
Abstract
Structural domains in proteins are the basic units to form various proteins. In the protein's evolution and functioning, domains play important roles. But the definition of domain is not yet precisely given and the update cycle of structural domain databases is long. The automatic algorithms identify domains slowly, while protein entities with great structural complexity are on the rise. Here, we present a method which recognizes the compact and modular segments of polypeptide chains to identify structural domains and contrast some data sets to illuminate their effect. The method combines support vector machine (SVM) with K-means algorithm. It is faster and more stable than most current algorithms and performs better. It also indicates that when proteins are presented as some Alpha-carbon atoms in 3D space, it is feasible to identify structural domains by the spatially structural properties. We have developed a web-server, which would be helpful in identification of structural domains (http://vis.sculab.org/~huayongpan/cgi-bin/domainAssignment.cgi).
Similar content being viewed by others


Introduction
As the basic units, the structural domain of proteins constitutes differently functional and structural proteins1. Recognition and prediction of structural domain is a crucial first step of structure and function prediction on proteins2. Identification of structural domain is also an important part of functional and structural experiments3,4 and a vital part of target selection process in structural genomics5 and drug discovery6,7. However, since the concept of structural domain was proposed in Wetlaufer DB's research8, 1973, a precise and consistent definition about structural domain has not yet been concluded.
An overview of all varied definitions9 about structural domain includes 6 characteristics as follow: the structural domain is usually compact (I) and/or stable (II), contains hydrophobic cores (III), folds independently of the rest of the protein structure (IV), combines with others in the evolution (V) and performs some specifically biological functions (VI). These 6 main characteristics involve 4 key aspects in proteomics: Characteristic (I, III) depict the structural properties of protein10; Characteristic (II, IV) are related to molecular dynamics11; Characteristic (V) is connected with phylogenetic analysis12,13,14 about domains; then, the function of protein13 is concerned in Characteristic (VI). Due to the diversity and complexity of protein structures, experts are not unanimous about the definition of structural domain, which poses a challenge to develop automatic methods to recognize structural domains.
Since proteins with great structural complexity are on the rise, it is pressing to update the structural domain databases and improve the performance of the off-the-shelf algorithms or develop new ones. The SCOP15 and CATH16 databases are the gold standards17 and often regarded as the data sets for comparing and evaluating different algorithms. But the update cycle of structural domain databases is long in general. And some divergences exist among different domain databases, since the experts and methods usually make different domain assignments based on their own understanding of structural domain.
Many automatic algorithms are constantly emerging to identify domains18 as correctly as possible and exactly annotate the structures of new proteins. PDP19 is mainly based on the assumptions of domain compactness. When the structure of a protein is not compact enough, it splits proteins into two domains illogically. DomainParser20,21 is based on graph theory and finds the minimal cut through domain interfaces, but rarely splits secondary structure elements between domains. Even it misses small domains or domains with low secondary structure ratio. PUU22 hypothesizes the domain is an autonomously folding unit, so that usually identifies more domains than experts. DDOMAIN22 maximizes the intra-domain contacts on a normalized contact-based domain-domain interaction profile. Its limitations is the assumption that each structural domain is a continuous segment, but some domains are consisted of some different segments. CA23 and SS23 algorithms, based on the alpha-carbon atoms and secondary structure elements respectively, use the average-linkage clustering algorithm to attain a dendrogram, then assign domains by cutting it. They are simple and fast, but neglect the spatially structural properties of domains.
Although various structural domain databases and computational approaches have been used to assist the identification of domains, each of them has its intrinsic limitations as mentioned above. In this study, we presented a hybrid method by combining 2 algorithms: support vector machine (SVM) and K-means to identify structural domains. It took full advantage of the spatially structural properties of proteins - the density and modularity of segments during partition. In terms of the correctness on domain assignment and execution speed, the hybrid method outran some off-the-shelf algorithms. When we evaluated different algorithms on the data sets, the performance varied with different data sets. Then, we also contrastively analyzed the data sets, in order to illuminate the effect of the data sets. Finally, we have developed a webserver, which would be helpful in identification of structural domains (http://vis.sculab.org/~huayongpan/cgi-bin/domainAssignment.cgi).
