
Abstract
Quantitative real-time PCR (qRT-PCR) is commonly used to measure gene expression to further explore gene function, while suitable reference genes must be stably expressed under different experimental conditions to obtain accurate and reproducible data for relative quantification. Taxol or paclitaxel is an important anticancer compound mainly identified in Taxus spp. The molecular mechanism of the regulation of taxol biosynthesis is current research goal. However, in the case of Taxus spp., few reports were published on screening suitable reference genes as internal controls for qRT-PCR. Here, eight reference genes were selected as candidate reference genes for further study. Common statistical algorithms geNorm, NormFinder, BestKeeper, ΔCt, and RefFinder were used to analyze the data from samples collected from a cell line of Taxus × media under various experimental conditions and from tissues of Taxus chinensis var. mairei. The expression patterns of TcMYC under salicylic acid treatment differed significantly, with the best and worst reference genes in the cell line. This study screened out suitable reference genes (GAPDH1 and SAND) under different treatments and tissues for the accurate and reliable normalization of the qRT-PCR expression data of Taxus spp. At the same time, this study will aid future research on taxol biosynthesis-related genes expression in Taxus spp., and can also be directly used to other related species.
Similar content being viewed by others

Introduction
Yew (Taxus spp.) is a medicinal tree distributed in many regions of the world. The antitumor drug paclitaxel (taxol) can be extracted from Taxus spp. Many researchers have reported that taxanes, including taxol, baccatin III, and 10-deacetylbaccatin III, can be extracted from the needles, stems, roots, and bark of yew trees1,2,3. Moreover, studies have shown that the presence of inductive treatments, such as coronatine (COR), methyl jasmonate (MeJA), salicylic acid (SA), and ethylene (ETH) can greatly enhance taxol content in Taxus spp.4,5,6,7. Furthermore, previous research indicates that abscisic acid (ABA) plays a critical role in ozone-induced taxol production by T. chinensis suspension cell-cultures8.
The molecular mechanism of the regulation of taxol biosynthesis is our research objective. Thus, we have previously reported on some of the genes encoding enzymes involved in taxol synthesis and transcription factors regulating such synthesis9,10,11. To investigate the expression of these taxol biosynthesis-related genes in Taxus spp., the qRT-PCR method was used commonly owing to its cost effectiveness, sensitivity, reproducibility, and simplicity. Although RNA-seq can yield information on gene expression, qRT-PCR has been usually used to investigate the gene expression pattern with relatively small number and to evaluate gene expression results of RNA-seq. In general, the two most common methods for qRT-PCR are absolute and relative quantification12. Absolute quantification is usually used to determine gene copy number, and relative quantification is commonly used to detect the transcripts level changes of target genes under different experimental conditions13. However, to obtain reproducible and reliable results, some experimental conditions must be satisfied in relative quantification qRT-PCR, such as gene specific primers, high-quality RNA without genomic DNA, and suitable reference genes, which have serious implications for the normalization of data14.
An ideal reference gene should have relatively stable expression across different developmental stages, distinct cell types, and various experimental conditions15. Therefore, it is necessary that stable reference genes are evaluated for each species under analysis; in this way, they can be assumed applicable to any experimental conditions in relative quantification qRT-PCR method16,17,18. However, the expression of many common reference genes changes under different conditions of time and space15. It is unlikely that a suitable universal reference gene will be adequate for different experiments. For instance, the expression of 18S rRNA was stable in rice but has different results in papaya19,20. Actin was expressed stable in the study of tomato virus infection but could not be a reliable reference gene in cucumber under salinity stress or in papaya under numerous experimental condition20,21,22. Therefore, it is necessary to select and optimize reference genes for qRT-PCR according to the experimental material and treatments to improve the accuracy of qRT-PCR analysis and interpretation14,23,24.
Although there are many studies related to the function of genes encoding key enzymes involved in taxol biosynthesis, at present, there are only a few reference genes that are used in taxane-biosynthesis-related gene expression; for example, GAPDH (encoding Glyceraldehyde-3-phosphate dehydrogenase), TBC41 (3,5-epimerase-4-reductase), and 18S rRNA (18S ribosomal RNA)25,26,27. Actin, one of the most stable reference genes, was screened and evaluated across different tissues and abiotic stress conditions in many plants28,29,30,31,32, and was also used as the reference gene to investigate the expression pattern of genes involving in Taxus spp.28. Whether these reference genes are stable and suitable to different experimental conditions, such as MeJA, COR, and ABA, has not been evaluated. With unstable reference genes, it is difficult to get accurate results and reveal the mechanism of taxol synthesis. However, proper evaluation for stability of stable reference genes for qRT-PCR analysis in Taxus spp. is still lacking.
