BD Biosciences) using anti-CD45.1 (A20) and anti-CD45.2 (104) antibodies (BioLegend), detecting Ly5.1 and Ly5.2, respectively.
Cell culture
The BM stromal cells isolated by collagenase treatment were maintained in α-MEM GlutaMax (Gibco) containing 10% FBS, 10% horse serum (Gibco) and 100 U/ml penicillin/streptomycin (Wako Chemicals, Osaka, Japan) for 7 days. The cells were starved in α-MEM GlutaMax containing 0.1% BSA, 10 μM HA130 (an inhibitor of autotaxin; Calbiochem) and 100 U/ml penicillin/streptomycin for 12 hr to exclude hematopoietic cells. The starved cells were stimulated with 1-oleoyl LPA (Avanti Polar Lipids, Alabaster, AL) at a final concentration of 10 μM by adding an equivalent volume of medium containing 20 μM 1-oleoyl LPA and incubated for 12 hr.
Quantitative reverse transcription-PCR
For preparation of cDNA templates from the cultured cells and sorted cells, total RNA was isolated with the RNAqueous-micro kit (Ambion, Austin, TX) or the RNeasy mini kit (Qiagen, Valencia, CA) and subjected to oligo-dT- and random hexamer-primed reverse transcription with the Primescript enzyme (Takara Bio, Otsu, Japan). Quantitative PCR was performed using a LightCycler 480 instrument (Roche Diagnostics) with the SYBR Premix ExTaq II (Takara Bio) and KAPA SYBR Fast qPCR Kit (Kapa Biosystems, Wilmington, MA). The mRNA levels were normalized to Hprt1 or Rn18s as a standard housekeeping gene. The primer sequences are listed in Supplementary Table S1. The PCR program was as follows: denaturation at 95 °C for 30 sec and 50 cycles of amplification consisting of denaturation at 95 °C for 10 sec and annealing and extension at 60 °C for 20 sec.
ELISA
At days 0 and 9 after 5-FU administration, the femurs were flushed with 500 μl of ice-cold PBS or 300 μl of ice-cold PBS containing 1% NP40 and a protease inhibitor cocktail to measure the protein levels of CXCL12 or SCF, respectively. The fluids were centrifuged at 500 × g for 5 min. Then, the supernatant was subjected to ELISAs for CXCL12 and SCF using the DuoSet ELISA kits (R&D systems) according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as the mean ± SEM and analyzed using GraphPad Prism 6 software (GraphPad Software). All data were combined from two or three independent experiments. The two-tailed unpaired Welch’s t-test, log-rank test or two-way ANOVA followed by Bonferroni’s post-hoc test was used for comparisons between 2 groups. One-way ANOVA followed by Tukey’s post-hoc test was used for comparisons among 3 groups. Values of P < 0.05 were considered statistically significant.
Results
In mouse BM, LPA4 is predominantly expressed in PDGFRα+ stromal cells
First, we examined the expression level of Lpar4 mRNA in various populations of mouse BM cells. Lin+ mature hematopoietic cells, Lin−Sca-1+c-Kit+ (LSK) HSPCs, CD34+ LSK hematopoietic progenitor cells (HPCs) and CD34− LSK HSCs were sorted from the BM cells harvested by flushing tibias and femurs (Fig. 1A). In addition, CD45−Ter119−CD31+ endothelial and CD45−Ter119−CD31− stromal cells were sorted from the BM cells obtained by collagenase treatment of the flushed bones (Fig. 1B). Hematopoietic cells, including Lin+ cells and HSCs and endothelial cells expressed little or no detectable Lpar4 mRNA, whereas HSPCs and HPCs expressed low levels of Lpar4. Notably, the Lpar4 mRNA expression level was quite high in the stromal cells (Fig. 1C). Based on the PDGFRα and Sca-1 expression levels, the stromal cells can be divided into three subsets: PDGFRα−Sca-1−, PDGFRα+Sca-1− and PDGFRα+Sca−1+ stromal cells (Fig. 1D). It has been reported that the PDGFRα+Sca-1− and PDGFRα+Sca-1+ subsets include CAR cells and MSCs, respectively23,26. Furthermore, the PDGFRα−Sca-1− subset was reported to consist mainly of osteoblasts26. Interestingly, PDGFRα+Sca-1− and PDGFRα+Sca-1+ stromal cells expressed 10–to 15-fold higher levels of Lpar4 mRNA than the parental unfractionated (CD45−Ter119−CD31−) stromal cells (Fig. 1E). These results demonstrate that the cells expressing Lpar4 mRNA in the mouse BM are predominantly PDGFRα+ stromal cells.

