
Abstract
The influenza A virus RNA polymerase cleaves the 5′ end of host pre-mRNAs and uses the capped RNA fragments as primers for viral mRNA synthesis. We performed deep sequencing of the 5′ ends of viral mRNAs from all genome segments transcribed in both human (A549) and mouse (M-1) cells infected with the influenza A/HongKong/1/1968 (H3N2) virus. In addition to information on RNA motifs present, our results indicate that the host primers are divergent between the viral transcripts. We observed differences in length distributions, nucleotide motifs and the identity of the host primers between the viral mRNAs. Mapping the reads to known transcription start sites indicates that the virus targets the most abundant host mRNAs, which is likely caused by the higher expression of these genes. Our findings suggest negligible competition amongst RdRp:vRNA complexes for individual host mRNA templates during cap-snatching and provide a better understanding of the molecular mechanism governing the first step of transcription of this influenza strain.
Similar content being viewed by others

Introduction
Influenza viruses belong to the family Orthomyxoviridae, consisting of enveloped viruses that contain single-stranded negative-sense segmented RNA genomes. Among the influenza virus genera (A, B and C), influenza A virus (IAV) is the most virulent and causes significant worldwide mortality and morbidity. The genome of IAV consists of eight segments, which encode at least 11 known proteins (reviewed in1). Upon internalization, the viral genome segments are released into the cytoplasm as vRNPs, which are transported into the nucleus using the importin α/importin β pathway. At an early step of the IAV replication cycle, the viral RNA-dependent RNA polymerase (RdRp) produces viral mRNAs, which have features of cellular mRNAs, including a 5′ cap structure and a poly(A) tail2,3,4,5,6,7. During the replication phase, the viral RdRp produces complementary RNAs (cRNAs) from the vRNAs, which are then used as templates to produce progeny vRNAs. These vRNAs serve as templates for the synthesis of more viral mRNAs or are exported to become incorporated into new virus particles. Notably, the viral RdRp is required for both of these steps.
One of the most intriguing aspects of IAV transcription is that synthesis of viral mRNAs is dependent on capped RNA primers derived from host pre-mRNAs. During this process, known as “cap-snatching”, the viral RdRp interacts with the C-terminal domain of host RNA polymerase II (RNAP II) to gain access to the 5′ cap structure of nascent mRNAs8. Following RdRp endonucleolytic activity, the capped RNA fragments prime viral mRNA transcription9,10,11,12,13,14,15,16. Because the excised capped RNA fragments also contain 10–15 nucleotides downstream of the cap, the priming activation of viral transcription yields products that are genetic hybrids of host and virus mRNAs; as a result, sequence heterogeneity is found at the host-derived 5′ ends of viral mRNAs5,6,7. As cleavage of caps by IAV also involves the destruction of the pre-mRNAs, which potentially induces RNAP II degradation, the antagonist properties of RdRp on host RNAP II transcription have been implicated in host genes shut-off (reviewed in17).
The IAV RdRp is a heterotrimeric complex of three subunits: polymerase basic protein 1 (PB1), polymerase basic protein 2 (PB2) and polymerase acidic protein (PA). PB1 contains conserved motifs typical of RdRp and possesses the polymerization activity18,19,20,21. The PB2 and PA subunits are involved in the initiation of transcription, by binding to and cleaving capped host pre-mRNAs, respectively22,23,24,25,26,27,28,29,30,31. In addition to these three RdRp core proteins, the nucleoprotein (NP) interacts with the PB1 and PB2 subunits, binds RNA and is required for transcription of viral genes32,33,34. Finally, the RdRp also requires a vRNA template to carry out cap-snatching of host pre-mRNAs35. The vRNAs contain 13 nucleotides at the 5′ end and 12 nucleotides at the 3′ end, which are highly conserved between all eight segments across all influenza A strains36. These nucleotides form a partially base-paired vRNA promoter, which has been proposed to interact with IAV RdRp subunits37.
