
Abstract
The establishment of a (set of) stably expressed reference gene(s) is required to normalize transcription data. Polyploidy is very common in the plant kingdom, but it is not necessarily the case that a reference gene which works well at the diploid level will also work well at the polyploid level. Here, ten candidate reference genes are compared in the context of gene transcription in the genus Chrysanthemum. The robustness of some, but not all, of these was shown to be high across ploidy levels. MTP (metalloprotease) and ACTIN (actin) were the most stable in diploid and tetraploid C. nankingense, while PSAA (photosynthesis-related plastid gene representing photosystem I) and EF-1α (elongation factor-1α) were the most stable in tetraploid and hexaploid C. zawadskii. EF-1α and PGK (phosphoglycerate kinase) was the best combination for the complete set of four taxa. These results suggest that when making cross-species comparison of transcript abundance involving different ploidy levels, care needs to be taken in the selection of reference gene(s).
Similar content being viewed by others


Introduction
The measurement of specific transcript abundance, particularly in response to external treatments or to genetic manipulation, is a feature of many plant studies1,2. A frequently used platform for this purpose is the quantitative real time PCR (qPCR) assay, thanks to its low cost, sensitivity, flexibility and scalability2. The accuracy of the platform is heavily dependent on an appropriate choice of reference gene(s) used to normalize transcript abundances. The transcription of an ideal reference gene must be stable both in time and space and be unaffected by any treatment or genetic manipulation involved2,3,4. A number of genes have been used for this purpose, even though it is clear that some of them are not as stable as is required5,6. Therefore, it is necessary to select suitable reference genes that are steadily expressed in the relevant experimental conditions before determining the expression pattern of a target gene by qRT-PCR3,7. The two algorithms geNORM8 and NormFinder9 have been developed as a means to identify optimal sets of reference gene(s) in specific experimental conditions10,11,12.
Polyploidy is widespread among the angiosperms, so much so that all lineages reflect at least one polyploidization event. Some species, referred to as cryptic polyploids, were long believed to be genuine diploids, but have turned out to harbour duplicated genome segments, so are thought to represent decayed polyploids. Combining two independent genomes within a single nucleus has profound effects on both DNA sequence and the transcriptome, a phenomenon commonly referred to as “genomic shock” which was predicted by McClintock13. Research has shown that polyploid formation is typically followed by large-scale changes in gene expression14,15. Although some reference genes have been shown to be effective over a wide range of developmental and environmental conditions within a species, little attention has been paid to whether the same reference gene(s) can be used in species comparisons in which variation in ploidy level is involved.
In a previous study, we investigated the expression dynamics of 21 selected reference genes in Arabidopsis diploid and polyploid species using published transcriptome data to provide a control dataset for gene expression studies of natural variations, in particular Arabidopsis tetraploid species16. However, suitable reference genes for use in cross-species comparisons within the genus without transcriptome data have never been methodically identified. The genus Chrysanthemum harbors species ranging in ploidy from diploid (2n = 2x = 18) to decaploid (2n = 10x = 90)17,18,19 and at least three ancient whole genome duplication (WGD) events have occurred during their evolution20,21. Here we report variation in the transcription of a set of standard reference genes in the leaves of diploid and tetraploid C. nankingense and tetraploid and hexaploid C. zawadskii by quantitative real-time RT-PCR using geNORM and NormFinder analysis.
Results
Performance of the primers
A total of ten genes were selected as candidate reference genes in leaves (the third and fourth true leaves from the shoot apex without any treatment) from diploid (2x) and tetraploid (4x) C. nankingense and 4x and hexaploid (6x) C. zawadskii. The primer and amplicon characteristics of these ten genes are summarized in Table 1. Melting curve (Fig. 1a) and PAGE (Fig. 1b) analyses showed that each primer pair amplified a single PCR product of the expected size. Analysis using the LinRegPCR program showed that amplification efficiency ranged from 0.8926 to 0.9952 and that the correlation coefficients ranged in value between 0.9922 and 0.9985 (Table 1).

Amplicon dissociation curves and PAGE analysis.