LPA4 expression in the BM.
(A) Representative flow cytometry gating of Lin+ mature hematopoietic cell, Lin−Sca-1+c-Kit+ HSPC, CD34+Lin−Sca-1+c-Kit+ HPC and CD34−Lin−Sca-1+c-Kit+ HSC populations. The BM cells were obtained by flushing tibias and femurs and were analyzed by flow cytometry. (B) Representative flow cytometry gating of CD45−Ter119−CD31+ endothelial cell and CD45−Ter119−CD31− stromal cell populations. The BM cells were obtained from femurs and tibias by collagenase treatment and were analyzed by flow cytometry. (C) Comparison of Lpar4 mRNA expression in Lin+ cells, HSPCs, HPCs, HSCs, endothelial cells and stromal cells by quantitative RT-PCR analysis (n = 6). ND: not detected. (D) Three stromal cell subsets based on PDGFRα and Sca-1 expression. A representative FACS plot shows PDGFRα−Sca-1−, PDGFRα+Sca-1− and PDGFRα+Sca-1+ cell populations. (E) Comparison of Lpar4 mRNA expression in PDGFRα−Sca-1−, PDGFRα+Sca-1− and PDGFRα+Sca-1+ cells by quantitative RT-PCR analysis (n = 6). ND: not detected.
We also examined mRNA expression levels of other LPA receptors in HSPCs and the three subsets of stromal cells. In HSPCs, Lpar4 mRNA expression level was the lowest among six receptors (Supplementary Fig. S1A). About the stromal cells, Lpar1 and Lpar6 mRNA were ubiquitously expressed in all subsets, while Lpar2, Lpar3 and Lpar5 mRNA were undetectable under our experimental conditions (Supplementary Fig. S1B-D).
LPA4-deficient mice have decreased HSPC number in the BM and spleen
Because the stromal cells form an important constituent of the perivascular niche, we analyzed hematopoietic parameters of LPA4-deficient mice under homeostatic conditions, including total BM cellularity and numbers of hematopoietic stem/progenitor cells and mature cells. The total BM cellularity was normal in LPA4-deficient mice (Fig. 2A). The numbers of CD3+CD4+, CD3+CD8+ and B220+ lymphocytes in the BM were also unchanged, but the number of CD11b+Gr-1+ granulocytes/monocytes was significantly higher in the BM of LPA4-deficient mice than in that of WT mice (Supplementary Fig. S2A-G). We further observed that the number of HSPCs was significantly lower in LPA4-deficient mice than in WT mice (Fig. 2B). However, the number of HSCs, which are defined as CD34− LSK (Fig. 1A) or CD41−CD48−CD150+ LSK cells (Fig. 2C), was normal in LPA4-deficient mice (Fig. 2D and E, respectively). Therefore, we reasoned that the HPC number is decreased in these mice (Fig. 2F). Next, we examined whether the decrease in HPC number was due to impaired cell cycle progression of HSCs and HSPCs. However, their cell cycle status in LPA4-deficient mice was normal (Fig. 2G-I).

Normal steady-state hematopoiesis in LPA4-deficient mice.
