
Abstract
The high pressure structures, metallization and superconductivity of recently synthesized H2-containing compounds (H2S)2H2 are elucidated by ab initio calculations. The ordered crystal structure with P1 symmetry is determined, supported by the good agreement between theoretical and experimental X-ray diffraction data, equation of states and Raman spectra. The Cccm structure is favorable with partial hydrogen bond symmetrization above 37 GPa. Upon further compression, H2 molecules disappear and two intriguing metallic structures with R3m and Im-3m symmetries are reconstructive above 111 and 180 GPa, respectively. The predicted metallization pressure is 111 GPa, which is approximately one-third of the currently suggested metallization pressure of bulk molecular hydrogen. Application of the Allen-Dynes-modified McMillan equation for the Im-3m structure yields high Tc values of 191 K to 204 K at 200 GPa, which is among the highest values reported for H2-rich van der Waals compounds and MH3 type hydride thus far.
Similar content being viewed by others

Introduction
Closed shell systems (e.g., H2O, H2S, CH4, SiH4, GeH4, NH3BH3, Ar, Kr and Xe) can absorb additional H2 molecules under high pressure conditions, forming a series of new H2-containing stoichiometric compounds, such as H2O(H2)1, (H2S)2H22, CH4(H2)43, SiH4(H2)24,5, GeH4(H2)26, NH3BH3(H2)x7,8, Ar(H2)29, Kr(H2)410 and Xe(H2)811. Given their high amount of hydrogen, these compounds are potential energy storage materials and high-temperature superconductors. For example, CH4(H2)4 with 33.4 wt% high hydrogen content is a potentially high hydrogen storage material12. Metallic hydrogen is one of the most important physical problems in high-pressure research, which is believed to be a room-temperature superconductor13,14. However, the metallization of hydrogen is still under debate in laboratories15,16. Hydrogen-rich compounds, such as GaH317, GeH318, Si2H619,20, SiH421,22,23,24, GeH425, KH626, CaH627, SiH4(H2)228 and GeH4(H2)229 are extensively explored as alternatives because their metallization can occur at relatively lower pressures through chemical pre-compressions, which present high temperature superconductivity. H2-containing compounds SiH4(H2)2 and GeH4(H2)2 have been predicted to have high Tc values of 98 K to 107 K at 250 GPa and 76 K to 90 K at 250 GPa (with the Coulomb parameter μ* = 0.1–0.13), respectively28,29. For the MH3 (M = Ga, Ge, Si) type hydride, Tc is estimated theoretically to be 76 K to 86 K (μ* = 0.1−0.13) at 160 GPa17, 140 K (μ* = 0.13) at 180 GPa18 and 139 K to 153 K (μ* = 0.1−0.13) at 275 GPa19, respectively. Theoretical predictions also revealed that the Tc of sodalite-like CaH6 at 150 GPa reached 220 K to 235 K (μ* = 0.1−0.13)27. Considering that H2 is the most abundant substance in the universe, the behavior of H2-containing compounds over a broad P–T range is important for planetary sciences.
Recently, Strobel et al. reported that mixing hydrogen sulfide (H2S) and hydrogen (H2) can form a stoichiometric compound (H2S)2H2 near 3.5 GPa, which is characteristic of a rotationally disordered structure2. X-ray diffraction (XRD) results have indicated that this structure belongs to the tetragonal space group I4/mcm. The experimental Raman data indicated that an ordering process occurred with structural transformation above 17 GPa. A candidate structure with space group I222 was proposed by ab initio density functional theory (DFT) calculations through structural optimizations. However, obtaining the most stable structure only by structural relaxation is difficult because of potential energy barrier. Another unexpected structure can possibly become stable instead.
We have taken a new route in this study using the recently fast-developed evolutionary algorithm Universal Structure Predictor: Evolutionary Xtallography (USPEX)30,31,32 to explore the high-pressure ordered crystal structures of (H2S)2H2. We found that a triclinic (P1) structure is energetically favorable. Motivated by the metallization and potential high-temperature superconductivity in (H2S)2H2, higher pressure structures have been extensively explored. Our calculations reveal a metallization pressure of 111 GPa and a high Tc of 200 K at 200 GPa. The lower metallization pressure is within the reach of current diamond–anvil techniques, which will inevitably stimulate extensive experiments.