The analysis of taxol biosynthesis-related gene expression in tissues or under various inducing treatments can provide important clues to elucidate gene functions involved in taxol biosynthesis, which would be helpful in improving taxol content. Therefore, in this study, a set of eight candidate genes were evaluated under different inductive conditions and in various tissues of Taxus spp., aiming to provide stable reference genes that can help to achieve in accurate, relative quantification qRT-PCR analysis of gene expression to understand taxol-biosynthesis-related gene expression patterns and functions.
Results
Primers performance for amplifying the candidate reference genes
By agarose gel electrophoresis, the PCR products of eight candidate reference genes were detected, and each obtain a single-specific expected size band (Fig. 1a). Consistently, a single peak was identified in the qRT-PCR melting curve analysis with each tested gene primer pairs (Fig. 1b). These results indicated that the test gene primer pairs had high specificity (Fig. 1a,b). Each primer pair amplification efficiency values varied between 92.5% and 103.9% with R2 ≥ 0.98 (Table 1). Thus, all the candidate genes primers used in this study were specific and effective.

Amplification specificity of primers using qRT-PCR. (a) Cropped agarose gel of eight candidate reference genes PCR products, and full-length gel is presented in Supplementary Fig. S1. (b) Uniformity of melting curves of amplified product for each candidate reference gene. The (a) was generated by ChemiDoc Touch Gel Imaging System (version 1708370; https://www.bio-rad.com/), the (b) was generated by LightCycler480 software (version 1.2.0.169; https://lifescience.roche.com/en_cn.html).
Expression profile of the candidate reference genes
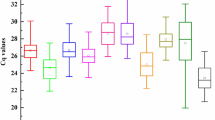
Firstly, the distribution of cycle threshold (Ct) values of the eight candidate reference genes in all treatment and tissues samples was analyzed (Fig. 2). As shown on Fig. 2, the Ct values of candidate genes from each sample showed a high variation, which indicated that the expression of the highest was 18S rRNA (6.92–8.98), while the lowest was ARP2 (19.55–25.22).

Ct values of eight candidate reference genes in all samples from various treatments and tissues. Boxes indicate the 25th and 75th percentiles in all samples; lines across the boxes represent the median; and the whisker caps represent the maximum and minimum values. The figure was generated by GraphPad Prism 9 (version 9.0.0; https://www.graphpad.com/).
According to mean value, standard deviation (SD), and coefficient of variation (CV) of Ct values, among the eight reference genes, GAPDH2 (CV = 12.72%) showed the lowest stability, while SAND (CV = 4.90%) and ARP2 (CV = 4.67%) showed relatively higher stability.
Analysis of expression stability candidate reference genes
In this study, geNorm analysis indicated that the two most stable genes after the treatments of elicitors were as follows: in ABA treatment, RCBL (M = 0.189) and GAPDH1 (M = 0.189); in COR treatment, ARP2 (M = 0.298) and SAND (M = 0.298); in MeJA treatment, GAPDH1 and ARP2 (M = 0.198); similarly, 18S rRNA and GAPDH1 were the most stable genes inducing with SA (M = 0.180); while GAPDH1 and Actin were the most stable genes after ETH treating (M = 0.262). On the other hand, the reference genes, 18S rRNA and SAND were the most stable (M = 0.346) across all tissues studied (Fig. 3, Supplementary Table S1).

Gene expression stability and ranking of eight reference genes under different treatments and tissues based on geNorm. Expression stability value (M) for each reference gene was obtained and graphed. ABA, abscisic acid; COR, coronatine; MeJA, methyl jasmonate; SA, salicylic acid; ETH, ethylene. The figures were generated by geNorm (version 3.4; http://medgen.ugent.be/~jvdesomp/genorm/).
NormFinder analysis showed that GAPDH1 (SV = 0.025) and RCBL (SV = 0.096) were the most stable under ABA treatment, thus confirming the results obtained by geNorm analysis. However, the results of these two programs were not all the same. Under COR treatment, the most suitable genes were 18S rRNA (SV = 0.146) and ARP2 (SV = 0.173); the most stable genes under MeJA and SA treatments were GAPDH1 (SV = 0.097; SV = 0.117) and SAND (SV = 0.130; SV = 0.081), while under ETH treatment, Actin (SV = 0.129) and 18S rRNA (SV = 0.139) were the most stable. As for the tissues studied, the expression of 18S rRNA (SV = 0.052) and SAND (SV = 0.120) were the most stable in all of them, as was the case when geNorm was used for the analysis. Moreover, TBC41 was the most unstable under ABA, COR, and ETH treatments, while Actin and RBCL was the most unstable under SA treatment and in the different tissues, respectively (Supplementary Table S1).