(a) Melting curve analysis of the ten reference genes. (b) PAGE analysis of the amplicons. Marker from top to bottom: 300 bp, 200 bp and 100 bp. The samples in each PAGE separation are ordered from left to right as EF-1α, PP2A, GAPDH, ACTIN, MTP, SKIP16, PGK, TIP41, TUB and PSAA.
Transcript abundances
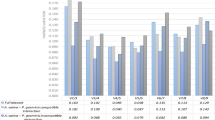
Within a taxon, the Ct value of each candidate gene varied very little among either the four biological replicates or the three technical replicates. These results suggested that the use of different biological replicate did not cause a significant difference. The lowest Ct was 16.8 (4x C. nankingense, EF-1α) and the highest 26.3 (6x C. zawadskii, PP2A). Across all templates, EF-1α was the most abundantly transcribed gene, accompanied by the lowest mean Ct (16.9), followed by PSAA (17.9), GAPDH (18.1), ACTIN (18.8) and TUB (22.4); PP2A was the least abundantly transcribed gene, accompanied by the highest mean Ct (26.0), followed by TIP41 (25.3), PGK (25.1), MTP (23.8) and SKIP16 (23.3) (Fig. 2). However, none of the reference genes were uniformly transcribed in both ploidy forms of either C. nankingense or C. zawadskii. Therefore, it was necessary to evaluate the reference genes for normalization under the different ploidy level and species.

Amplification curves: the X axis represents the PCR cycle number (0–40).
The red line represents the threshold fluorescence at which the Ct was determined.
Stability of the references genes
A geNorm based analysis, carried out to determine which of the reference genes would be most suitable in each of the four taxa, produced the ranking based on M values, as depicted in Fig. 3. On the basis that an M value of <0.5 indicates stability, MTP, ACTIN, PGK, TUB, GAPDH and EF-1α were considered to be stable in the contrast 2x vs 4x C. nankingense, while PSAA, EF-1α, PP2A, ACTIN, PGK and GAPDH were all stable for 4x vs 6x C. zawadskii. The least stable gene in the comparison 2x vs 4x C. nankingense was PSAA, even though this gene was stable in the contrast 4x vs 6x C. zawadskii. The most stable genes across all four taxa were EF-1α and PGK, while PSAA, SKIP16 and TIP41 all performed poorly (Fig. 3). The pairwise variation parameter V calculated by geNorm predicts the optimal number of reference genes required. A Vn/Vn + 1 ratio <0.15 is taken to indicate that no additional reference gene is necessary22,23. Here, the optimal number of genes was two in every case (Fig. 3). NormFinder analysis generated a very similar ranking to the GeNorm output. The combinations MTP/ACTIN, EF-1α/PSAA and EF-1α/PGK were predicted to deliver the most reliable level of normalization (Fig. 4).

Average expression stability values (M) of the ten reference genes and the determination of the optimal number of reference genes required for normalization.
Pairwise variation (Vn/n + 1) was analyzed between reference genes n and n + 1 to optimize normalization in the contrasts 2x vs 4x C. nankingense (red), 4x vs 6x C. zawadskii (green) and between all four taxa (blue).

Venn diagram to identify stably transcribed reference genes in Chrysanthemum spp. and their stability values calculated using NormFinder.
A comparison was made between transcript abundances in 2x C. nankingense as estimated from qPCR (2−ΔΔCt method24) with those derived from RNA-Seq analysis (NCBI SRA accession: SRP049642, be measured by reads per kilobase of exon model per million mapped reads, RPKM25). Two methods produced the comparable transcript abundances, especially for ACTIN, EF-1α and GAPDH (Fig. 5). Hence, the Ct value could represent the relative transcript abundance in some extent. Indubitably, the transcript abundance (as well as the Ct) of ACTIN, EF-1α and PGK was similar (the second stable) in the contrasts 2x and 4x C. nankingense, 4x and 6x C. zawadskii and all four taxa, respectively (Fig. 2). To test the importance of the choice of reference gene(s), the relative abundances of ACTIN, EF-1α and PGK transcript were quantified, based on normalization carried out with either a stable or an unstable reference gene. When normalized on the basis of the three most stable reference genes, transcript abundance proved to be stable in the contrasts 2x vs 4x C. nankingense (MTP), 4x vs 6x C. zawadskii (PSAA) and all four taxa (EF-1α); conversely, when normalized on the basis of the least stable reference genes, this was no longer the case (Fig. 6).