Results
The performance of the hybrid method
Based on the density and modularity of alpha-carbon atoms in 3D space, the hybrid method would correctly identify structural domains and catch up with some best currently available algorithms as shown in Table 1,2,3. As a whole, all algorithms had similar results with quite small differences, but the hybrid method had a narrow lead over the others by success rates of 77% (Table 1) and 82% (Table 3) on ASTRAL SCOP data set and Benchmark_3 respectively. From the perspective of execution time in Table 2, the hybrid method was about 2.5 times faster than CA algorithm and over 40 times faster than DDomain and DomainParser2 algorithms.
In detail, the hybrid method performed well on ASTRAL SCOP data set with success rates of 80%, 71%, 83%, 49%, 77% for single-domain, 2-, 3-, 4- and overall, on the Benchmark_2 were 93% for single-, 72% for 2-, 92% for 3-, 25% for 4-domain, 80% for overall and on the Benchmark_3 were 91% for single-, 75% for 2-, 95% for 3-, 33% for 4-domain, 82% for overall. However, it fell behind PDP algorithm on the Benchmark_2 by 3% on the overall as shown in Table 2, which was caused by domain assignments on the chains with 2- and 4-domain. In the Benchmark_2, there were only 4 chains with 4-domain, it was less statistically meaningful and was dismissed here. Moreover, we also found that PDP algorithm performed unsteadily on the chains with 2-domains, which achieved 62%, 84% and 76% on ASTRAL SCOP data set, Benchmark_2 and Benchmark_3 respectively. The fluctuation was caused by the unstable proportion of different Classes (all alpha, all beta, alpha/beta and alpha+beta in SCOP database) in 3 data sets as shown in Figure 1. The proportion of alpha/beta class in ASTRAL SCOP data set was more than the others and the proportion of alpha+beta class in the Benchmark_2 was the highest. That indicated the PDP algorithm preferred to correctly identify domains on the alpha+beta class proteins, so that it was more outstanding only on the Benchmark_2. In contrast, the hybrid method did not be affected by the fluctuant proportion of different Class proteins. In addition, the hybrid method was also about 10 times faster than PDP algorithm. Thus, the hybrid method not only could correctly and quickly identify structural domains, but also kept a solid performance.

The proportion of chains with 2-domain belonging to different Classes.
The (a), (b), (c), (d) classes represent all alpha, all beta, alpha/beta and alpha+beta proteins respectively. The proportion of c class on 3 data sets fluctuates dramatically, but the proportion of d class on the Benchmark_2 rapidly increases. That affects the evaluation of algorithms.
Contrastive analysis of different data sets
As shown in Table 1,2,3, the performance of all algorithms varied with different data sets, especially the percentages of correct identification on the Benchmark_2 and Benchmark_3 were obviously higher than those on ASTRAL SCOP data set. Then, we contrastively analyzed the data sets extracted from SCOP, CATH v3.5 and Pfam v27.0 databases and the Benchmark_2, Benchmark_3 (described in Methods). There were 2 main influences on the evaluation of algorithms: the distribution of polypeptide chains and the scale covering the sample space in different data sets.
The distribution of chains' proportions and quantities of the chains with single-, 2-, 3- and 4-domain in the data sets were showed in Table 4. In the ASTRAL SCOP 30, CATH v3.5 and Pfam v27.0, the proportion of chains with single-domain was over twice that of the rest chains. However, it was approximately equal with the proportion of chains with 2-domain and about twice that of the rest chains with 3- and 4-domain in the Benchmark_2 and Benchmark_3. Except the unbalanced distribution in data sets, the scale covering the sample space was also dramatically different. As shown in Figure 2, ASTRAL SCOP data set involved more Classes, Folds, Superfamilies and Families and was similar to SCOP database. However, the number of different Folds and Superfamilies involved in ASTRAL SCOP data set was over 8 times more than those involved in the Benchmark_2 and Benchmark_3. And the number of Families in ASTRAL SCOP data set was more than the others by a further factor of 15 (see Supplementary Table S1). Thus, ASTRAL SCOP data set was appropriate to train the algorithms.