(A-F) Measurement of hematopoietic parameters in the BM. Total BM cellularity (A), HSPC number (B), HSC numbers defined as CD34− LSK and CD41−CD48−CD150+ LSK cells (D,E, respectively) and HPC number (F) in the BM (n = 9–11). Representative flow cytometry gating of CD41−CD48−CD150+ LSK HSC population is shown in (C). (G) Cell cycle status assessed through Ki-67 and DAPI staining. (H,I) Cell cycle status of HSPCs (H) and HSCs (I) in the BM (n = 9). (J and K) HSPC number (J) and HSC number (K) in the spleen (n = 8–9). (L) FACS analyses of donor-derived cells in PB three months after BM transplantation. (M) Analyses of BM chimeras of WT and LPA4-deficient mice reconstituted with Ly5.1 WT BM (n = 8–9). (N) Analyses of BM chimeras of Ly5.1 WT mice reconstituted with WT or LPA4-deficient BM (n = 7–8). *P < 0.05.
To further characterize HSPCs of LPA4-deficient mice, we assessed the hematopoietic colony-forming capacity of the BM cells cultured with cytokines. The colony numbers of LPA4-deficient mice were slightly lower than those of WT mice (Supplementary Fig. S3). However, considering the intrinsically smaller number of HSPCs in the BM of LPA4-deficient mice (Fig. 2B), the colony-forming capacity seemed to be comparable between WT and LPA4-deficient mice. In addition, the expression levels of various transcripts in HSPCs were measured. Among the genes examined, Cdk2 and Spi1 (encoding PU.1) were down-regulated significantly in HSPCs of LPA4-deficient mice compared with those of WT mice (Supplementary Fig. S4).
We next examined the numbers of Lin−IL7Rα+Flt3+ common lymphoid progenitors (Supplementary Fig. S2H), Lin−Sca-1−c-Kit+CD34+FcRγII-IIIhigh granulocyte and macrophage progenitors and Lin−Sca-1−c-Kit+CD34−FcRγII-IIIlow megakaryocyte and erythrocyte progenitors (Supplementary Fig. S2I), all of which are differentiated from HPCs. These cell numbers were comparable between the two genotypes (Supplementary Fig. S2J). However, the ratio of granulocyte and macrophage progenitors to HSPCs was significantly increased in LPA4-deficient mice (Supplementary Fig. S2K). In mice, adult hematopoiesis occurs not only in the BM but also in the spleen27. By analyzing the spleen, we found that LPA4-deficient mice have significantly decreased numbers of both HSCs and HSPCs (Fig. 2J-K).
To investigate the contribution of LPA4 to BM reconstitution, WT and LPA4-deficient mice were lethally irradiated and transplanted with WT or LPA4-deficient BM. Three months after transplantation, the chimerism of the recipient mice was determined in PB cells (Fig. 2L). The results showed no significant difference in the percentages of donor cells among these BM chimeras (Fig. 2M-N). Together, these results suggest that LPA4 regulates the homeostasis of HSPCs in the BM and spleen.
LPA4-deficient mice show a delay in hematopoietic recovery after myelosuppression
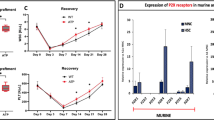
To investigate the function of LPA4 under myelosuppression, mice were injected with 5-FU or were sublethally irradiated. LPA4-deficient mice displayed significantly higher lethality than WT mice in both myelosuppression models (Fig. 3A-B). After 5-FU administration, the numbers of red blood cells and platelets in PB were reduced similarly in both genotypes (Supplementary Fig. S5A-B). However, the numbers of white blood cells in LPA4-deficient mice were reduced earlier than those in WT mice (Supplementary Fig. S5C). At day 10 after 5-FU administration, LPA4-deficient mice had a significantly reduced HSPC number in the BM, although the total BM cellularity was unaffected by the drug (Fig. 3C-D).

Impaired hematopoietic recovery of LPA4-deficient mice after 5-FU administration.