Transcript abundance as estimated from qPCR analysis and from RNA-Seq data.
The X axis represents relative transcript abundance from RNA-Seq and the Y axis the relative transcript abundance estimated from qPCR.

Relative transcript abundances of ACTIN, EF-1α and PGK across species and ploidy levels.
Normalization performed using single either stable or unstable reference genes in the contrasts. (a) Relative abundance of ACTIN transcript in 2x vs 4x C. nankingense (reference genes PSAA/MTP), (b) relative abundance of EF-1α transcript in 4x vs 6x C. zawadskii (SKIP16/PSAA) and (c) relative abundance of PGK transcript between all four taxa (PSAA/EF-1α).
Application of the qRT-PCR protocol to evaluate the expression of RAD51 gene
To demonstrate the usefulness of the validated candidate reference genes, the transcription levels of RAD51 in both C. nankingense and C. zawadskii were estimated using qPCR (the isolation and primer details provided in Supplementary information). Current knowledge of the functions of the RAD51 was known to be a key pathway in cells for the homologous recombination and repair of DNA damage26. This gene was chosen because polyploidization events in the genus Chrysanthemum are associated with a certain range of DNA repair disorder27, which can be expected for a high transcript abundance of RAD51 in 4x C. nankingense and 6x C. zawadskii. However, when normalized using a variety of reference genes, there was a substantial degree of divergence in its estimated relative transcript abundance. Based on the unstable reference genes PSAA and SKIP16, the level of transcription of RAD51 was almost the same in 2x and 4x C. nankingense and similarly between 4x and 6x C. zawadskii. In contrast, estimates based on normalization using the most stable reference gene combination (i.e., ACTIN and EF-1α), suggested that there was a more than two fold difference in transcript abundance in the higher ploidy species (Fig. 7).

Relative abundance of RAD51 transcript, estimated from qPCRs based on either stable or unstable reference genes.
(a) The contrast 2x vs 4x C. nankingense, using as reference genes either PSAA or MTP/ACTIN, (b) the contrast 4x vs 6x C. zawadskii, using as reference genes either SKIP16 or PSAA/EF-1α.
Discussion
The diversity of reference gene expression in different Chrysanthemum species and ploidy levels
The qPCR platform has been used in many experiments designed to monitor the transcription response to external treatment and/or genetic manipulation28. The reference genes used in these experiments have been chosen on the basis that they encode house-keeping proteins which are unlikely to regulate by the treatments imposed. Examples are genes involved in glucose metabolism (GAPDH and PSAA), in protein degradation (MTP), in the regulation of phosphorylation (PP2A), in determining cytoskeletal structure (TUB and ACTIN), in protein synthesis (EF-1α) or binding (SKIP16); others include genes encoding protein kinase (PGK) and the tonoplast intrinsic protein TIP414. Here, the stability of ten widely used reference genes was tested in two ploidy forms of each of two Chrysanthemum species. Even though each of these genes is known to be stably transcribed throughout development and under a range of environmental conditions within a species4,29,30,31, it became clear that comparisons across species or between ploidy levels within a species would require more than one reference gene (Fig. 3 and Fig. 4). PSAA was the most stable single gene in the comparison between 4x and 6x C. zawadskii, but performed poorly in the comparison 2x vs 4x C. nankingense. Meanwhile, EF-1α and PGK were stably transcribed at all three ploidy levels across the two species, which implies that these would represent a good choice of reference gene for inter-species and inter-ploidy level comparisons (Fig. 6 and Fig. 7).
Possible mechanisms for variation in reference gene stability
Comparisons between different Arabidopsis thaliana accessions (and some tetraploid Arabidopsis spp.) have shown that the transcription of the genes encoding a pentatricopeptide repeat protein and F-box protein are rather variable and particularly so in contrasts made between a diploid and a tetraploid accession. Chrysanthemum species vary in ploidy level from 2x to 10x, but large volumes of transcriptomic date have yet to be assembled for these taxa. The present qPCR data demonstrated that the reliability of some genes used for qPCR reference purposes is questionable when they are used in comparisons across species or across ploidy levels (Figs 6 and 7). The data underline the need for care to be taken in the selection of reference genes for specific polyploid combinations.