PA-mediated cleavage occurs at a phosphodiester bond 10–15 nt downstream of the cap structure of host pre-mRNAs. The N-terminal region of PA adopts a folding similar to resolvases and type II restriction endonucleases and possesses the endonuclease activity29,31. In vitro studies have suggested that the N-terminal region of PA preferentially cleaves RNA at a phosphodiester bond 3′ end of a guanine (G) residue, although it is unknown if the same preference occurs in vivo with the complete RpRp complex38. The IAV RdRp can also use the dinucleotide AG as primer to initiate complementary RNA synthesis in vitro39,40,41. Other studies have proposed that the viral endonuclease cleaves host mRNA after a purine residue; IAV RdRp was found to preferentially use CA-terminated capped fragments to initiate complementary RNA synthesis and add a G residue directed by the complementary C residue located at the 3′ end of the vRNA template, which has the sequence 3′-UCG7,10,42,43. Initiation by the addition of a C directed by the G at position 3 in the vRNA template has also been observed44,45. To substantiate these findings and to clarify the IAV endonuclease sequence specificity, a more extensive study of the host-derived primers used during viral transcription in infected cells is needed.
In this study, we investigated the characteristics of the host-derived sequences located at the 5′ end of the mRNAs of the clinical human isolate A/HongKong/1/1968 (H3N2) to determine whether RNA selection occurs during cap-snatching of host pre-mRNAs by the viral RdRp. To this end, we performed high-throughput sequencing of the host-derived sequences located at the 5′ ends of the eight IAV mRNAs early after infection of both human lung epithelial (A549) and mouse kidney epithelial (M-1) cells. We investigated the nature of nucleotide motifs enriched in the host primers and observed noticeable differences in both the length distributions and the identity of the host primers used by the eight viral mRNAs. Despite these differences, our analysis suggests that most of the host primers originate from highly abundant host mRNAs. Overall, our results suggest a new layer of complexity within the A/HongKong/1/1968 (H3N2) cap-snatching mechanism, wherein cellular RNPs could be used to recruit the RdRp:vRNA complexes to specific sets of genes/pre-mRNAs.
Results
High-throughput sequencing of the 5′ UTR of influenza A/HongKong/1/1968 (H3N2) virus mRNA and extraction of the heterogeneous sequences
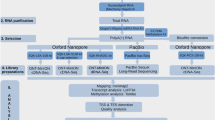
We performed high-throughput sequencing of the host-derived 5′ ends located on the eight mRNAs of A/HongKong/1/1968 (H3N2) following infection of both human lung epithelial (A549) and mouse kidney epithelial (M-1) cells. To obtain information about host pre-mRNAs used mainly at the earliest step of the viral replication cycle (i.e. during early viral mRNA synthesis), both cell lines were infected by A/HongKong/1/1968 (H3N2) at high MOI and polyadenylated mRNA was extracted four hours after infection. We then selectively ligated an RNA oligonucleotide to the 5′-ends of capped mRNAs, reverse-transcribed the mRNAs using a poly-dT primer and amplified each IAV cDNA by PCR using the 5′ RACE primer and gene-specific primers located just downstream of the translation initiation codons. A subsequent round of PCR amplification was performed with primers containing Illumina adapter sequences and barcodes for multiplexing the samples. To avoid PCR over-amplification, the number of PCR cycles was kept to the minimum required to observe a band by agarose gel electrophoresis. The PCR products were gel-extracted and submitted for Sanger sequencing, which allowed us to confirm the identities of the sequences (Fig. 1). All the cDNAs were subsequently mixed and sent for high-throughput sequencing using the Illumina HiSeq 2000 System, which produced 154,826,647 reads of 100 nucleotides (nts) in length from the library.

Library of IAV transcripts prepared for deep sequencing by Illumina HiSeq.