The proportions of Classes, Folds, Superfamilies and Families of 4 different data sets.
The rader map draws the proportions of Classes, Folds, Superfamilies and Families of the ASTRAL SCOP data set, Benchmark_2 and Benchmark_3 compared with SCOP database. The whole area (the blue part) is the full sample space in SCOP database. The red, green and purple regions become smaller in order, which indicates that the scales covering the sample space of ASTRAL SCOP data set, the Benchmark_2 and Benchmark_3 are smaller.
Discussion
Comparison of some domain identification methods
Here we just compared the domain identification methods: PDP19, DDOMAIN22, DomainParser20,21, CA23 algorithm with the hybrid method. Currently, these 4 methods performed best on domain identification.
PDP19 was mainly based on the principles of domain compactness. When recognizing the structural domains, PDP tried to cut at all possible sites in the polypeptide chain19. It would be time-consuming. PDP also defined a domain as a set of continuous protein segments, but some domains contained 2 or more uncontinuous segments. DDOMAIN22 had the same limitation, when it partitioned domains by maximizing the intra-domain contacts on the domain-domain interaction profile.
DomainParser20,21 and CA23 algorithm were based on graph theory and formulated the protein as a network, in which each node represented the residue and each edge represented the residue-residue interaction. Then, the problem of domain assignment was solved by cutting the network. Under the assumption that residue-residue contacts were denser within a domain than between domains, DomainParser20,21 and CA23 algorithm rarely splitted the secondary structure elements between domains. In addition, the network simplification of protein 3D structure can cause the loss of some spatial information and hinder investigations into the steric effects of protein24. Thus, from the perspective of 3D structure, partitioning the polypeptide chains directly was the biggest difference between the hybrid method with others, thereby the hybrid method can capture the spatially structural properties of protein.
The Benchmarks constructing process
Through repeating the similar process of constructing the Benchmark_2 and Benchmark_3 as Holland et al.'s work25, we found that the distribution of polypeptide chains with different number of domains changed dramatically and most chains with great complex structure were filtered out.
For constructing the benchmarks, the criteria were: 1) sequence identity was no more than 30%; 2) the chains were assigned with the same number of domains in SCOP, CATH and Pfam databases; 3) domain overlap (described in Methods) was higher than the threshold. Then, the threshold was set 5% ~ 95% by the increments of 5%. In Figure 3(a), the number of chains decreased rapidly with the increase of threshold and most chains with >3-domain were filtered out before being checked domain overlap (the 3rd step), since the divergences between databases occurred on the chains with complex structure firstly. It also reconfirmed that experts firstly gave different domain assignments on the chains with more complex structure. Although the number of chains was declining, in Figure 3(b) the proportion of the chains with single-domain was rising.

The distribution of polypeptide chains at different threshold.
(a) The number of chains with different number of structural domains under different thresholds. The number of chains with single-, 2- and 3-domain (the blue, red and green polylines) changes more gently than that of chains with 4-domain represented by the purple polyline. (b) The proportion of chains with different number of structural domains under different thresholds. The proportion of chains with single-domain (the blue polyline) increases towards 99% but the others (the red, green and purple polylines) descend continuously. Finally, the proportion of chains with 4-domain decreases to about 0%.
With domain overlap rising, the domain assignments of chains in the Benchmark_3 were more consistent in different databases (illuminated in Data sets, Methods), so that the Benchmark_3 were used to analyze the domain assignments given by the hybrid method in detail. However, most chains with great complex structure were also filtered out, therefore, the construction of a data set including proteins with complex structure and high domain overlap is our next task.