Microarray and next generation sequencing technology based experiments have delivered plenty of evidence to show that transcription levels vary between species, between ploidy levels and between tissue types32,33. In some cases, this variation is not revealed by standard qPCR experiments where transcription is normalized using a reference gene(s), although its detection can be improved by spiking with mRNA in proportion to the genomic DNA concentration (for transcripts per cell method)34. An alternative approach expresses abundances on a per transcriptome basis after quantifying the size of the transcriptome (after quantifying whole transcriptome size)35. These methods will greatly increases the size and complexity of an RT-PCR experiment. A more direct approach, as taken here, is to identify a set of stable reference genes (stable in the sense that their transcripts form a constant proportion of the transcriptome taken across species/ploidy levels). Special emphasis has been concentrated on the possibility of comparison across species and ploidy levels using reference gene(s). Although the reference genes identified here may be less appropriate for experiments involving other species, ploidy levels, tissues or treatments, a rigorous guide for identifying good reference genes has been been followed, in line with the “Minimum Information for publication of Quantitative real-time PCR Experiments” guidelines36,37.
Polyploidy, in which two or more genomes are combined into a single nucleus, induces a range of both genomic and transcriptomic changes19,38. The pattern of expression and even the function of a significant number of genes can be materially altered by the genomic shock accompanying polyploidization13. These changes provide a source of variation for natural selection to act upon, with the result that polyploidization has been and continues to be, a major driver of evolution in the flowering plants39. For example, genes involved in energy, metabolism, cellular biogenesis and plant hormonal regulation are up regulated in some allotetraploids, frequently within one or a few generations and can result in an altered phenotype and ecology38,40,41,42. The process could in principle result in the loss of stability at the polyploid level of a reference gene which operates well at the diploid level, although this is unlikely for genes which act in basal processes such as glycolytic pathway (eg. PGK), structure (eg. ACTIN) and normal physiological function (eg. EF-1α). However, the exact regulatory mechanisms underlying these reference genes in polyploids are still poorly characterized. Therefore, determining the transcriptional regulatory mechanisms will be the next important step.
Methods
Plant materials and growth conditions
Plants of diploid (2x) and tetraploid (4x) C. nankingense and 4x and hexaploid (6x) C. zawadskii were obtained from the Chrysanthemum Germplasm Resource Preserving Centre, Nanjing Agricultural University, China. (32°05′N, 118°8′E, 58 m altitude). All plants were propagated by cuttings and grown in a greenhouse held at 22°C during the day and at a minimum of 15°C during the night. The relative humidity varied from 70 to 75% and no artificial light was given.
RNA isolation and cDNA synthesis
Harvesting of leaves (the third and fourth true leaves from the shoot apex) was carried out in the morning of a sunny day (April 4, 2014, 10:00). Within a maximum of 5 min, the excised leaves were snap-frozen in liquid nitrogen and stored at −80°C until required. Total RNA was extracted using the TRIzol reagent (Takara, Japan). Before reverse transcription, total RNA was treated with RNase-free DNase I (Takara) at 37°C for 30 min to remove any contaminating genomic DNA. The integrity of the RNA preparations was assessed by agarose gel electrophoresis and the concentration of each sample measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, USA). Only samples showing an A260/A280 ratio of 1.9–2.0 and an A260/A230 ratio >2.0 were used for further analysis. To compare relative transcript abundances estimated from both qPCR and RNA-Seq, total RNA extracted from 2x C. nankingense was subjected to the RNA-Seq procedure, using an Illumina (San Diego, CA, USA) HiSeqTM 2000 device, following the manufacturer's protocols. Briefly, mRNA was purified from total RNA by the addition of beads coated with oligo (dT). A fragmentation buffer was added to generate fragments in the size range 100–400 nt and these served as the template for the synthesis of the cDNA first strand, achieved by random hexamer priming. The cDNA second strand was produced using a SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen, USA), purified by passing through a QiaQuick PCR extraction kit (Qiagen, Germany), The products were ligated with one another via the incorporation of sequencing adapters and after agarose gel electrophoresis, a suitable size range of fragments was selected. The resulting library was pair-end sequenced and the subsequent transcriptome assembly was achieved using the Trinity software package17. The qRT-PCR products (exactly) matching each of the ten candidate reference genes were identified using the local Blast module of BioEdit v7.0.9.0. Transcript abundances were obtained by a count of the RPKM25.