(A) Survival curves of LPA4-deficient and WT mice after 5-FU administration (n = 8). (B) Survival curves of LPA4-deficient and WT mice after X-ray irradiation (n = 7). (C,D) Kinetics of total BM cellularity (C) and HSPC number (D) in the BM after 5-FU administration (n = 6–11). (E,F) Survival curves of BM chimeric mice after 5-FU administration. (E) Analyses of Ly5.1 WT mice reconstituted with WT or LPA4-deficient BM (n = 9). (F) Analyses of WT and LPA4-deficient mice reconstituted with Ly5.1 WT BM (n = 8-9). *P < 0.05. **P < 0.01. ***P < 0.001.
As described above, HPCs and stromal cells express Lpar4 mRNA in the BM (Fig. 1C). To determine which cell type is responsible for the hypersusceptibility of LPA4-deficient mice to 5-FU, we used the BM chimeric mice shown in Fig. 2M,N. When WT mice transplanted with WT or LPA4-deficient BM cells were treated with 5-FU, the lethality was indistinguishable between these two classes of chimeric mice (Fig. 3E). In contrast, when we administered 5-FU to WT and LPA4-deficient mice that had undergone transplantation of BM from WT donors, the LPA4-deficient recipients were significantly more susceptible to 5-FU than WT recipients (Fig. 3F), recapitulating the results observed with naïve mice. Histological observations of the lung, liver, colon and small intestine 7 days after 5-FU administration revealed that little or no injury occurred in these organs of WT and LPA4-deficient mice (data not shown). Thus, these results suggest that LPA4 expressed in stromal cells is involved in hematopoietic recovery after myelosuppression.
LPA4 deficiency in BM stromal cells impairs the production of HSPC proliferation factors
CXCL12 and SCF, important cytokines for the HSPC maintenance and proliferation, are produced predominantly by BM stromal cells28,29. Because LPA4-deficient mice displayed reduced numbers of HSPCs and impaired extramedullary hematopoiesis, we evaluated the CXCL12 and SCF protein levels in the BM of LPA4-deficient mice. Under homeostatic conditions, there was no significant difference in the protein levels of CXCL12 between LPA4-deficient and WT mice (Fig. 4A). The protein level of SCF was too low to be detected. At day 9 after 5-FU administration, LPA4-deficient mice had a significantly lower level of SCF protein in the BM than did WT mice (Fig. 4B) and the production of CXCL12 protein tended to be impaired in LPA4-deficient mice (Fig. 4C). These in vivo data suggest that LPA4 in the stromal cells regulates the SCF protein expression under myelosuppression. Consistent results were obtained with cultures of primary BM stromal cells. The LPA4-deficient stromal cells expressed a significantly lower level of Scf mRNA than did WT cells (Fig. 4D). In addition, the LPA4-deficient stromal cells also showed a significant reduction in the mRNA level of tenascin-c (TN-C) (Fig. 4F), an extracellular matrix protein that promotes HSPC proliferation30. When we stimulated stromal cells with LPA, upregulation of the TN-C mRNA level was observed in both WT and LPA4-deficient cells (Fig. 4F). In contrast, the expression levels of Scf and Cxcl12 mRNA were unaffected by LPA stimulation (Fig. 4D-E). Together, these results suggest that LPA4 in stromal cells regulates the production of proliferation-promoting factors for HSPCs.

Reduced production of SCF and TN-C in LPA4-deficient stromal cells.
(A) The amount of CXCL12 per femur from naïve LPA4-deficient and WT mice (n = 9). (B,C) The amounts of SCF (B) and CXCL12 (C) per femur from LPA4-deficient and WT mice (n = 13–14) at day 9 after 5-FU administration. (D-F) Quantitative RT-PCR for Scf (D) Cxcl12 (E) and TN-C (F) mRNA expression in isolated BM stromal cells from LPA4-deficient and WT mice that were treated with or without LPA (n = 9–10). *P < 0.05 vs. WT.