Human (A549) and mouse (M-1) cells were infected with the A/HongKong/1/1968 (H3N2) strain of IAV for four hours. The mRNA was extracted, an RNA oligonucleotide (yellow box) was ligated to the 5′ end and the mRNAs were reverse-transcribed using a poly-dT primer. PCR amplification was performed on each IAV cDNA using the 5′ RACE primer and a gene-specific primer that annealed 20–40 nucleotides downstream of the ATG codon (highlighted in red). The region corresponding to the heterogeneous host-derived 5′ end of viral mRNA is shown in black. Four-nucleotide identifier tags (blue boxes) were added to each library for multiplex sequencing and adapter sequences for sequencing (red boxes) were added onto the ends of the DNA.
The reads were divided into their respective host and viral groups based on their barcodes and the sequences of the non-coding regions at the 5′ ends of each viral mRNA. Approximately 52.5% of the reads obtained did not include sequences corresponding to any of the eight IAV non-coding regions. Because the sequences on IAV mRNA used for amplification have a low GC content (45–50%), a relatively low primer annealing temperature had to be used during PCR amplification, which might have resulted in co-amplification of unrelated host mRNA. Overall, we obtained 28.7 and 44.8 million reads corresponding to IAV mRNA from human and mouse cells, respectively. The inset of Figure 2 shows the proportion of each read for the different IAV mRNAs obtained from human cells (similar data derived from mouse cells are presented in Supplementary Fig. 1). Unequal amounts of reads were obtained for each transcript, which likely reflects the differences in the amount of DNA mixed before deep sequencing.

Length distribution of the heterogeneous sequences obtained from infected A549 cells.
All sequences representing each of the eight IAV transcripts isolated from infected A549 cells and located between the ligated RACE primer and position G2 on IAV mRNA are included. Inset: proportion of IAV sequences corresponding to each transcript obtained following high-throughput sequencing.
Analysis of the sequences between the ligated RACE primer and position G2 on IAV mRNA (herein referred to as “heterogeneous sequences” and indicated by the white characters on a black background in Fig. 1) indicated that 91.8% of the heterogeneous sequences had lengths ranging from 10 to 15 nt, with main peaks at 11–12 nts and that the length distribution was similar in the samples derived from both hosts (Fig. 2 and Supplementary Fig. 1). A small population of sequences was shorter than 10 nt (about 6.7%) and likely represents the result of amplification of degraded RNA. For this reason, they were removed from subsequent analyses. Interestingly, the length distributions varied between the different transcripts. We observed main peaks at 11 nt for the mRNAs coding for PB1 and PB2 and at 12 nt for those coding for NA, NP, PA, NS (Fig. 2). Similar variations were also observed in IAV mRNAs isolated from mouse cells (Supplementary Fig. 1), suggesting differences in viral RdRp-mediated cleavage and/or host mRNAs used during cap-snatching of this influenza strain.
Analysis of the heterogeneous sequences located at the 5′ end of the eight viral mRNAs
To determine whether the heterogeneous sequences contain specific RNA motifs, we calculated the nucleotide frequencies observed from 15 nt upstream of the G2 and up to 5 nt downstream of G2 for all IAV mRNAs (Fig. 3 and Supplementary Table 1). Sequences that appeared only once (1.4%) were removed from this and all subsequent analyses, to avoid the contribution of mutations that might have been generated by the protocols used. While the virus-encoded sequence was conserved (5′ GC[G/A]AAA), the heterogeneous sequences showed a high degree of divergence in sequence, consistent with what we observed by Sanger sequencing of the amplicons (Fig. 1). Despite variability, we observed a nonrandom distribution of the nucleotide frequencies within the heterogeneous sequences. The nucleotide immediately upstream of G2 was a purine in the majority of the sequences (67.5 +/− 2.0%). While an A was the enriched residue in most of the transcripts, PB2 mRNA exhibited a preference for G. We also observed a preference for C (63.7 +/− 6.7%) and G (56.9 +/− 5.7%) residues at location -2 and -3 upstream of G2 in all IAV mRNAs, with the exception of those coding for PB1 and PB2. By analyzing these three positions together, we observed a preference for a GCA trinucleotide at the 3′ end of the heterogeneous sequence in those mRNAs (Supplementary Fig. 2). For PB2 mRNAs, the G residue located immediately upstream of G2 was preceded predominantly by a GCA motif, causing a shift in the position of the GCA trinucleotide as compared to the other IAV mRNAs and producing a heterogeneous sequence for this IAV mRNA terminating with a GCAG tetranucleotide as the most abundant motif.