The details of domain assignments
It is helpful to study the domain assignments made by the hybrid method. We were concerned with the results on the Benchmark_3, because all the chains in which had the same number of domains and approximately consistent domain boundaries with structural domain databases. There were 29 incorrect domain assignments including 5, 13, 1, 2 on chains with single-, 2-, 3- and 4-domain. In the first 18 incorrect assignments, we found the alpha-carbon atoms in the chains scatter so chaotically in 3D space that the hybrid method failed.
For the chains with multi-domain, the hybrid method was always inclined to undercut. Here were two more clear cases with incorrect domain assignments: PDB:1DJZ chain A and PDB:1CWV chain A. The PDB:1DJZ chain A contained 3 domains, but was assigned with 2 domains by the hybrid method in Figure 4. Whether this chain was assigned with 2 or 3 domains, the modularity was visually obvious. But if the chain sequence information was taken into account, the hybrid method would correctly assign. Another case PDB:1CWV chain A contained 5 domains indeed, which was only assigned with 4 domains by the hybrid method and all other current algorithms. In Figure 5(a), the 4 different colored segments showed more obvious modularity in relation to each other than the 5 colored segments in Figure 5(b). Therefore it easily led to the incorrect domain assignment only in consideration of protein spatial structure. These improvements would be made in the future work.

PDB 1DJZ chain A domain assignment.
(a) PDB 1DJZ chain A, assigned with 2 domains by the hybrid method. (b) Actual domain assignment in the Benchmark_3 with 3 domains.

PDB 1CWV chain A domain assignment.
(a) PDB 1CWV chain A, assigned with 4 domains by the hybrid method. (b) Actual domain assignment in the Benchmark_3 with 5 domains.
Surprisingly there were 3 cases with 3-domain (PDB:1KSI chain A, PDB:1D0G chain T, PDB:1DCE chain A) that could not be partitioned correctly by all current methods. However, all of them were assigned correctly by the hybrid method. The hybrid method was better at partitioning the chains with 3-domain than other algorithms. For example, PDB:1KSI was a eukaryotic copper-containing amine oxidase. In Figure 6, the 3 different colored segments displayed a certain modularity in 3D space. Though the modularity in PDB:1KSI chain A was not as obvious as PDB:1CWV chain A, it was enough to correctly identify structural domains by the hybrid method. Nevertheless, there were a few flaw between 2 domain assignments in Figure 6. However, the domain assignments on PDB:1D0G chain T was approximately in agreement with the assignment in the Benchmark_3 as shown in Figure 7.

PDB 1KSI chain A domain assignment.
(a) PDB 1KSI chain A, assigned with 3 domains by the hybrid method. (b) Actual domain assignment in the Benchmark_3 with 3 domains. The different colored regions between (a) and (b) is clear.

PDB 1D0G chain T domain assignment.
PDB:1D0G chain T was approximately in agreement with the assignment in the Benchmark_3.
In the present study, although the performance of all algorithm varied with different data sets, the hybrid method performed well. In particular, some chains assigned correctly only by the hybrid method. Based on the spatially structural properties of the chains, the method could grasp some intrinsic properties of structural domains -- most structural domains correspond to more compact and modular segments of chains. However, identification of structural domain is still an intractable challenge. The main issue is also not solved that experts and researchers cannot give a clear and precise definition of structural domain. In addition, the construction of a gold benchmark to evaluate various algorithms is in progress. There is still a long way to go.
Methods
Domain overlap
Domain overlap18 between two domain assignments was measured by calculating the maximum fraction of residues in the entire chain for which assignments made by a given method and the reference method were identical.
Feature extraction
With alpha-carbon atoms representing amino acid residues, a protein is simplified as a group of nodes in 3D space. There are 3 kinds of features extracted during domain assignment – the density of nodes, the dispersion and the number of nodes. The number of nodes denotes the number of residues in the segment splitted out. The 2 other kinds of features are the following in detail, which are the spatially structural properties.