Results
Variable-cell structure prediction simulations with 1 to 4, 6 and 8 H3S formula units per cell (f.u./cell) at pressures between 20 and 300 GPa were performed. A group of new structures was uncovered under high pressure—triclinic P1 (8 f.u./cell), orthorhombic Cccm (16 f.u./cell), trigonal R3m (3 f.u./cell) and cubic Im-3m (2 f.u./cell) as shown in Fig. 1. The lattice parameters of these structures at different pressures are also listed in Table S1 of the supplementary information.

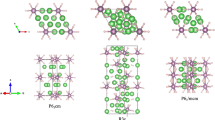
High-pressure crystal structures of (H2S)2H2.
(a) P1 structure normal to the (001) plane, (b) P1 structure normal to the (010) plane, (c) Cccm structure normal to the (001) plane, (c) Cccm structure normal to the (100) plane, (e) R3m and (f) Im-3m. Large spheres represent S and small spheres denote H atoms, respectively.
Enthalpy difference curves (relative to the Cccm structure) for different structures are presented in Fig. 2. P1 is the most stable structure below 37 GPa, which has lower energy than that of the proposed experimental I222 structure2. This P1 structure consists of an ordered H-bonded H2S network and two anti-parallel hydrogen molecules occupying the interstitial sites. One molecule lies at z = 0, whereas the other lies at the z = 1/2 plane (Figs. 1a and 1b). The Cccm structure is energetically stable between 37 and 111 GPa. Unlike the P1 structure, partial hydrogen bond symmetrization is formed in the Cccm structure. This structure consists of 3D H2S networks that trap two different types of hydrogen molecules in channels: one type is arranged such that the head-tail hydrogen molecule (H2–H2) alignment follows the c-axis, whereas the other type is characterized by parallel hydrogen molecule (H1–H1) configuration pointing to the b-axis (Figs. 1c and 1d). The absence of any imaginary frequency also confirms the dynamic stability of the P1 and Cccm phases (see supplementary Fig. S1).

Calculated enthalpies per H3S unit as the function of pressure.
Calculated enthalpy curves for various structures relative to our predicted Cccm structure as a function of pressure. The decomposition enthalpies into S+3/2H2 and H2S+1/2H2 are also plotted.
The lattice angles α = 90.12°, β = 89.85° and γ = 90.15° is approximately equal to 90° for the P1 structure at 20 GPa (see supplementary Table S1), which can thus be viewed as a slightly distorted orthorhombic lattice. This condition can explain the experimental suggestion of an orthorhombic structure. We compared the XRD patterns of the P1 structure with that of the experimental pattern in Fig. 3a. A theoretical pattern was obtained with the same X-ray wavelength of 0.39796 Å as the experimental one. The simulated XRD patterns for the P1 structures agree well with those for the experiment data2. The calculated equation of states (EOS) for the P1 structure was also compared with experimental one in Fig. 3b. Good agreement between the theoretical and experimental results provides another support for the validity of the P1 structure. Given that hydrogen atoms are undetectable by XRD patterns, the Raman spectrum of P1 is presented for comparison with the experimental results (see supplementary Fig. S2). Two H2 vibron-peaks are well reproduced and the two low-frequency features agree qualitatively between theory and experiment. This Raman result, together with the XRD and EOS evidence, support our predicted P1 structure in (H2S)2H2.

Calculated XRD and EOS compared with experiment data.
(a) The simulated XRD patterns and (b) calculated EOS for the P1 structures compared with experiment data (ref. 2). (c) Various interatomic distances of P1, Cccm, R3m and Im-3m structures with pressure.
The Cccm structure transforms to a denser R3m structure above 111 GPa with a volume collapse of 9.2% on further compression (Fig. 3b). The R3m structure shows a disappearing H2 molecule and forms a pyramidal H3S molecular unit as depicted in Fig. 1e. A high-symmetry Im-3m structure becomes favorable above 180 GPa. This structure is characterized by two S atoms located at a simple body centered cubic lattice and H atom located symmetrically between the S atoms (Fig. 1f). The hydrogen bond symmetrization completed in the R3m→Im-3m transition is observed. The coordination number of the S atom increased from three to six with the formation of hydrogen-shared SH6.