Sequence variation present in the heterogeneous sequences located at the 5′-end of viral mRNAs obtained from infected A549 cells.
For each transcript, the top panel shows the relative nucleotide frequency at each position (log scaled) and the bottom panel shows the Logo representation of the nucleotide variation. The nucleotides are numbered according to the host/virus junction (blue line), defined as the phosphodiester bond located upstream of position G2 on IAV mRNA. The grey bars represent the percentage of the population of reads included in the calculation. Only sequence reads that appeared at least twice were used in the analysis. For all IAV mRNAs, only nucleotide frequencies observed from 15 nt upstream of the G2 and up to 5 nt downstream of G2 are represented. All motifs are represented in the 5′ to 3′ orientation.
Further upstream of this motif, the heterogeneous sequences showed a bias towards G/C nucleotides for all IAV mRNA leaders (63.8 +/− 9.4%). Specifically, the fragments used to prime most IAV mRNAs showed a preference for G-rich primers, while those used to prime PB1 mRNA were C/U-enriched (Fig. 3 and Supplementary Table 1). Sequence motifs were comparable between human and mouse -derived samples for each transcript, suggesting a common bias towards similar sequences in each species (Supplementary Table 2 and Fig. 3–4). Finally, an enrichment in adenines just upstream of the G/C-rich region was observed. This correlates with the 5′-ends of most of the heterogeneous sequences (ranging from 9 to 15 nt in length), as represented by the grey bars showing the percentage of the population of reads included in the calculation (Fig. 3 and Supplementary Fig. 3). This enrichment likely reflects the preference for this nucleotide during transcription initiation by RNAP II46 and also provides evidence that our procedure to ligate an RNA oligonucleotide selectively to the 5′-end of 5′-capped mRNAs was effective.

Human sequences and genes used by the eight viral transcripts.
(a) Heatmap representation showing the distribution of the heterogeneous sequences (vertical axis) associated with each IAV transcript. (b) Heatmap representation showing the distribution of the host genes (vertical axis) potentially used by each IAV transcript. The heterogeneous sequences/host genes are hierarchically clustered. Data in each column have been normalized to the maximum value in that column. To simplify the representation, only sequences/genes that appears at least 1000 times were used.
When we compared the heterogeneous sequences directly, our results indicated a minimal overlap in their identities among the different viral mRNAs (Fig. 4a and Supplementary Fig. 5a). Using pair-wise comparisons of the populations of sequences with χ2 tests, our analysis revealed significant differences among the heterogeneous sequences located at the 5′ end of the eight IAV mRNAs, with all p-values smaller than 10−15. One possibility to explain these divergences is that our sequencing sampling depth might have been insufficient. To estimate the complexity of the sequencing libraries, we calculated rarefaction curves for each mRNA group. Supplementary Fig. 6 and 7 show rarefaction curves representing the number of unique heterogeneous sequence variants as a function of the number of reads obtained for both human and mouse-derived samples. For all samples, the rarefaction curves approached a plateau corresponding to their respective number of unique variants, indicating that the sampling depth was sufficient and that more sampling is unlikely to yield additional sequence variants. Taken together, the observed divergences in length distributions, nucleotide frequencies and sequence identities strongly suggest that the different vRNA templates of this IAV strain use different host mRNAs during cap-snatching and/or transcription initiation of viral mRNAs.