The density of nodes
In 3D space, most structural domains in protein correspond to those regions containing more intensive nodes. Because of the high density of nodes in some regions, these regions are like clusters which indicate the structural domain is compact and forms a hydrophobic core. Thus, the density of nodes is considered as a feature to describe the structural domain, which is equal to the total number of alpha-carbon atoms in the cluster divided by the approximate volume.
The dispersion
Though the nodes in the same domain are close, the nodes in the different structural domains are widely separated. The dispersion of nodes in the protein with multi-domain is high, which also indicates the modularity. PCA (Principle component analysis) can measure the degree of dispersions by calculating the variances in 3 mutually orthogonal directions. Thus, we employ this feature to decide whether the chain is the one with single-domain or with multi-domain. In addition, after the chain is determined as the one with multi-domain, K-means algorithm can recognize the domains by taking advantage of their modularity in protein.
The hybrid method
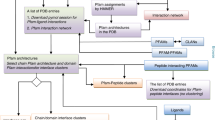
After feature extraction, the hybrid method is based on the spatially structural properties – high density and modularity of nodes to identify the structural domains. We combines the two-stage SVM (support vector machine) with K-means algorithm to construct the hybrid method. The flowchart of the whole process is shown on Figure 8.

The flowchart of the hybrid method.
The hybrid method combines two-stage SVM with K-means to identify the structural domains. At first, it extracts the features for modelling. Then, it trains two-stage SVM model. At the two-stage SVM, the features Den, Dis and NN represent the density, dispersion and the number of nodes. The numbers (1, 2, 3) in Den2a, Dis2a, NN2a, Den3a, Dis3a and so on are the number of segments in the chain, which are tentatively partitioned. The alphabet (a, b, c) represents the 1st, 2nd or 3rd segment. In the 1st-Stage SVM model, 9 features are used, but 6 of them about 2 segments are kept and some new features about 3 segments are added into the 2nd-Stage SVM model. Finally, K-means model completes domain assignments.
The 1st-Stage SVM model: There are 9 features used to train 1st-Stage SVM model. 3 of them are the density, the dispersion and the number of nodes in the whole chain. In order to determine if the chains have single domain or multiple domains, we try to partition the chain into 2 segments by K-means algorithm (K = 2). Then, the 6 other features are the density, the dispersion and the number of nodes in 2 segments respectively as shown in Figure 8. Based on 9 features, the 1st-Stage SVM model is trained on the 1st training set (see detail in Data set). Then, all chains are divided into 2 kinds – the one with single-domain and with multi-domain.
The 2nd-Stage SVM model: Based on 1st-Stage SVM model, all the chains classified as the ones with multi-domain are considered as the input of the 2nd-Stage SVM model. And the 2nd-Stage SVM model is aimed at distinguishing chains with 2- and >2-domain. Then, we keep using 6 features about 2 segments in 1st-Stage SVM model and add some new features. As 1st-Stage SVM model, the chains are also tentatively partitioned into 3 segments by K-means algorithm (K = 3). The density, the dispersion and the number of nodes in 3 segments respectively are regarded as the new features as shown in Figure 8. Finally, the 2nd-Stage SVM model uses 15 features and is trained on the 2nd training set (see detail in Data sets, Methods). K-means algorithm partitions the chains judged as the ones with 2-domain into 2 segments. The other chains are further analyzed by K-means algorithm in next procedure.
K-means model for optimizing modularity
After going through the two-stage SVM model, the chains with >3-domain are divided from the data sets. Then, K-means randomly selects K(>3) nodes as seeds and automatically cluster nodes into groups corresponding to structural domains. The modularity of clusters plays an important role in the clustering process. The modularity contains intra-cluster and inter-cluster cohesiveness. Optimizing modularity by the following formulas can find the optimal K value.