The decomposition enthalpies into S + H2 and H2S + H2 are examined to check the phase stability of (H2S)2H2 under pressure (Fig. 2). The structures of P63m, C2/c and Cmca for H233, I41/acd34 and β-Po35 for S, P2/c, Pc, Pmc21, P-1 and Cmca for H2S36 in their corresponding stable pressures are adopted. The most stable phases of (H2S)2H2 have lower enthalpy than S + H2 and H2S + H2 over the pressure range of 20 GPa to 300 GPa. Thus, the (H2S)2H2 crystal remains stable against decomposition in our studied pressure range. The calculated pressure–volume data of (H2S)2H2 is also shown in Fig. 3b. The volume collapses with 1.5% for P1 to Cccm transition and 9.2% for Cccm to R3m transition, suggesting a first-order phase transition character, whereas the volume change is continuous for R3m to Im-3m transition, supporting a second-order nature.
The pressure dependence of the nearest intramolecular and intermolecular bond distances are plotted in Fig. 3c to understand the molecular interactions observed in (H2S)2H2. Four types of intramolecular H–H bond lengths are nearly equal with distances of approximately 0.75 Å and slightly longer than that 0.74 Å in solid H2 at 20 GPa, indicating a weak interaction between H2S and H2. The two H–H bond lengths increased, whereas the other two decreased with increasing pressure, consistent with the observed redshift and blueshift of H–H Raman vibrons2, respectively. The hydrogen bond (H2···S2) is also relatively weak with distance of 2.01 Å at 20 GPa. The strength of the hydrogen bond gradually increases with increasing pressure and significantly elongates one of the H2–S1 covalent bonds for a single molecule. The length of the H3–S1 bond (symmetrization hydrogen bond) is 1.59 Å, the H4–S1 covalent bond is 1.39 Å and the H4···S2 bond is 1.84 Å in the Cccm structure at 40 GPa, indicating a strong hydrogen bond. The length of the strong hydrogen bond decreases, whereas the length of the H4–S1 bond slightly increases with increasing pressure. Notably, the length of the H1–H1 bond decreases to 0.744 Å, whereas the length of the H2–H2 bond increases to 0.757 Å at 110 GPa (Fig. 3d), indicating a slightly increased interaction between S and H2 under pressure. The R3m structure is reconstructive with the disappearing H2 molecule and forms new H-S bonds at 111 GPa. Thus, a large kinetic barrier is formed for the transformation from Cccm to R3m. The length of the H1–S1 bond increases with increasing pressure, whereas the length of the hydrogen bond (H1···S2) decreases. Thus, the hydrogen bond symmetrized completely accompanied by the R3m→Im-3m phase transition at a pressure of 180 GPa. The nearest H–H distance at 110 GPa in the R3m structures is 1.599 Å, much longer than the H–H bond length of ~1.2 Å in CaH6 which is characteristic of a weak covalent bond27. The H–H distance decreases slowly and becomes 1.433 Å at 300 GPa with increasing pressure, indicating that H and H atoms do not present any bond.
The electronic band structure and projected density of states (DOS) for all (H2S)2H2 structures were also explored. The P1 structure is an insulator with an indirect band gap of 3.34 eV at 20 GPa (see supplementary Fig. S3). The bands are flat, indicating that the P1 structure is a typical molecular crystal. The band gap for the Cccm structure decreases with increasing pressure and closes at approximately 110 GPa. Given the deficiency of DFT associated with generalized gradient approximation (GGA), the calculated band gaps are likely to be underestimated. Therefore, the Cccm structure should be an insulator in its stable pressure range. Notably, both the R3m and Im-3m structures are good metals with a large DOS at the Fermi level, as shown in Fig. 4. The projected DOS clearly indicates strong hybridizations of S and H orbitals. Energy band structures near the Fermi level for both R3m and Im-3m are characterized as “flat band-steep band”, indicating that they may be good superconductors37. The predicted metallization pressure of 111 GPa is significantly lower than the currently suggested pressure for the metallization of bulk molecular hydrogen38.

Electronic band structure and density of states (DOS).
Electronic band structure and atom-projected DOS for (a) R3m at 130 GPa and (b) Im-3m at 200 GPa along the selected high symmetry lines, where the dotted lines at zero indicate the Fermi level.