For qPCR experiments, the cDNA first strand was synthesized from the total RNA preparations described above. A 1 μg aliquot of total RNA was treated with ultraclean Oligo(dT)18 and SuperScript III Reverse Transcriptase (Takara), following the manufacturer's protocol. To confirm the absence of contaminating genomic DNA, the cDNA was amplified using primers recognizing a segment of Chrysanthemum flowering locus T-like protein (FT) gene-specific promoter (Forward primer sequence: 5′-AGCTGTAGCGTAGCGCAAGT–3′, Reverse primer: 5′-CAAACGCAATAACAAGAGCA–3′, GenBank: KF752603.1, PCR product size: 171 bp).
PCR primers and qPCR
Ten commonly used reference genes were selected (Table 1), based on their performance across development and under different photoperiod treatments in either C. lavandulifolium4 or C. nankingense17. Primers were designed and optimized using a standard set of design criteria (e.g., target 3′-untranslated region for primer design, no hairpin/dimer/false priming/cross dimer structures, primer Tm = 55–60°C, length 18 to 25 bases and GC content between 40 and 60%), which generate a unique and short PCR product (between 80 and 250 bp) of the expected length to facilitate multiparallel qPCR using a standard PCR program.
The qPCR design, calculations and statistics used followed the MIQE guidelines36,37. Each 20 μL PCR contained 5 μL diluted cDNA, 10 μL 2 × SYBR® Premix Ex TaqTM II (Takara), 0.6 μL of each primer (10 μM) and 3.8 μL ddH2O. Primers and qPCR reagents were mixed and a standard volume was aliquoted into each reaction of MicroAmp optical 8-tube strip (0.2 ml) to minimize the number of pipetting steps. To minimize the risk of contamination, all PCR operation performed in a clean environment free of dust under a positive airflow hood and check for DNA contamination of primer (by PCR reactions on no template (water) controls, NTC)43.
The reactions were performed in a Mastercycler ep realplex 2 S device (Eppendorf, Germany). To counteract the possibility of systematic variation across the PCR block, the reactions were fullly randomized within different 8-tube strips. Reactions based on the same primer pair were run together and one strip per block was used as an inter-run calibrator to assess the plate-to-plate variation43. The temperature cycling regime comprised an initial denaturation step (95°C/2 min), followed by 40 cycles of 95°C/30 s, 55–58°C (primer-dependent, see Table 1)/30 s, 72°C/30 s. A melting curve analysis (60–95°C, Table 1) and 10% PAGE gel electrophoresis (19:1 acrylamide: bisacrylamide, 7.5 M urea, 50 mM Tris-borate-EDTA, pH 7.8, fragments visualized by silver staining) was conducted following each assay to confirm amplification specificity. Four biological replicates per taxon were analysed and each sample was represented by three technical replicates.
Statistical analysis
The LinRegPCR program44 was used to calculate the threshold cycle (Ct) and the PCR efficiency and correlation coefficients for each primer pair. Stability was assessed using both geNorm and NormFinder software. The former calculates a stability value (M) for each gene8, with an M value <0.5 being taken as an indicator of stable transcription45. The latter provides a direct measure of the variation using an ANOVA-based model and ranks the candidate genes accordingly9.
To determine the influence of the choice of reference genes on normalization outcome, the second stable gene in the comparisons 2x vs 4x C. nankingense, 4x vs 6x C. zawadskii and between all four taxa, was taken as the target and their transcript abundances normalized using either a stable or an unstable reference gene. RAD51 was known to be a key pathway in cells for the homologous recombination and repair of DNA damage26. The transcription of RAD51 was also quantified using both stable and unstable reference genes (the isolation and primer details provided in Supplementary information). Fold changes in transcript abundance were calculated using the 2−ΔΔCt method24.