Discussion
In this study, we found that LPA4-deficient mice showed hypersusceptibility to myelosuppressive stresses, likely due to impaired stress recovery of the HSPC number in the BM. This impairment was associated with reduced production of SCF in the BM of LPA4-deficient mice. Bone marrow chimeric mice showed that the target cells of LPA4 signaling were of non-hematopoietic origin. Therefore, it was consistent that the LPA4 deficiency in the BM stromal cell cultures suppressed the expression levels of Scf and also TN-C. SCF potently regulates HSPC proliferation and differentiation through the receptor c-Kit31. Previously, PDGFRα+Sca-1− cells were shown to produce SCF in the BM and contribute to the formation of the perivascular niche23. TN-C is an extracellular matrix protein that is predominantly produced by PDGFRα+ stromal cells in the BM and regulates HSPC proliferation through integrin α930. The expression of TN-C was reportedly upregulated after myelosuppressive stress30. Similarly to LPA4-deficient mice, TN-C-deficient mice were hypersensitive to lethal myelosuppression and showed a delay of hematopoietic recovery30. Thus, we assume that the lethality to myelosuppressive stress was dependent, at least in part, on LPA4 from the stromal cells. Because Lpar4 mRNA was predominantly expressed in PDGFRα+Sca-1+/ PDGFRα+Sca-1− stromal cells in the BM, LPA4 may regulate the function of PDGFRα+ cells by affecting the production of HSPC proliferation factors, although the underlying molecular mechanisms remain to be elucidated.
To date, BM mesenchymal stromal cells have been reported to express at least two G protein-coupled receptors (GPCRs), parathyroid hormone/parathyroid hormone-related peptide receptor (PTH1)32 and prostaglandin E receptor 4 (EP4)33, that support HSPC proliferation. Activation of PTH1 resulted in the upregulation of the protein expression of Jag1 in α1(I) collagen-expressing osteoblastic cells and then induced HSPC proliferation through Notch1. Meanwhile, EP4 activation enhanced the mRNA expression of various mitogenic protein genes, as well as Jagged1, in ALCAM–Sca-1+ mesenchymal progenitor cells. It is interesting to note that both GPCRs are coupled to Gs protein for these phenotypes25. However, we observed that LPA4 is coupled to G12/13 protein not but to Gs, Gi/o or Gq protein in mouse C3H10T1/2 mesenchymal cells (Keisuke Yanagida and S.I., manuscript in preparation). Consistently, LPA4-deficient stromal cells showed no change in Jagged1 expression level (Supplementary Fig. S6). Thus, our results suggest that LPA4 controls the HSPC niche via a novel GPCR signaling pathway in BM stromal cells. Because Lpar1 and Lpar6 mRNA were also expressed in PDGFRα+ stromal cells in the BM, LPA1, LPA4 and LPA6 may exert redundant functions in the hematopoiesis-supporting activity. Indeed, these three LPA receptors were reported to couple to G12/13 protein15.
In steady-state hematopoiesis, LPA4-deficient mice showed mildly reduced numbers of HPCs in the BM. Moreover, we detected a severe reduction in HSPCs in the spleen. It is likely, therefore, that the hypersusceptibility to myelosuppression of LPA4-deficient mice was also partly caused by these “basal” impairments of steady-state hematopoiesis in addition to the attenuated emergency hematopoiesis. As far as we know, no gene-targeted knockout mouse lines have been reported to have hematopoietic phenotypes similar to that of LPA4-deficient mice, although gene targeting in mice has allowed investigators to reveal hematopoietic functions of various genes.
In relation to the abnormal BM hematopoiesis in LPA4-deficient mice, we would like to note that both the ratios of granulocyte and macrophage progenitors to HSPCs and the absolute number of granulocyte/monocytes were mildly increased in the BM of LPA4-deficient mice compared with WT mice. In line with the phenotypes, we found that Spi1 gene (encoding PU.1) was down-regulated significantly in HSPCs of LPA4-deficient mice compared with those of WT mice. PU.1 is the transcription factor that suppresses early granulocytic development in adult mice34. Together, these results suggest that LPA4 deficiency promotes the differentiation of HPCs into granulocyte and macrophage progenitors, also contributing to the reduced numbers of HPCs.