The calculated phonon dispersion curves and projected phonon DOS for R3m and Im-3m structures are presented in Fig. 5. The absence of any imaginary frequency indicates their dynamical stability. The low-frequency bands come mainly from the vibrations of the S atom, whereas higher-frequency modes are mostly related to the H atoms. The electron phonon coupling (EPC) λ, logarithmic average phonon frequency ωlog and the Eliashberg phonon spectral function α2F(ω)39 are investigated to explore the possible superconductivity of (H2S)2H2. The resulting λ of R3m is 2.07 at 130 GPa, indicating a fairly strong EPC; the ωlog calculated from the phonon spectrum is 1125.1 K. The calculated λ of Im-3m is 2.19 at 200 GPa, whereas the ωlog is 1334.6 K. Using the calculated ωlog and commonly accepted values of the Coulomb pseudopotential μ* (0.1–0.13)40 from the Allen-Dynes-modified McMillan equation41, the superconducting transition temperature Tc for R3m was obtained in the range of 155 K to 166 K at 130 GPa. Similarly, the calculated Tc for the Im-3m structure at 200 GPa reaches high values of 191 K to 204 K. Tc decreases nearly linearly with pressure (184 K at 250 GPa and 179 K at 300 GPa for μ* = 0.13) at an approximate rate (dTc/dP) of −0.12 K/GPa for the Im-3m structure.

Phonon properties and Eliashberg spectral function.
Phonon dispersion curves, phonon density of states (PHDOS) projected on S and H atoms and Eliashberg spectral function α2F(ω) together with the electron-phonon integral λ(ω) for (a) R3m at 130 GPa and (b) Im-3m at 200 GPa.
Discussion
Motivated by the metallization and potential high-temperature superconductivity in hydrogen-rich materials of (H2S)2H2 or H3S, high-pressure structures at a wide pressure range of 20 GPa to 300 GPa were extensively explored. Four order structures under high pressure have been predicted: P1 (stable below 37 GPa), Cccm (stable at 37 GPa–111 GPa), R3m (stable at 111 GPa–180 GPa) and Im-3m (stable above 180 GPa). The R3m and Im-3m structures are good metals above 111 GPa. The estimated Tc of the Im-3m phase at 200 GPa is 191 K–204 K, which are among the highest values reported for H2-rich van der Waals compounds28,29 and MH3 (M = Ga, Ge, Si)-type hydride17,18,19 thus far. In a survey of literature, many similarities in structures, metallization and superconductivity for the MH317,18,19 are observed. They all have a high-symmetry cubic structure under high pressure, with high superconducting transition temperature. The lowest Tc value is 86 K for GaH3. They are also good metals at an experimentally accessible pressure, with the highest metallization pressure at 190 GPa for Si2H6. Although the crystal structure-predicting methods implemented by USPEX code are efficient for global structural convergence, it is limited by the sizes of the simulation cell employed. Given that the number of energy minima on the potential-energy surface grows exponentially with increasing cell size, making an extensive structural search is impossible. Thus, we cannot rule out the possibility that the actual high-pressure phase of (H2S)2H2 with larger unit cells may exist.
Notably, no H2 molecules were observed in the R3m and Im-3m structures. However, H2 molecules still exist up to 300 GPa for other H2-containing compounds28,29,42, such as SiH4(H2)2, GeH4(H2)2 and Ar(H2)2, although the intramolecular H–H bond was stretched under high pressure. We calculated the electron localization function (ELF) to understand the bonding nature in the R3m and Im-3m structure (Fig. 6). The ELF value for the S–H bonds is close to 1.0, which suggests a strong polar covalent bond. The ELF value between the nearest H–H is very low, indicating the absence of covalent bond characteristics between hydrogen atoms. This scenario is different from MH3 (M = Ga, Ge, Si)17,18,19, where the charge transfer from M to H atoms forms a strong ionic character. We propose a possible formation process of covalent bond in the R3m structure as follows: the H2 molecules dissociated to hydrogen atoms under high pressure and the hydrogen atoms then cooperated with sulfur atoms to form a strong polar covalent bond. This strong bond makes the H and S atoms tightly bound to form a dense H3S structure, yielding the reduction of its volume.

Calculated ELF of (H2S)2H2.
The calculated ELF of (a) R3m with isosurface value of 0.73, (b) R3m for the (011) plane, (c) Im-3m with isosurface value of 0.73, (d) Im-3m for (100) plane.