References
Gachon, C., Mingam, A. & Charrier, B. Real-time PCR: what relevance to plant studies? J Exp Bot 55, 1445–1454 (2004).
Udvardi, M. K., Czechowski, T. & Scheible, W.-R. Eleven golden rules of quantitative RT-PCR. Plant Cell 20, 1736–1737 (2008).
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K. & Scheible, W. R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139, 5–17 (2005).
Fu, J., Wang, Y., Huang, H., Zhang, C. & Dai, S. Reference gene selection for RT-qPCR analysis of Chrysanthemum lavandulifolium during its flowering stages. Mol Breeding 31, 205–215 (2013).
Suzuki, T., Higgins, P. & Crawford, D. Control selection for RNA quantitation. BioTechniques 29, 332–337 (2000).
Thellin, O. et al. Housekeeping genes as internal standards: use and limits. J Biotechnology 75, 291–295 (1999).
Czechowski, T., Bari, R. P., Stitt, M., Scheible, W. R. & Udvardi, M. K. Real-time RT-PCR profiling of over 1400 Arabidopsis transcription factors: unprecedented sensitivity reveals novel root-and shoot-specific genes. Plant J 38, 366–379 (2004).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, research0034 (2002).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64, 5245–5250 (2004).
Radonić, A. et al. Guideline to reference gene selection for quantitative real-time PCR. Biochem Bioph Res Co 313, 856–862 (2004).
Løvdal, T. & Lillo, C. Reference gene selection for quantitative real-time PCR normalization in tomato subjected to nitrogen, cold and light stress. Anal Biochem 387, 238–242 (2009).
Lin, Y. & Lai, Z. Reference gene selection for qPCR analysis during somatic embryogenesis in longan tree. Plant Sci 178, 359–365 (2010).
McClintock, B. The significance of responses of the genome to challenge. Sciences 226, 792–801 (1983).
Paun, O., Fay, M. F., Soltis, D. E. & Chase, M. W. Genetic and epigenetic alterations after hybridization and genome doubling. Taxon 56, 649–656 (2007).
Adams, K. L. & Wendel, J. F. Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8, 135–141 (2005).
Wang, H. et al. Reference genes for normalizing transcription in diploid and tetraploid Arabidopsis. Sci Rep 4, 6781 (2014).
Wang, H. et al. Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PloS One 8, e62293 (2013).
Yang, W., Glover, B. J., Rao, G. Y. & Yang, J. Molecular evidence for multiple polyploidization and lineage recombination in the Chrysanthemum indicum polyploid complex (Asteraceae). New Phytol 171, 875–886 (2006).
Wang, H. et al. Rapid genomic and transcriptomic alterations induced by wide hybridization: Chrysanthemum nankingense x Tanacetum vulgare and C. crassum x Crossostephium chinense (Asteraceae). BMC genomics 14, 902 (2013).
Malinska, H. et al. Similar patterns of rDNA evolution in synthetic and recently formed natural populations of Tragopogon (Asteraceae) allotetraploids. BMC Evol Biol 10, 291 (2010).
Barker, M. S. et al. Multiple paleopolyploidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Mol Biol Evol 25, 2445–2455 (2008).
Fernandez, P. et al. Comparison of predictive methods and biological validation for qPCR reference genes in sunflower leaf senescence transcript analysis. Plant Cell Rep 30, 63–74 (2011).
Maroufi, A., Van Bockstaele, E. & De Loose, M. Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol Biol 11, 15 (2010).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative CT method. Nat protoc 3, 1101–1108 (2008).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods 5, 621–628 (2008).
Carreira, A. & Kowalczykowski, S. C. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. P Natl Acad Sci USA 108, 10448–10453 (2011).
Wang, H. et al. Microsatellite polymorphism among Chrysanthemum sp. polyploids: the influence of whole genome duplication. Sci Rep 4, 6730 (2014).
Artico, S., Nardeli, S., Brilhante, O., Grossi-de-Sa, M. & Alves-Ferreira, M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC plant biology 10, 49 (2010).