Hematopoiesis during embryonic development occurs in the fetal liver and then, postnatally, the HSCs in the fetal liver migrate to the BM35, where CXCL12 is abundant and acts as an essential factor for HSC retention36. Indeed, the in vivo deletion of CXCL12 decreased and increased the number of HSCs in the BM and of that in the spleen and PB, respectively36. It is worth mentioning that compromised HSC homing was observed in the spleen of naïve LPA4-deficient mice, although the BM of these mice contains the normal number of HSCs as well as a normal level of CXCL12 protein. These results suggest that the total absolute number of HSCs in the LPA4-deficient fetal liver may be intrinsically reduced, resulting in the compromised extramedullary hematopoiesis observed in the adult. Alternatively, the abnormal angiogenesis during the embryogenesis of LPA4-deficient mice18 may inhibit the homing of HSCs from the liver to the spleen.
The LPA4-deficient stromal cells showed significant decreases in the basal mRNA expression levels of Scf and TN-C in vitro. These significant decreases were observed even after LPA treatment, although the expression of TN-C was comparably induced by LPA treatment in both genotypes, likely via LPA receptor(s) other than LPA4. Thus, LPA4 signaling in stromal cells may indirectly regulate the intrinsic capacity for the production of SCF and TN-C by affecting unknown processes such as cellular differentiation.
Myelosuppression is a life-threatening adverse event observed during anticancer treatments such as chemotherapy and radiotherapy37. Currently, granulocyte colony-stimulating factor is used to promote granulocytic recovery after such anticancer treatments38. In the present study, we reveal that LPA4 facilitated the regeneration of HSPCs after myelosuppression in mice. It is possible that selective agonism of LPA4 could promote multi-lineage hematopoietic recovery and protect cancer patients from myelosuppression.
Additional Information
How to cite this article: Igarashi, H. et al. The lysophosphatidic acid receptor LPA4 regulates hematopoiesis-supporting activity of bone marrow stromal cells. Sci. Rep. 5, 11410; doi: 10.1038/srep11410 (2015).
References
Tokumura, A. A family of phospholipid autacoids: occurrence, metabolism and bioactions. Prog Lipid Res 34, 151–184 (1995).
Choi, J. W. et al. LPA receptors: subtypes and biological actions. Annu Rev Pharmacol Toxicol 50, 157–186 (2010).
Okudaira, S., Yukiura, H. & Aoki, J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 92, 698–706 (2010).
Sano, T. et al. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J Biol Chem 277, 21197–21206 (2002).
Hosogaya, S. et al. Measurement of plasma lysophosphatidic acid concentration in healthy subjects: strong correlation with lysophospholipase D activity. Ann Clin Biochem 45, 364–368 (2008).
van Meeteren, L. A. et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol 26, 5015–5022 (2006).
Tanaka, M. et al. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J Biol Chem 281, 25822–25830 (2006).
Davenport, A. P. et al. International Union of Basic and Clinical Pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev 65, 967–986 (2013).
Hecht, J. H., Weiner, J. A., Post, S. R. & Chun, J. Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. J Cell Biol 135, 1071–1083 (1996).
An, S., Bleu, T., Hallmark, O. G. & Goetzl, E. J. Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J Biol Chem 273, 7906–7910 (1998).
Bandoh, K. et al. Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. J Biol Chem 274, 27776–27785 (1999).
Noguchi, K., Ishii, S. & Shimizu, T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem 278, 25600–25606 (2003).
Lee, C. W., Rivera, R., Gardell, S., Dubin, A. E. & Chun, J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J Biol Chem 281, 23589–23597 (2006).
Yanagida, K. et al. Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. J Biol Chem 284, 17731–17741 (2009).
Yanagida, K. & Ishii, S. Non-Edg family LPA receptors: the cutting edge of LPA research. J Biochem 150, 223–232 (2011).
Yanagida, K., Kurikawa, Y., Shimizu, T. & Ishii, S. Current progress in non-Edg family LPA receptor research. Biochim Biophys Acta 1831, 33–41 (2013).