The predicted high Tc for (H2S)2H2 or H3S in the order of ~200 K is very encouraging, which prompted us to study the underlying superconducting mechanism. The corresponding Eliashberg spectral function α2F(ω) and integrated λ as a function of the frequency for the R3m at 130 GPa and Im-3m at 200 GPa are presented in Fig. 5. S vibrations in the frequency region below 15 THz contribute approximately 33.1% in total λ for the R3m structure, whereas H vibrations above 18 THz contribute 66.9% of λ. The low-frequency S vibrations (< 18 THz) and high-frequency H vibrational mode (> 20 THz) contribute 18.4% and 82.6% to the EPC, respectively, for the Im-3m structure. This result highlights that the H atoms play a significant role in superconductivity, although the S vibrations at low frequencies are also contributors. This physical mechanism is similar to GaH318 and GeH4(H2)229, but different from SiH4(H2)228 where the strong interactions between H2 and SiH4 molecules vibrations dominate the superconductivity.
Notably, the calculated metallization pressure of (H2S)2H2 (111 GPa) is within the diamond anvil cell capability. The DFT calculation is well known to underestimate the band gap because of its systematic defects for treating the excited states. Thus, the actual metallization pressure for (H2S)2H2 is likely higher than the DFT estimated values in this study. Nevertheless, the calculated metallization pressure for (H2S)2H2 is approximately one-third of the suggested pressure for the metallization of solid hydrogen38. The estimated maximal Tc of 200 K in (H2S)2H2 is higher than those predicted for most hydrogen-containing compounds, such as SnH4(H2)228, GeH4(H2)229 and MH3 (M = Ga, Ge, Si)17,18,19. Our findings represent a significant step toward the understanding of the high pressure behavior of metallic hydrogen and hydrogen-rich hydrides, which can stimulate future high-pressure experiments on structural and conductivity measurements.
Methods
The high-pressure crystal structure of (H2S)2H2 were explored by merging the evolutionary algorithm and ab initio total-energy calculations, as implemented in the USPEX code30,31,32. Structures (population size: 10–60 structures, increasing with system size) were produced randomly in the first generation. The subsequent generation is created from 60% of the lowest-enthalpy structures of the preceding generation. New structures are created by heredity (60%), permutation (10%) and lattice mutation (30%) operations. The best structure of each generation is also carried over to the next generation. The calculation stops when the best structure does not change for more than 20 generations.
The underlying structure relaxations are performed using density functional theory within the Perdew-Burke-Ernzerhof parameterization of the GGA43, as implemented in the Vienna ab initio simulation package VASP code44. The all-electron projector augmented wave method45 is adopted with the core radii of 0.8 a.u. for H (1s2) and 1.5 a.u. for S (3s23p4). Brillouin zone (BZ) sampling using a grid of spacing of 2π × 0.05 Å−1 and a plane-wave basis set cutoff of 500 eV are sufficient for the initial search over structures. However, we recalculated the enthalpy curves with higher accuracy using the energy cutoff 800 eV and k-point mesh of 2π × 0.03 Å−1. These values were selected to ensure that the total energy are well converged to be better than 1 meV/atom.
Lattice dynamics and superconducting properties were calculated using density functional perturbation theory46 and the plane-wave pseudopotential method with Vanderbilt-type ultrasoft potentials47, as implemented in the QUANTUM-ESPRESSO code48. Convergence tests provide a suitable value of 80 Ry kinetic energy cutoff. The q-point mesh in the first BZ of 3 × 3 × 5 for R3m and 8 × 8 × 8 for Im-3m structures are used in the interpolation of the force constants for the phonon dispersion curve calculations. A denser k-point mesh 16 × 16 × 24 for R3m and 32 × 32 × 32 for Im-3m structures are adopted to ensure k-point sampling convergence with a Gaussians width of 0.03 Ry, which approximates the zero width limits in the calculation of EPC parameter λ.
References
Vos, W. L., Finger, L. W., Hemley, R. J. & Mao, H.-K. Novel H2-H2O clathrates at high pressures. Phys. Rev. Lett. 71, 3150–3153 (1993).
Strobel, T. A., Ganesh, P., Somayazulu, M., Kent, P. R. C. & Hemley, R. J. Novel Cooperative Interactions and Structural Ordering in H2S-H2 . Phys. Rev. Lett. 107, 255503 (2011).