The SD results from BestKeeper analysis showed that ARP2 was the most stable reference genes inducing by ABA and RBCL gene under COR and ETH treatments, while 18S rRNA was the most suitable reference gene under MeJA and SA treatments, and in the different tissues sampled (Supplementary Table S1).
Here, ΔCt analysis showed that RBCL, 18S rRNA, GAPDH1, and Actin, were the most stabilized genes under ABA, COR, MeJA, and ETH treatments, respectively, while SAND was the most stable reference gene under SA treatment and across tissues (Supplementary Table S1).
Finally, the stability of the eight candidate reference genes was compared based on the RefFinder tool which integrates the outputs of geNorm, NormFinder, BestKeeper, and the comparative Ct methods. According RefFinder analysis, SAND ranked as the most stabilized and TBC41 as the least stabilized gene among all samples (Table 2). In addition, the results indicated that RBCL/GAPDH1, 18S rRNA/ARP2, GAPDH1/ARP2, GAPDH1/SAND, and Actin/GAPDH1, were the most stable reference gene combinations under ABA, COR, MeJA, SA, and ETH treatments, respectively, while the combination 18S rRNA/SAND/ARP2 was most suitable in the different tissues.
Optimal number of reference genes under different experimental conditions
The optimum number of reference genes to suit specific experimental conditions can be provided by the geNorm software. When the Vn/Vn+1 ratio is less than 0.15, it indicates that the optimum number is n33. In this study, all values of the Vn/Vn+1 ratio under ABA, MeJA, COR, SA, and ETH treatments were lower than 0.15, indicating that two reference genes were suitable. Conversely, all values of the Vn/Vn+1 ratio in different tissues were greater than 0.15, with the lowest value calculated at 0.154 (V4/V5), thus suggesting that the three stable reference genes were suitable (Fig. 4).

Optimal number of reference genes in different samples from various treatments and tissues using geNorm. Pairwise variation (V) was calculated to obtain the minimum number of reference genes required for normalization under each experimental condition. ABA, abscisic acid; COR, coronatine; MeJA, methyl jasmonate; SA, salicylic acid; ETH, ethylene. The figure was generated by GraphPad Prism 9 (version 9.0.0; https://www.graphpad.com/).
TcMYC expression pattern under SA treatment
The TcMYC gene (accession number KC878013) was selected to examine its expression level under SA treatment with the two sets of the best and worst reference genes. As Fig. 5 shows, the expression pattern of TcMYC did not significantly change under SA treatment for 2 h with the use of the combination of GAPDH1 and SAND as reference genes. Moreover, after induction by SA treatment for 6–48 h, there was no significant difference in the transcripts of TcMYC, which decreased and remained at a relatively stable level, about 0.5-fold that of the control (0 h), with GAPDH1/SAND used as reference genes. Although the results seemed very similar, the expression pattern of TcMYC was different with GAPDH1 or SAND alone used as the unique reference gene, relative to when both GAPDH1 and SAND were used as reference genes. For example, the transcripts of TcMYC were significantly fewer after induction by SA treatment for 12 h when compared with 2 h, with GAPDH1/SAND used as reference genes. However, there was no significant difference in the transcripts of TcMYC for 2–24 h with GAPDH1 or SAND as reference gene. Furthermore, the transcript level of TcMYC was more inaccurate with the worst reference gene, namely, TBC41. With both GAPDH1 and SAND used as reference genes, the transcripts of TcMYC decreased significantly and remained at a relatively stable level, about 0.5-fold that of the control (0 h) after induction by SA treatment for 6–48 h. The quantification of the expression level of TcMYC with TBC41, at all these sampling-time points of SA treatment changed clearly at 2, 6 and 12 h; at these timings, the transcripts of TcMYC at 2 h of SA treatment was clearly lower than that of the control (0 h), and they also were clearly lower at 6 and 12 h than that of 48 h.

Relative quantification of TcMYC expression in T. × media cells induce by salicylic acid with two stable reference genes, GAPDH1 and SAND, separately or in combination, and the unstable reference genes TBC41. The figure was generated by GraphPad Prism 9 (version 9.0.0; https://www.graphpad.com/).