The ck, ni, center are the kth-cluster's center, ith node, the center of the whole chain respectively. The d(x,y) represents the Euclidean distance between x and y.
Assessment of correctness
The hybrid method is evaluated by the same assessing method as Feldman's work. Firstly, if the number of domains in a chain determined by the hybrid method is the same as the one in test set, domain overlap will be further calculated. If not, it is considered wrong. Secondly, if domain overlap is not lower than 75%, then this domain assignment is supposed to be correct.
Data sets
In this study, there are 3 datasets: the Benchmark_2, Benchmark_3 (constructed by Holland et al.25) and a non-redundant ASTRAL SCOP data set in which chains with greater than 30% sequence identity are removed. The chains in Benchmark_2 and Benchmark_3 are filtered by some rigorous criteria. The Benchmark_2 prefers the chains with the same number of domains from SCOP, CATH. The Benchmark_3 further meets the demands of domain overlap between domain assignments. Therefore, the domain assignments in the Benchmark_3 are more consistent with SCOP and CATH databases. Here, we only attain the half of Benchmark_2 and Benchmark_3 from a website pdomain (http://pdomains.sdsc.edu/v2/dataset.php), on which there are 156 and 135 chains available.
In the ASTRAL SCOP data set, there are 9,500 structural domains and 7,135 diverse chains. Only 7077 chains are available to download from the current PDB databank (Protein Data Bank). The distribution of chains with different number of domains (5,322 for 1-, 1,341 for 2-, 304 for 3-, 73 for 4-, 37 for >4-domain) is heavily unbalanced, so we screen 2 subsets as training sets for the two-stage SVM, where the chains in the Benchmark_2 and Benchmark_3 are also filtered out.
For training the 1st-Stage SVM, the 1st training set contains all chains with multi-domain and the same number of chains with single domain selected randomly. We repeat random selection and train SVM algorithm for 9 times. Then 9 predictions on the ASTRAL SCOP data set are used to vote and decide if the chain is the one with single-domain or multi-domain. For training the 2nd-Stage SVM, based on all chains with multi-domain correctly predicted, the 2nd training set includes all chains with >2-domain and the same number of ones with 2-domain. We still randomly select and train for 9 times. The final prediction is also voted by 9 predictions.
In addition, CATH v3.5 (Http://www.cathdb.info/) and Pfam v27.0 (Ftp://ftp.ebi.ac.uk/pub/-databases/Pfam) are downloaded for repeating the similar process of constructing the Benchmark_2 and Benchmark_3, in which the number of chains with single-, 2-, 3-, 4- and >4-domain is 77,455, 32,428, 6,090, 2,495, 1,108 and 139,152, 33,892, 6,500, 2,495, 1,687 respectively.
References
Chen, H. et al. Mechanical perturbation of filamin A immunoglobulin repeats 20–21 reveals potential non-equilibrium mechanochemical partner binding function. Sci. Rep. 3, 1642 (2013).
Lewis, T. E. et al. Genome3D: a UK collaborative project to annotate genomic sequences with predicted 3D structures based on SCOP and CATH domains. Nucleic Acids Res. 41, D499–D507 (2013).
Kryshtafovych, A. et al. Challenging the state of the art in protein structure prediction: Highlights of experimental target structures for the 10th Critical Assessment of Techniques for Protein Structure Prediction Experiment CASP10. Proteins 82, 26–42 (2014).
Guo, J., Ren, H., Ning, L., Liu, H. & Yao, X. Exploring structural and thermodynamic stabilities of human prion protein pathogenic mutants D202N, E211Q and Q217R. J. Struct. Biol. 178, 225–232 (2012).
Weisser, H. et al. An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. 12, 1628–1644 (2013).
Hu, Q.-H. et al. Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase. Sci. Rep. 3, 2475 (2013).
Sato, M., Sawahata, R., Sakuma, C., Takenouchi, T. & Kitani, H. Single domain intrabodies against WASP inhibit TCR-induced immune responses in transgenic mice T cells. Sci. Rep. 3, 3003 (2013).