Somayazulu, M. S., Finger, L. W., Hemley, R. J. & Mao, H. K. High-Pressure Compounds in Methane-Hydrogen Mixtures. Science 271, 1400–1402 (1996).
Strobel, T. A., Somayazulu, M. & Hemley, R. J. Novel Pressure-Induced Interactions in Silane-Hydrogen. Phys. Rev. Lett. 103, 065701–065704 (2009).
Wang, S. B., Mao, H. K., Chen, X. J. & Mao, W. L. High pressure chemistry in the H2-SiH4 system. Proc. Natl. Acad. Sci. USA 106, 14763–14767 (2009).
Strobel, T. A., Chen, X.-J., Somayazulu, M. & Hemley, R. J. Vibrational dynamics, intermolecular interactions and compound formation in GeH4-H2 under pressure. J. Chem. Phys. 133, 164512–164519 (2010).
Lin, Y., Mao, W. L. & Mao, H. Storage of molecular hydrogen in an ammonia borane compound at high pressure. Proc. Natl. Acad. Sci. USA 106, 8113 (2009).
Chellappa, R. S., Somayazulu, M., Struzhkin, V. V., Autrey, T. & Hemley, R. J. Pressure-induced complexation of NH3BH3-H2 . J. Chem. Phys. 131, 224515–224519 (2009).
Loubeyre, P., Letoullec, R. & Pinceaux, J.-P. Compression of Ar(H2)2 up to 175 GPa: A new path for the dissociation of molecular hydrogen? Phys. Rev. Lett. 72, 1360 (1994).
Kleppe, A. K., Amboage, M. & Jephcoat, A. P. New high-pressure van der Waals compound Kr(H2)4 discovered in the krypton-hydrogen binary system. Sci. Rep. 4, 4989 (2014).
Somayazulu, M. et al. Pressure-induced bonding and compound formation in xenon–hydrogen solids. Nat. Chem. 2, 50–53 (2010).
Mao, W. L. & Mao, H. K. Hydrogen storage in molecular compounds. Proc. Natl. Acad. Sci. USA 101, 708 (2004).
Wigner, E. & Huntington, H. B. On the Possibility of a Metallic Modification of Hydrogen. J. Chem. Phys. 3, 764–770 (1935).
Ashcroft, N. W. Metallic Hydrogen: A High-Temperature Superconductor? Phys. Rev. Lett. 21, 1748 (1968).
Eremets, M. I. & Troyan, I. A. Conductive dense hydrogen. Nat. Mater. 10, 927–931 (2011).
Zha, C.-S., Liu, Z. & Hemley, R. J. Synchrotron Infrared Measurements of Dense Hydrogen to 360 GPa. Phys. Rev. Lett. 108, 146402 (2012).
Gao, G. et al. Metallic and superconducting gallane under high pressure. Phys. Rev. B 84, 064118 (2011).
Abe, K. & Ashcroft, N. W. Quantum disproportionation: The high hydrides at elevated pressures. Phys. Rev. B 88, 174110 (2013).
Jin, X. et al. Superconducting high-pressure phases of disilane. Proc. Natl. Acad. Sci. USA 107, 9969–9973 (2010).
Flores-Livas, J. A. et al. High-Pressure Structures of Disilane and Their Superconducting Properties. Phys. Rev. Lett. 108, 117004 (2012).
Chen, X.-J. et al. Pressure-Induced Metallization of Silane. Proc. Natl. Acad. Sci. USA 105, 20–23 (2008).
Eremets, M. I., Trojan, I. A., Medvedev, S. A., Tse, J. S. & Yao, Y. Superconductivity in Hydrogen Dominant Materials: Silane. Science 319, 1506–1509 (2008).
Chen, X.-J. et al. Superconducting Behavior in Compressed Solid SiH4 with a Layered Structure. Phys. Rev. Lett. 101, 077002 (2008).
Martinez-Canales, M. et al. Novel Structures and Superconductivity of Silane under Pressure. Phys. Rev. Lett. 102, 087005–087004 (2009).
Gao, G. et al. Superconducting High Pressure Phase of Germane. Phys. Rev. Lett. 101, 107002–107004 (2008).
Zhou, D. et al. Ab initio study revealing a layered structure in hydrogen-rich KH6 under high pressure. Phys. Rev. B 86, 014118 (2012).