Discussion
Many reference genes have been used to investigate gene expression in a number of plants, including Actin31,32, GAPDH25, Ubiquitin34, and 18S rRNA26. There is not one single stable and suitable gene for all experimental conditions, such as different tissues and development periods, as well as stress treatments17,35,36. Previous studies reported the selection and evaluation work of stable and suitable reference genes in plants is very important15,29,35,37,38, however, reports about the evaluation of stable reference genes in Taxus spp. are scarce. In this study, the transcripts of eight candidate reference genes changed greatly under corresponding experimental conditions and in different tissues; furthermore, stable reference genes and their combinations differed by the algorithms. These variations among the five tools are an expected result due to the differences in statistical algorithms used in each program39. In order to overcome their individual limitations and ensure a more reliable evaluation of the data, it was suggested that the suitable reference genes were chosen according to the results of RefFinder, which takes into consideration the rankings of the other four software methods (geNorm, NormFinder, BestKeeper, ΔCt method). Moreover, it was also suggested to consider the reference gene or their combinations stability obtained by at least three programs39,40.
Currently, the consensus is that multiple reference genes are more accurate and reliable than any single gene for revealing the gene expression level41,42. The suitable reference genes number can be confirmed with the pairwise variation (V value) analyzed by geNorm. By default, when Vn/Vn+1 < 0.15, the suitable reference genes number is n. In this study, all V2/V3 values were less than 0.15 under ABA, COR, MeJA, SA, and ETH treatments, indicating that using two reference genes were suitable under these experimental conditions. The Vn/Vn+1 values of various tissues were larger than 0.15, indicating that a new reference gene should be added in examining gene expression patterns in tissues of Taxus spp., besides the top two reference genes given by geNorm. The reason for this observation might be the large differences among tissue types; similar results were reported for maritime pine (Pinus pinaster Aiton)14. Three stable reference genes, 18S rRNA, SAND, and ARP2, were suggested for use in future investigations of gene expression in different tissues in Taxus spp.
The traditional reference gene, 18S rRNA, was considered one of the least stable reference genes among common reference genes analyzed in many plants37,43,44. For example, 18S rRNA was evaluated and performed poorly in 29 samples different developmental processes and stress treatments (cold, heat, and salt) as well as organs in Lilium28. Moreover, in strawberry (Fragaria × ananassa), it was not recommended for normalization under specific experimental conditions due to low stability45. In contrast, the performance of 18S rRNA was not the worst in this study, where it ranked first in the evaluation for RefFinder’s stability under COR treatment and across tissues, as per RefFinder. Although TBC41 performed more stable in T. × media cell cultures elicited by MeJA and M-β-cyclodextrin, separately or in combination46, it was less stable in this study under ABA, COR, MeJA and ETH treatments. These results clearly showed that it is necessary to evaluate reference genes before starting relative quantification, according to the experimental conditions.
Some novel reference genes were selected in the current study with better performance compared to the common housekeeping genes under specific conditions. A previous study reported that SAND1 was the optimal reference gene for roots and leaves from plants of Gentiana macrophylla exposed to abiotic stress47. Similarly, we found that SAND also was the stable reference gene in different tissues of Taxus spp. Moreover, RBCL was the most suitable reference gene under ABA treatment, while ARP2 also showed better performance among eight candidate reference genes under COR and MeJA treatments.
Taxol production by Taxus cells was induced upon incubation of the cell culture at the optimal SA concentration5,48. Our results indicated that the expression of TcMYC was down-regulated after SA treatment. This suggested that TcMYC played negative regulating role in the complex signaling network of SA-induced taxane biosynthesis. Moreover, evaluation of the expression of TcMYC under SA treatment (Fig. 4), definitely proved that gene expression results were more accurate with stable reference genes than with unstable reference genes.
Recently, high-throughput sequencing technology has been developed rapidly and has been already widely used in genomics and functional genomics. Thus, a great deal of genomes and transcriptome sequences and gene expression data have been generated by sequencing, that are helpful and effective to quickly screen ideal internal reference genes. With the genome and transcriptome sequences, some new reference genes have been explored and widely used with higher expression stabilities49,50,51,52,53. This can benefit to evaluate new and common candidate reference genes in Taxus. To examine the transcripts level of a few target genes, RNA-seq method costs are higher compared to qRT-PCR. Moreover, qRT-PCR is also usually used to verify the results of unigene expression of RNA-seq. In the present study, the sequences of novel reference genes, RBCL, ARP2, and SAND, were obtained from transcriptome data, and performed better than other traditional reference genes. If economy permits, using transcriptome data to obtain the most suitable reference genes is a feasible and time-saving strategy that not only provides sequences, but additionally, it seems highly convenient for screening good housekeeping genes.