Yung, Y. C., Stoddard, N. C. & Chun, J. LPA receptor signaling: pharmacology, physiology and pathophysiology. J Lipid Res 55, 1192–1214 (2014).
Sumida, H. et al. LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood 116, 5060–5070 (2010).
Liu, Y. B. et al. LPA induces osteoblast differentiation through interplay of two receptors: LPA1 and LPA4 . J Cell Biochem 109, 794–800 (2010).
Uccelli, A., Moretta, L. & Pistoia, V. Mesenchymal stem cells in health and disease. Nat Rev Immunol 8, 726–736 (2008).
Zon, L. I. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature 453, 306–313 (2008).
Mendelson, A. & Frenette, P. S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med 20, 833–846 (2014).
Omatsu, Y. et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 33, 387–399 (2010).
Méndez-Ferrer, S. et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010).
Hoffman, C. M. & Calvi, L. M. Minireview: complexity of hematopoietic stem cell regulation in the bone marrow microenvironment. Mol Endocrinol 28, 1592–1601 (2014).
Morikawa, S. et al. Prospective identification, isolation and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J Exp Med 206, 2483–2496 (2009).
Wolber, F. M. et al. Roles of spleen and liver in development of the murine hematopoietic system. Exp Hematol 30, 1010–1019 (2002).
Sugiyama, T., Kohara, H., Noda, M. & Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988 (2006).
Heissig, B. et al. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell 109, 625–637 (2002).
Nakamura-Ishizu, A. et al. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood 119, 5429–5437 (2012).
Kent, D. et al. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin Cancer Res 14, 1926–1930 (2008).
Calvi, L. M. et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846 (2003).
Ikushima, Y. M. et al. Prostaglandin E(2) regulates murine hematopoietic stem/progenitor cells directly via EP4 receptor and indirectly through mesenchymal progenitor cells. Blood 121, 1995–2007 (2013).
Dakic, A., Wu, L. & Nutt, S. L. Is PU.1 a dosage-sensitive regulator of haemopoietic lineage commitment and leukaemogenesis? Trends Immunol 28, 108–114 (2007).
Mazo, I. B., Massberg, S. & von Andrian, U. H. Hematopoietic stem and progenitor cell trafficking. Trends Immunol 32, 493–503 (2011).
Tzeng, Y. S. et al. Loss of Cxcl12/Sdf-1 in adult mice decreases the quiescent state of hematopoietic stem/progenitor cells and alters the pattern of hematopoietic regeneration after myelosuppression. Blood 117, 429–439 (2011).
Kulkarni, S., Ghosh, S. P., Hauer-Jensen, M. & Kumar, K. S. Hematological targets of radiation damage. Curr Drug Targets 11, 1375–1385 (2010).
Dale, D. C. Colony-stimulating factors for the management of neutropenia in cancer patients. Drugs 62 Suppl 1, 1–15 (2002).
Acknowledgements
We thank Ms Keiko Yamashita and Mr Atsushi Sekiba for technical assistance and Dr. Satoshi Eguchi (Department of Medical Biology, Akita University Graduate School of Medicine) for suggestions on FACS experiments. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (S.I.).
Author information
Authors and Affiliations
Contributions
H.I. and S.I. designed research, analyzed data and wrote the paper; H.I. performed experiments; N.A., T.O.-N. and D.Y. designed research and discussed the data.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Igarashi, H., Akahoshi, N., Ohto-Nakanishi, T. et al. The lysophosphatidic acid receptor LPA4 regulates hematopoiesis-supporting activity of bone marrow stromal cells. Sci Rep 5, 11410 (2015). https://doi.org/10.1038/srep11410
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11410
This article is cited by
-
SOX17-mediated LPAR4 expression plays a pivotal role in cardiac development and regeneration after myocardial infarction
Experimental & Molecular Medicine (2023)
-
The lysophospholipase D enzyme Gdpd3 is required to maintain chronic myelogenous leukaemia stem cells
Nature Communications (2020)
-
Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6
Nature (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