Mapping the heterogeneous sequences to known transcription start sites (TSS) and identification of the targeted host genes
To identify the origin of the heterogeneous sequences and to gain information about sequences further downstream of the cleavage site, we attempted to map the heterogeneous sequences on the human genome. However, because the length of the heterogeneous sequences was small (10–15 nt), we were not able to successfully use standard procedures for gene identification. As an alternative approach, we decided to map the fragments to regions surrounding TSS, since the fragments originate from the 5′ ends of capped host mRNAs. To this end, we used TSS previously reported for A549 cells derived by Gene Identification Signature (GIS) paired-end ditag (PET) sequencing, as part of the ENCODE Transcriptome Project. Using these genomic locations, we mapped the heterogeneous sequences to windows located from −100 to +100 nt around the TSS using standard alignment procedures. Additionally, for each fragment, we selected the sequence closest to the TSS and whenever more than one solution resulted from this filtering step, we selected one randomly. Altogether, we mapped 56.2% of the heterogeneous sequences to 132,764 TSS associated with 13,229 poly-A mRNAs expressed in A549 cells.
Using this strategy, we found that the heterogeneous sequences derived from the A/HongKong/1/1968 (H3N2) mRNAs mapped to different sets of genes (Fig. 4b). We observed significant differences among the eight IAV mRNAs by pair-wise comparisons of the populations of genes using χ2 tests (p-values smaller than 10−15). This is in agreement with the difference in sequence identity we observed and also suggests that the different vRNA templates are associated with different host pre-mRNAs during cap-snatching. We then performed enrichment analysis of Gene Ontology (GO) terms to assess whether the different vRNA templates target different biological processes. Despite significant differences among the genes associated with the eight viral vRNAs, pair-wise comparisons of the lists of GO terms using χ2 tests indicated no significant differences (all p-values were greater than 0.05; Fig. 5). To account for bias due to the gene set we used, we also performed a GO terms enrichment analysis from a host mRNA expression profile obtained from mock-infected cells. Comparison of GO terms corresponding to genes targeted by the virus and those associated with expression profiles from mock-infected cells indicated that cap-snatching by all A/HongKong/1/1968 (H3N2) transcripts targets genes that are most abundant and likely highly expressed (i.e. no significant difference was observed in the enrichment of GO terms between host genes used by A/HongKong/1/1968 (H3N2) vs. host mRNA expression profiles obtained from mock-infected cells). Similar results were obtained from the IAV mRNAs isolated from mouse cells (Supplementary Fig. 5b and 8).

Enrichment of Gene Ontology (GO) terms corresponding to the genes used by influenza A/HongKong/1/1968 (H3N2) virus cap-snatching in human cells.
Data are presented as level 3 GO categorization for biological process. The GO terms are hierarchically clustered. Data in each column have been normalized to the maximum value in that column. To account for bias due to the gene set we used, mRNA expression profile of mock infected cells (i.e. “MOCK) was used for the GO terms enrichment analysis.
Identification of nucleotides enriched in host pre-mRNAs downstream of the heterogeneous sequences
To determine whether a specific RNA motif exists at the cleavage site, we used the results of the mapping of the heterogeneous sequences to known TSS to calculate nucleotide frequencies for host mRNA locations downstream of the 3′ end of the heterogeneous sequences. The positions located immediately downstream of the heterogeneous sequences showed an enrichment in G and C nucleotides (Fig. 6). Because we previously observed a preference for a CA dinucleotide at the 3′ end of the heterogeneous sequences (immediately upstream), we then calculated the frequencies of the four-nucleotide motifs composed of the last two nucleotides of the heterogeneous sequences and the first two nucleotides located immediately downstream. Using the frequencies of all tetranucleotides present in the regions located −100 to +100 nt around the TSS as negative controls, we calculated that the CA|GC motif was the most enriched in our dataset (Supplementary Fig. 9). However, this enrichment was not uniform among all viral mRNAs. Similar results were observed in the sequences derived from the viral mRNAs isolated from mouse cells, where the C[A/G]|GC motif was the most enriched as compared to all tetranucleotides present in the regions located −100 to +100 nt around the TSS of the mouse genome (Supplementary Fig. 10). These findings suggest that such a motif might be preferentially used by A/HongKong/1/1968 (H3N2) during cap-snatching and viral transcription in these two hosts.