In conclusion, this is the first study in which a set of candidate reference genes was analyzed in terms of their expression stability in Taxus spp. Although the final ranking of reference gene of five different statistical algorithms showed slightly different, however by combining and analyzing the data together, GAPDH1 and SAND are most stable reference genes under all experimental conditions. This study also provides a directive foundation for further analyses of gene expression involved in the complex mechanisms of the taxol-biosynthesis pathway.
Methods
Plant materials and stress treatments
Taxus chinensis var. mairei plants were grown in soil pots for 5 years in the greenhouse of the Chinese Academy of Forestry. The tissue samples including roots, stems, leaves, phloems, and xylems were collected immediately frozen in liquid nitrogen, and then stored at − 80 °C for RNA extraction. These samples were collected from three different trees as biological replicates.
A Taxus × media cell line was selected as material to be induced by elicitors, which grown on B5 solid medium as described previously54. For inducing treatments, after filter-sterilized, the elicitor (ABA, COR, MeJA, SA, and ETH) was added to the medium before solidification, with the final concentration of these elicitors was 100 μM, 1 μM, 100 μM, 100 μM, and 30 μM, respectively10,55,56,57. Cells were harvested at 0, 2, 6, 12, 24, and 48 h after each treatment. All cell samples were also collected immediately frozen in liquid nitrogen and stored at − 80 °C. Cells in three culture flasks were collected as biological replicates.
Total RNA extraction and cDNA synthesis
Total RNA was extracted from the samples of tissues and cells line samples with the Column Plant RNAout 2.0 kit (TIANDZ, Beijing, China), which was treated with DNase I to remove the genomic DNA. The total RNA quantity and the purity were assessed with NanoDrop 8000 spectrophotometer (Thermo Fisher SCIENTIFIC, Waltham, MA, USA) and electrophoresis on 1.5% agarose gels. Using the FastQuant RT kit with gDNase (TIANGEN, Beijing, China) cDNA synthesis was performed with 1 μg total RNA. For use as templates for RT-PCR and qRT-PCR, cDNA samples were diluted four-fold in sterile water and stored at − 20 °C.
Candidates reference gene selection and primers design
There were 23 candidate reference genes were selected and designed primers with Primer premier 6 software for the qRT-PCR experiments, including 17 reference genes were obtained from our previous RNA-seq data (unpublished), and six reference genes previously had been used in studies of gene expression in Taxus spp. However, of these, there were eight reference genes were screened out, which produced the single predicted band and a unique absorption peak though RT-PCR and qRT-PCR reaction (Fig. 1, Table 1). Among the eight candidate reference genes, ARP2 (encoding actin-related protein) (accession number MK281332), GAPGH1 (accession number MK281329), RBCL (Ribulose-1,5-bisphospate carboxylase/oxygenase) (accession number MK281331), and SAND (Sand protein) (accession number MK281330), were selected from our previous transcriptomic data (unpublished), and the reference genes Actin, TBC41, 18S rRNA, and GAPDH2 previously had been used in studies of gene expression in Taxus spp.25,26,27,28, while they were not evaluated in different tissues and different treatments, such as ABA, COR, MeJA, SA, and ETH involved in this study. The specificity of primer pairs was verified by electrophoresis on 1.5% agarose gels and analysis of the dissociation (melt) curves. According to the standard curve method, the PCR amplification efficiencies and regression coefficients (R2) of each primer pair were calculated using a cDNA sample with serial four-fold serial dilution, which was reverse transcripted from the total RNA of T. × media cell.
The target gene TcMYC was selected to confirm the expression difference with stable and unstable reference genes under SA treatment. The primer pairs of TcMYC were synthesized according to our previous study9 (Table 1).
RT-PCR and qRT-PCR assay
RT-PCR analyses were performed with a reaction volume of 25 μL containing: 12.5 µL 2 × Taq PCR Master Mix (TIANGEN, Beijing, China), 3 µL of cDNA, 1 μL of gene-specific primer pairs (10 μM), and 7.5 μL sterile ddH2O. The amplification condition was 3 min at 94 °C for initial denaturation; 30 cycles of 30 s at 94 °C, 30 s at 60 °C, and 1 min at 72 °C, and the final extension at 72 °C for 5 min, finally, take 6 μL products for 1.5% agarose gel electrophoresis detection.