Wang, H., Tse, J. S., Tanaka, K., Iitaka, T. & Ma, Y. Superconductive sodalite-like clathrate calcium hydride at high pressures. Proc. Natl. Acad. Sci. USA 109, 6463–6466 (2012).
Li, Y. et al. Superconductivity at 100 K in dense SiH4(H2)2 predicted by first principles. Proc. Natl. Acad. Sci. USA 107, 15708–15711 (2010).
Zhong, G. et al. Structural, Electronic, Dynamical and Superconducting Properties in Dense GeH4(H2)2 . J. Phys. Chem. C 116, 5225–5234 (2012).
Oganov, A. R. & Glass, C. W. Crystal structure prediction using ab initio evolutionary techniques: Principles and applications. J. Chem. Phys. 124, 244704–244715 (2006).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How Evolutionary Crystal Structure Prediction Works—and Why. Acc. Chem. Res. 44, 227–237 (2011).
Lyakhov, A. O., Oganov, A. R., Stokes, H. T. & Zhu, Q. New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 184, 1172–1182 (2013).
Pickard, C. J. & Needs, R. J. Structure of phase III of solid hydrogen. Nat. Phys. 3, 473–476 (2007).
Degtyareva, O. et al. Novel chain structures in group VI elements. Nat. Mater. 4, 152–155 (2005).
Luo, H., Greene, R. G. & Ruoff, A. L. β-Po phase of sulfur at 162 GPa: X-ray diffraction study to 212 GPa. Phys. Rev. Lett. 71, 2943–2946 (1993).
Li, Y., Hao, J., Liu, H., Li, Y. & Ma, Y. The metallization and superconductivity of dense hydrogen sulfide. J. Chem. Phys. 140, 174712 (2014).
Simon, A. Superconductivity and Chemistry. Angew. Chem., Int. Ed. 36, 1788–1806 (1997).
Azadi, S. & Foulkes, W. M. C. Fate of density functional theory in the study of high-pressure solid hydrogen. Phys. Rev. B 88, 014115 (2013).
Allen, P. B. Neutron Spectroscopy of Superconductors. Phys. Rev. B 6, 2577–2579 (1972).
Ashcroft, N. W. Hydrogen Dominant Metallic Alloys: High Temperature Superconductors? Phys. Rev. Lett. 92, 187002 (2004).
Allen, P. B. & Dynes, R. C. Transition temperature of strong-coupled superconductors reanalyzed. Phys. Rev. B 12, 905–922 (1975).
Yao, Y. & Klug, D. D. High-pressure phase transition and metallization in Ar(H2)2 . Phys. Rev. B 83, 020105 (2011).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Baroni, S., de Gironcoli, S., Dal Corso, A. & Giannozzi, P. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys. 73, 515–562 (2001).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892 (1990).
Paolo, G. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502 (2009).
Acknowledgements
This work was supported by the National Basic Research Program of China (No. 2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (No. IRT1132), National Natural Science Foundation of China (Nos. 51032001, 11204100, 11074090, 10979001, 51025206 and 11104102), National Found for Fostering Talents of basic Science (No. J1103202), Specialized Research Fund for the Doctoral Program of Higher Education (20120061120008 and 20110061120007) and China Postdoctoral Science Foundation (2012M511326 and 2013T60314). Parts of calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.
Author information
Authors and Affiliations
Contributions
T.C. and D.F.D. designed research. D.F.D. and Y.X.L. performed research. D.F.D., F.B.T., D.L., Z.H.Z., H.Y.Y., X.L.H., B.B.L. and W.J.T. analyzed the data. D.F.D. and T.C. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Duan, D., Liu, Y., Tian, F. et al. Pressure-induced metallization of dense (H2S)2H2 with high-Tc superconductivity. Sci Rep 4, 6968 (2014). https://doi.org/10.1038/srep06968
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06968
This article is cited by
-
Full-bandwidth anisotropic Migdal-Eliashberg theory and its application to superhydrides
Communications Physics (2024)
-
Superconductivity in the Janus WSH Monolayer
Journal of Superconductivity and Novel Magnetism (2024)
-
Magnetic flux trapping in hydrogen-rich high-temperature superconductors
Nature Physics (2023)
-
Absence of near-ambient superconductivity in LuH2±xNy
Nature (2023)
-
Hopes raised for room-temperature superconductivity, but doubts remain
Nature (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