Analysis of nucleotide sequences enriched at the cleavage site in targeted human pre-mRNAs.
Logo representation of the last three nucleotides of the heterogeneous sequence and the next three nucleotides found in the host pre-mRNA downstream of the heterogeneous sequences. The nucleotides are numbered according to the heterogeneous sequences/pre-mRNA junction (blue line). The “ALL” dataset represents the sum of all the individual datasets. All motifs are represented in the 5′ to 3′ orientation.
Discussion
Using information derived from cloning and sequencing of a small number of IAV mRNAs generated in vitro or extracted from infected cells, several hypotheses have been proposed on the sequence specificity of the IAV endonuclease and subsequent initiation of viral mRNA transcription by host capped oligonucleotides1. However, the gene origin of the host leaders and information of the sequences downstream from the cleavage sites remained unknown. Here, we performed high-throughput sequencing of the 5′ ends of total viral mRNA isolated from both human (A549) and mouse (M-1) cells infected with A/HongKong/1/1968 (H3N2) to investigate the characteristics of the 5′ terminus of viral mRNA early after infection.
Analysis of the heterogeneous sequences located at the 5′ end of the eight viral mRNAs indicated that most had lengths ranging from 10 to 15 nt, with main peaks at 11-12 nts. This result is consistent with previous studies showing that IAV RdRp generally cleaves host mRNA 10–15 nt downstream of the cap9,11,15,38,47,48,49. Interestingly, the host-derived sequences in both PB1 and PB2 mRNAs were smaller by one nucleotide (peaking at 11 nt) when compared to the other viral mRNAs. This was equally apparent in viral mRNA isolated from A549 and M-1cells and could be related to the difference at position 4 in the 3′ end of PB1 and PB2 vRNA templates (C vs. U in all other segments) in the IAV strain used. This difference could also explain the observed disparity in the nucleotide frequency distributions of the PB1 and PB2 mRNA host leaders as compared to the other six transcripts. Unlike the strain used in this study, most IAV strains have a C at position 4 in all three polymerase segments (including PA); hence it would be interesting to investigate the role of this nucleotide in host leader selection.
Although these sequences were heterogeneous, nucleotide frequency distributions were not random. For all of IAV mRNAs, the host primers showed a content bias towards G/C nucleotides. Although it is possible that high GC content might be used as a determinant during cap-snatching, it is likely that this simply reflects the high GC content frequently reported around TSS50. In fact, calculation of nucleotide frequencies observed in the windows located from −100 to +100 nt around the TSS indicated a GC content of 63.5%, which is similar to the GC content calculated for the heterogeneous sequences (i.e. 63.8 +/− 9.4%). We observed an enrichment in purines at a location just upstream of G2. While an A was the preferred residue in most of the transcripts, PB2 mRNA exhibited a preference for G. These results are consistent with several studies that have shown a preference for A and G residues just upstream of G25,6,42,51,52,53. A GC motif preceded this purine in a large number of sequences, making GCA trinucleotide the most abundant motif found at the 3′ end of the heterogeneous sequences. This result is in agreement with previous reports, which found that primers with CA at their 3′-ends are preferred for transcription initiation in infected cells and in vitro and that CA-terminated capped fragments can be used for viral mRNA synthesis in vitro7,42,43,48,54. For PB2 mRNA, a GCAG tetranucleotide was found instead of a GCA trinucleotide. The observed shift in the GCA trinucleotide, as compared to the other IAV mRNAs, is in line with a prime-and-realign mechanism48,55 during PB2 mRNA synthesis; presumably, an initial addition of a G residue directed by the complementary C located at the 3′ end of the vRNA template is followed by a realignment of the nascent chain, which results in the observed duplication of the G residue. This might explain the enrichment in G observed at the location just upstream of G2.