Wetlaufer, D. B. Nucleation, rapid folding and globular intrachain regions in proteins. Proc. Natl. Acad. Sci. 70, 697–701 (1973).
Veretnik, S. & Shindyalov, I. Computational Methods for Domain Partitioning of Protein Structures. in Biological and Medical Physics, Biomedical Engineering: Computational Methods for Protein Structure Prediction and Modeling (ed. Xu, Y., Xu, D. & Liang, J.) 125–145 (Springer, New York, 2007).
Xu, D. & Zhang, Y. Ab Initio structure prediction for Escherichia coli: towards genome-wide protein structure modeling and fold assignment. Sci. Rep. 3, 1895 (2013).
Kaila, V. R., Wikström, M. & Hummer, G. Electrostatics, hydration and proton transfer dynamics in the membrane domain of respiratory complex I. Proc. Natl. Acad. Sci. 111, 6988–6993 (2014).
Ezkurdia, I. & Tress, M. L. Protein Structural Domains: Definition and Prediction. Curr. Protoc. Protein Sci. 66, 2.14.1–12.14.16 (2011).
Letunic, I., Doerks, T. & Bork, P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305 (2012).
Torruella, G. et al. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 29, 531–544 (2012).
Andreeva, A. et al. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 36, D419–D425 (2008).
Greene, L. H. et al. The CATH domain structure database: new protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res. 35, D291–D297 (2007).
Schaeffer, R. D., Jonsson, A. L., Simms, A. M. & Daggett, V. Generation of a consensus protein domain dictionary. Bioinformatics 27, 46–54 (2011).
Veretnik, S., Bourne, P. E., Alexandrov, N. N. & Shindyalov, I. N. Toward consistent assignment of structural domains in proteins. J. Mol. Biol. 339, 647–678 (2004).
Alexandrov, N. & Shindyalov, I. PDP: protein domain parser. Bioinformatics 19, 429–430 (2003).
Xu, Y., Xu, D. & Gabow, H. N. Protein domain decomposition using a graph-theoretic approach. Bioinformatics 16, 1091–1104 (2000).
Guo, J. T., Xu, D., Kim, D. & Xu, Y. Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res. 31, 944–952 (2003).
Holm, L. & Sander, C. Parser for protein folding units. Proteins 19, 256–268 (1994).
Feldman, H. J. Identifying structural domains of proteins using clustering. BMC Bioinformatics 13, 286 (2012).
Yan, W. et al. The construction of an amino acid network for understanding protein structure and function. Amino acids 46, 1419–1439 (2014).
Holland, T. A., Veretnik, S., Shindyalov, I. N. & Bourne, P. E. Partitioning protein structures into domains: why is it so difficult? J. Mol. Biol. 361, 562–590 (2006).
Acknowledgements
This study was funded by the National Natural Science Foundation of China (No. 21175095, 21375090) and the Science and Technology Innovation Seedling project in Sichuan Province (Grant No.20132004).
Author information
Authors and Affiliations
Contributions
Y.H. designed and performed the experiments and interpreted the data. M.Z. and M.L. conceived the research. Y.H., M.Z. and M.L. wrote the manuscript. Y.H. prepared figures 1–6. Y.W. and Z.X. built the web server and prepared the figures 7–8. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
cross-validation test
Supplementary Information
Dataset 1 ASTRAL SCOP
Supplementary Information
Dataset 2 Benchmark 2
Supplementary Information
Dataset 3 Benchmark 3
Supplementary Information
Dataset 4 Cath
Supplementary Information
Dataset 5 Pfam
Supplementary Information
Dataset 6
Supplementary Information
Dataset 7
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hua, Y., Zhu, M., Wang, Y. et al. A hybrid method for identification of structural domains. Sci Rep 4, 7476 (2014). https://doi.org/10.1038/srep07476
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep07476
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