Using a Roche Light Cycler 480 (Roche, Basel, Switzerland), qRT-PCR analyses were performed with a reaction volume of 10 μL containing: 5 µL SYBR FAST qRT-PCR Kit Master Mix (2×) (KAPA, Sigma-Aldrich, St. Louis, MI, USA), 1.2 µL of cDNA, 1.3 μL of gene-specific primer pairs (5 μM), and 2.5 μL sterile ddH2O. The condition of the reaction was 95 °C for 3 min, 45 cycles of 95 °C for 10 s, 60 °C for 20 s and 72 °C for 30 s to calculate Ct values. Three biological and technical replicates for the sample of each treatment and tissue were used in the experiments.
Gene expression stability analysis
Among the test reference genes of each sample, the Ct values were analyzed with Delta Ct (ΔCt), BestKeeper (version 1.0; https://www.gene-quantification.de/bestkeeper.html), geNorm (version 3.4; http://medgen.ugent.be/~jvdesomp/genorm/), and NormFinder (version 0.953; https://moma.dk/normfinder-software) methods16,31,58,59. Moreover, comprehensive evaluation was also carried out with the Ct values using the RefFinder tool (http://leonxie.esy.es/RefFinder/?type=reference). Using the 2−ΔΔCT method12 and one-way ANOVA (SAS 9.2 software), the TcMYC expression data and statistical analyses was analyzed.
Data availability
All data generated or analyzed during this study are included in this published article and its “Supplementary Information” Files.
References
Witherup, K. M. et al. Taxus spp. needles contain amounts of taxol comparable to the bark of Taxus brevifolia: Analysis and isolation. J. Nat. Prod. 53, 1249–1255 (1990).
Zocher, R. et al. Biosynthesis of taxol: Enzymatic acetylation of 10-deacetylbaccatin-III to baccatin-III in crude extracts from roots of Taxus baccata. Biochem. Biophys. Res. Commun. 229, 16–20 (1996).
He, C. T. et al. Ranscriptome profiling reveals specific patterns of paclitaxel synthesis in a new Taxus yunnanensis cultivar. Plant Physiol. Biochem. 122, 10–18 (2018).
Mirjalili, N. & Linden, J. C. Methyl jasmonate induced production of taxol in suspension cultures of Taxus cuspidata: Ethylene interaction and induction models. Biotechnol. Prog. 12, 110–118 (2010).
Rezaei, A., Ghanati, F. & Dehaghi, M. A. Stimulation of taxol production by combined salicylic acid elicitation and sonication in Taxus baccata cell culture. Int. Conf. Life Sci. Technol. 3, 193–197 (2011).
Li, S. T. et al. Transcriptional profile of Taxus chinensis cells in response to methyl jasmonate. BMC Genom. 13, 295 (2012).
Cusido, R. M. et al. A rational approach to improving the biotechnological production of taxanes in plant cell cultures of Taxus spp. Biotechnol. Adv. 32, 1157–1167 (2014).
Xu, M. J. et al. Abscisic acid plays critical role in ozone-induced taxol production of Taxus chinensis suspension cell cultures. Biotechnol. Prog. 27, 1415–1420 (2011).
Yang, Y. F. et al. Identification and characterization of MYC transcription factors in Taxus sp. Gene 675, 1–8 (2018).
Li, F. L., Qiu, D. Y., Ma, X. J., Liu, M. & Xu, D. R. Suppression of taxoid 14β-hydroxylase gene expression in Taxus × media via RNA interference. China Biotechnol. 12, 55–60 (2009) (in Chinese).
Yang, Y. F., Liu, H. W. & Qiu, D. Y. Characterization of AP2/ERF gene from Taxus baccata by using bioinformatics tools. Amino Acids Biotic Res. 36, 54–59 (2014) (in Chinese).
Livak, K. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Fleige, S. et al. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotech. Lett. 28, 1601 (2006).
Nolan, T., Hands, R. E. & Bustin, S. A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 1, 1559–1582 (2006).
He, M. J. et al. Selection of suitable reference genes for abiotic stress-responsive gene expression studies in peanut by real-time quantitative PCR. Electron. J. Biotechnol. 28, 76–86 (2017).
Silver, N., Best, S., Jiang, J. & Thein, S. L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7, 33 (2006).
Huis, R., Hawkins, S. & Neutelings, G. Selection of reference genes for quantitative gene expression normalization in flax (Linum usitatissimum L.). BMC Plant Biol. 10, 71 (2010).
Selim, M. et al. Identification of suitable reference genes for real time RT-PCR normalization in the grapevine-downy mildew pathosystem. Plant Cell Rep. 31, 205–216 (2012).