Gu, C. et al. Reference gene selection for quantitative real-time PCR in chrysanthemum subjected to biotic and abiotic stress. Mol Biotechnol 49, 192–197 (2011).
Takatsu, Y., Nishizawa, Y., Hibi, T. & Akutsu, K. Transgenic chrysanthemum (Dendranthema grandiflorum (Ramat.) Kitamura) expressing a rice chitinase gene shows enhanced resistance to gray mold (Botrytis cinerea). Sci Hortic 82, 113–123 (1999).
Yang, T., Stoopen, G., Thoen, M., Wiegers, G. & Jongsma, M. A. Chrysanthemum expressing a linalool synthase gene ‘smells good’, but ‘tastes bad’ to western flower thrips. Plant Biotechnol J 11, 875–882 (2013).
Loven, J. et al. Revisiting global gene expression analysis. Cell 151, 476–482 (2012).
Gan, X. et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477, 419–423 (2011).
Kanno, J. et al. “Per cell” normalization method for mRNA measurement by quantitative PCR and microarrays. BMC genomics 7, 64 (2006).
Coate, J. E. & Doyle, J. J. Quantifying whole transcriptome size, a prerequisite for understanding transcriptome evolution across species: an example from a plant allopolyploid. Genome Biol Evol 2, 534–546 (2010).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622 (2009).
Bustin, S. A. et al. MIQE precis: Practical implementation of minimum standard guidelines for fluorescence-based quantitative real-time PCR experiments. BMC Mol Biol 11, 74 (2010).
Chen, Z. J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu Revi Plant Biol 58, 377–406 (2007).
Leitch, A. R. & Leitch, I. J. Genomic plasticity and the diversity of polyploid plants. Science 320, 481–483 (2008).
Wang, J. et al. Genomewide nonadditive gene regulation in Arabidopsis allotetraploids. Genetics 172, 507–517 (2006).
Osborn, T. C. et al. Understanding mechanisms of novel gene expression in polyploids. Trends Geneti 19, 141–147 (2003).
Chen, Z. J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci 15, 57–71 (2010).
Rieu, I. & Powers, S. J. Real-time quantitative RT-PCR: design, calculations and statistics. The Plant cell 21, 1031–1033 (2009).
Ramakers, C., Ruijter, J. M., Deprez, R. H. L. & Moorman, A. F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett 339, 62–66 (2003).
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F. & Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8, R19 (2007).
Acknowledgements
We thank Dr Z. Jeffrey Chen (D. J. Sibley Centennial Professor of Plant Molecular Genetics, The University of Texas at Austin) for helpful discussions. The study was supported by the National Natural Science Foundation of China (31372100, 31272203, 31272202, 31272196, 31171987), the Program for New Century Excellent Talents in University of the Chinese Ministry of Education (NCET-10-0492, NCET-12-0890), the Natural Science Fund of Jiangsu Province (BK2012773, BK2011641), the Fundamental Research Funds for the Central Universities” (KYZ201147), a scholarship granted to Dr. Sumei Chen (File No. 201306855010, CSC, Ministry of Education, China).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: H.W., J.J. and F.Z. Performed the experiments: H.W., F.C. and S.C. Analyzed the data: H.W., J.J. and F.Z. Contributed reagents/materials/analysis tools: H.W. and J.J. Wrote the paper: H.W. J.J. All authors read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, H., Chen, S., Jiang, J. et al. Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp.. Sci Rep 5, 8094 (2015). https://doi.org/10.1038/srep08094
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08094
This article is cited by
-
Selection and validation of appropriate reference genes for RT-qPCR analysis of flowering stages and different genotypes of Iris germanica L
Scientific Reports (2021)
-
Transcriptome analysis of differentially expressed genes in chrysanthemum MET1 RNA interference lines
Physiology and Molecular Biology of Plants (2021)
-
Validation of reference genes for quantitative gene expression in the Lippia alba polyploid complex (Verbenaceae)
Molecular Biology Reports (2021)
-
Identification and validation of new reference genes for accurate quantitative reverse transcriptase-PCR normalization in the Antarctic plant Colobanthus quitensis under abiotic stress conditions
Polar Biology (2021)
-
Selection and validation of reference genes for quantitative gene expression normalization in Taxus spp.
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