By mapping the heterogeneous sequences to TSS previously reported for poly-A mRNAs expressed in A549 cells, we found that CA|GC was enriched at a location corresponding to the last two nucleotides of the heterogeneous sequences and the next two nucleotides of the host pre-mRNA. Because the G residue just downstream of the 3′ end of the heterogeneous sequences might correspond to the conserved G2, it is possible that a large number of cap-snatched fragments have a 3′ end corresponding to CAG and that the IAV RdRp endonucleolytic cleavage occurs between G and the downstream C. This is consistent with studies showing that IAV RdRp usually cleaves after a purine residue7,10,49, that IAV RdRp endonucleolytic cleavage is highly efficient for substrates that carry GC motifs56 and that the purified N-terminal domain of PA selectively cleaves after G residues of 5′-GC-3′ motifs38. However, because the vRNA template is required to activate endonuclease cleavage and transcription35, our results cannot exclude the possibility that the observed enrichment of this G residue is determined by the complementarity of the host primer to the 3′ end of the vRNA template. In addition to this G, our analysis indicates that heterogeneous sequences terminating with either CA or CAG are enriched. This supports the idea of base pairing to the 3′ end of the IAV genome to allow elongation from the host primers during IAV mRNA transcription initiation10,35,45,48,57. Specifically, primers with nucleotides complementary to the 3′ end of IAV vRNA template (i.e. CA, AG, GG and AGC) were used by IAV RdRp to transcribe IAV mRNA in vitro7,58,59,60. This enrichment of sequence repeats that are complementary to the 3′ end of the IAV RNA templates also supports a prime-and-realign mechanism for transcription of this IAV strain, as previously observed in the leader sequence of viruses that use cap-snatching48,61,62,63,64,65.
Unexpectedly, our results indicated a divergence in the host mRNAs/genes that are used by the eight viral transcripts and this was observed in both human- and mouse-derived samples. This was revealed by noticeable differences in length distributions, nucleotide motifs, identity of the heterogeneous sequences and mapping the reads to known TSS. This divergence can occur during cap-snatching and/or transcription initiation of viral mRNAs. Because they do not target the same host mRNAs, our results suggest negligible competition amongst RdRp:vRNA complexes for individual host mRNA templates during cap-snatching. Although the source of this divergence is unknown, it is likely associated with the vRNA template itself, or with cellular RNPs binding selectively to each of the eight vRNAs. It is thus tempting to suggest a new layer of complexity within the A/HongKong/1/1968 (H3N2) cap-snatching mechanism, wherein cellular RNPs could be used to recruit the RdRp:vRNA complexes to specific sets of genes/pre-mRNAs, or to selectively localize the RdRp:vRNA complexes. Such a model is supported by a recent observation showing that the different vRNAs localized in distinct sites in the nucleus66. Because, the length distributions, the nucleotide motifs for each A/HongKong/1/1968 (H3N2) mRNA and the shift observed for PB2 mRNA are very similar between the two cell lines tested, this also suggests conservation of such putative RdRp:vRNA complexes to efficiently cap-snatch the same motifs/host pre-mRNAs in these two hosts. Noteworthy, our analysis suggests that despite divergence in the host mRNAs/genes used during cap-snatching, the majority of the host primers originate from the most abundant genes, which are likely highly expressed. It is possible that by targeting these genes, transcription of A/HongKong/1/1968 (H3N2) mRNA modulates globally the expression of abundant host proteins, which might contribute to host shut-off.
In conclusion, we performed high throughput sequencing of A/HongKong/1/1968 (H3N2) mRNA from infected cells early after infection. Our analysis provided important information about the primers used to initiate viral mRNA transcription and sequence specificity of the viral endonuclease. In addition to substantiating several previous findings, this study provided evidence for vRNA template partitioning during cap-snatching by this IAV strain. Although two cellular systems were used for infection by A/HongKong/1/1968 (H3N2), further studies are needed to generalize our findings to other IAV strains, or other RNA viruses using cap-snatching to provide primers for viral transcription. More importantly, our findings might have far reaching consequences towards a better understanding of the molecular mechanism governing the first step of IAV transcription and the global inhibition of the expression of cellular genes.