Kim, B. R., Nam, H. Y., Kim, S. U., Kim, S. I. & Chang, Y. J. Normalization of reverse transcription quantitative PCR with housekeeping genes in rice. Biotechnol. Lett. 25, 1869–1872 (2003).
Zhu, X. et al. Evaluation of new reference genes in papaya for accurate transcript normalization under different experimental conditions. PLoS ONE 7, e44405 (2012).
Mascia, T., Santovito, E., Gallitelli, D. & Cillo, F. Evaluation of reference genes for quantitative reverse-transcription polymerase chain reaction normalization in infected tomato plants. Mol. Plant Pathol. 11, 805–816 (2010).
Wan, H. J. et al. Selection of appropriate reference genes for gene expression studies by quantitative real-time polymerase chain reaction in cucumber. Anal. Biochem. 399, 257–261 (2010).
Granados, J. M., Ávila, C., Cánovas, F. M. & Cañas, R. A. Selection and testing of reference genes for accurate RT-qPCR in adult needles and seedlings of maritime pine. Tree Genet. Genomes 12, 60 (2016).
Vanguilder, H. D., Vrana, K. E. & Freeman, W. M. Twenty-five years of quantitative PCR for gene expression analysis. Biotechniques 44, 619–626 (2008).
Patil, R. A., Kolewe, M. E., Normanly, J., Walker, E. L. & Roberts, S. C. Contribution of taxane biosynthetic pathway gene expression to observed variability in paclitaxel accumulation in Taxus suspension cultures. Biotechnol. J. 7, 418–427 (2012).
Xie, S., Xu, X. P., Zhang, M., Fu, C. H. & Yu, J. L. Cloning and function analysis of the TcNPR1 gene from Taxus chinesis. Plant Sci. J. 32, 383–391 (2014) (in Chinese).
Ramirez-Estrada, K. et al. Changes in gene transcription and taxane production in elicited cell cultures of Taxus × media and Taxus globosa. Phytochemistry 117, 174–184 (2015).
Li, Y. Y., Yang, Y. F., Liu, H. W., Wang, S. & Qiu, D. Y. Cloning and expression analysis of TcAPLs in Taxus chinensis. Chin. Traditional Herbal Drugs. 10, 112–119 (2015) (in Chinese).
Yang, Y. F. et al. Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Biol. Rep. 37, 507–513 (2010).
Huang, L. K. et al. Reference gene selection for quantitative real-time reverse-transcriptase PCR in orchardgrass subjected to various abiotic stresses. Gene 553, 158–166 (2014).
Wang, H. B. et al. Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp. Sci. Rep. 5, 8094 (2015).
Zhang, W. C. et al. Screening of reference genes in four woody-oil trees and spatio-temporal expression analysis of actin gene. Mol. Plant Breed. 14, 4576–4582 (2018) (in Chinese).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, research0034.1-research0034.11 (2002).
Yang, Y. F. et al. Expression patterns of some genes involved in tanshinone biosynthesis in Salvia miltiorrhiza roots. Ind. Crops Prod. 130, 606–614 (2019).
Chen, L. et al. Validation of reference genes for RT-qPCR studies of gene expression in banana fruit under different experimental conditions. Planta 234, 377–390 (2011).
Fernandes-Brum, C. N. et al. A panel of the most suitable reference genes for RT-qPCR expression studies of coffee: Screening their stability under different conditions. Tree Genet. Genome 13, 131 (2017).
Kundu, A., Patel, A. & Pal, A. Defining reference genes for qRT-PCR normalization to study biotic and abiotic stress responses in Vigna mungo. Plant Cell Rep. 32, 1647–1658 (2013).
Chao, W. S., Wang, H. X., Horvath, D. P. & Anderson, J. V. Selection of endogenous reference genes for qRT-PCR analysis in Camelina sativa and identification of FLOWERING LOCUS C allele-specific markers to differentiate summer- and winter-biotypes. Ind. Crops Products 129, 495–502 (2019).
Moreira, V. S. et al. Selection and validation of reference genes for quantitative gene expression analyses in various tissues and seeds at different developmental stages in Bixa orellana L. Physiol Mol. Biol. Plants 24, 369–378 (2018).
Kanakachari, M. et al. Evaluation of suitable reference genes for normalization of qPCR gene expression studies in brinjal (Solanum melongena L.) during fruit developmental stages. Appl. Biochem. Biotechnol. 178, 433–450 (2016).
Guo, J. L., Hui, L., Wu, Q. B., Xu, L. P. & Que, Y. X. The choice of reference genes for assessing gene expression in sugarcane under salinity and drought stresses. Sci. Rep. 4, 7042 (2014).
Mariot, R. F. et al. Selection of reference genes for transcriptional analysis of edible tubers of potato (Solanum tuberosum L.). PLoS ONE 10, e0120854 (2015).
Kong, Q. S. et al. Identification of suitable reference genes for gene expression normalization in qRT-PCR analysis in watermelon. PLoS ONE 9, e90612 (2014).
Lu, X. X., Liu, Y. Z., Zhao, L. C., Liu, Y. N. & Zhao, M. W. Selection of reliable reference genes for RT-qRT during methyl jasmonate, salicylic acid and hydrogen peroxide treatments in Ganoderma lucidum. World J. Microbiol. Biotechnol. 34, 92 (2018).
Zhang, Y. T. et al. Evaluation of suitable reference genes for qRT-PCR normalization in strawberry (Fragaria × ananassa) under different experimental conditions. BMC Mol. Biol. 19, 8 (2018).
Sabater-Jara, A. B. et al. Synergistic effect of cyclodextrins and methyl jasmonate on taxane production in Taxus × media cell cultures. Plant Biotechnol. J. 12, 1075–1084 (2014).
He, Y. H., Yan, H. L., Hua, W. P., Huang, Y. Y. & Wang, Z. Z. Selection and validation of reference genes for quantitative real-time PCR in Gentiana macrophylla. Front. Plant Sci. 7, 945 (2016).
Wang, Y. D., Wu, J. C. & Yuan, Y. J. Salicylic acid-induced taxol production and isopentenyl pyrophosphate biosynthesis in suspension cultures of Taxus chinensis var. mairei. Cell Biol. Int. 31, 1179–1183 (2007).
Czechowski, T., Stitt, M., Altmann, T., Udvardi, M. K. & Scheible, W. R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 139, 5–17 (2005).
Zhou, C. F. et al. Selection of reference genes for quantitative real-time PCR in six oil-tea camellia based on RNA. Mol. Biol. 47, 836–851 (2013).
Lin, F. et al. Genome-wide identification of housekeeping genes in maize. Plant Mol. Biol. 86, 543–554 (2014).
Yim, K. Y. et al. Using RNA-seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS ONE 10, e0136343 (2015).
Zhou, Z., Cong, P., Tian, Y. & Zhu, Y. Using RNA-seq data to select reference genes for normalizing gene expression in apple roots. PLoS ONE 12, e0185288 (2017).
Sun, G. L. et al. Deep sequencing reveals transcriptome re-programming of Taxus × media cells to the elicitation with methyl jasmonate. PLoS ONE 8, e62865 (2013).
Xiong, L., Ishitani, M. & Zhu, J. K. Interaction of osmotic stress, temperature, and abscisic acid in the regulation of gene expression in Arabidopsis. Plant Physiol. 119, 205–212 (1999).
Onrubia, M. et al. Coronatine, a more powerful elicitor for inducing taxane biosynthesis in Taxus media cell cultures than methyl jasmonate. J. Plant Physiol. 170, 211–219 (2013).
Chang, B. W. Metabolome and transcriptome analysis of TIAs biosynthesis in Catharanthus roseus induces by exogeno ethylene. https://kns.cnki.net/KCMS/detail/detail.aspx?dbcode=CDFD&dbname=CDFDLAST2017&filename=1016309236.nh&v=MTg4ODdmWnVSbUZ5bmdWcnJLVkYyNkdMQzRGOVBQcVpFYlBJUjhlWDFMdXhZUzdEaDFUM3FUcldNMUZyQ1VSN3E= (2016). (in Chinese)
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulatedtarget genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515 (2004).
Acknowledgements
We are thankful to Professor Z.W. Huang for his helpful in statistical analysis. This work was supported by a Grant for National non-profit Research Institutions of Chinese Academy of Forestry (CAFYBB2018SY009), National Natural Science Foundation of China (31570675), and the projects of the National Key Research and Development Program of China (2017YFD060070605).
Author information
Authors and Affiliations
Contributions
Y.Y. proposed the study. K.Z., Y.L. and Z.Y. collected samples, performed the qRT-PCR experimental analyses, produced the Tables and Figures for the manuscript, and also drafted the manuscript. W.F., D.C. L.J., and D.Q. revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, K., Fan, W., Chen, D. et al. Selection and validation of reference genes for quantitative gene expression normalization in Taxus spp.. Sci Rep 10, 22205 (2020). https://doi.org/10.1038/s41598-020-79213-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79213-1
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