Methods
Cells and viruses
M-1 (mouse kidney epithelium, ATCC) and A549 (human lung epithelium, ATCC) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10 % fetal bovine serum, 1 mM sodium pyruvate, 10% (v/v) non-essential amino acids (Gibco) and 2 mM L-glutamine. All cells were incubated at 37°C in the presence of 5% CO2. The H3N2 human influenza isolate A/Hong Kong/1/1968 was a clonal derivative that had been previously sequence characterized67. Viruses were grown in 10 day old specific pathogen free embryonated chicken eggs (Canadian Food Inspection Agency, Ottawa, Ontario). Viruses were titrated by plaque assay in MDCK cells as described previously68.
Library Preparation
M-1 and A549 cells were infected with IAV at an MOI of 2 pfu per cell. Cells were collected at 4 h post-infection, washed twice with 1X PBS and polyadenylated RNA was extracted using the Dynabeads mRNA Direct Kit (Ambion) according to manufacturer's instructions. Purified mRNA was subjected to 5′ rapid amplification of cDNA ends (RACE) using the ExactSTART Eukaryotic mRNA 5′ & 3′-RACE Kit (Epicentre Biotechnologies) according to manufacturer's protocol. Briefly, ~0.3 μg of mRNA was dephosphorylated to convert uncapped RNA into unligatable 5′-hydroxyl RNA. The mRNA was then treated with pyrophosphatase to remove the 5′ cap and an RNA oligo was ligated to the monophosphate mRNA. The 5′-oligo-tagged RNA was subjected to reverse transcription using MMLV reverse transcriptase (NEB) and amplified by 5 cycles of PCR using kit-provided primers. PCR products were cleaned using the Gel/PCR DNA Fragments Extraction Kit (Geneaid) and a fraction (~1/10th) of the product was used as a template for PCR using an IAV mRNA-specific primer located just upstream of the start codons of the genes. After about 25 cycles of PCR, products were gel-purified using the Gel/PCR DNA Fragments Extraction Kit (Geneaid) and a fraction (~1/25th) was used as template in another round of PCR (16–20 cycles) to add sequence identifier tags for multiplexing and adapter sequences compatible with the Illumina Sequencing platform. PCR products were gel-purified and verified by Sanger Sequencing (StemCore Laboratories, Ottawa, Canada). Products were then mixed in approximately equal proportions and a single sample was sent for deep sequencing using the Illumina HiSeq 2000 System (McGill University and Genome Quebec Innovation Centre, Montreal, Canada).
Analysis of the 5′ UTR of IAV mRNA
For each sequence, the name of the sequence, the composition in nucleotides and the sequencing quality score were stored in a database. Based on both their multiplexing tags and the transcript-specific sequences, the sequences were also divided into their respective host and viral mRNA groups. In-house Perl scripts were used for all data extraction and to obtain statistics on fragment length and nucleotide composition. As a first step, the sequences found between the primers used for PCR amplification were extracted. Then the heterogeneous sequences (i.e. upstream of G2) were extracted by searching for the conserved sequences corresponding to G2 to G12 on IAV mRNA and by accepting two mutations. To identify the origin of the heterogeneous sequences, in house Perl-scripts were used to synthesize an artificial genome composed of sequence windows comprised from -100 to +100 nt around the TSS from the hg19 and mm10 genomes of human and mouse, respectively. For TSS of A549 cells, data from the Gene Identification Signature (GIS) paired-end ditag (PET) sequencing as part of the ENCODE Transcriptome Project were used (GEO accession number: GSM1006902). For mouse cells, transcription start positions in the mm10 genome were obtained from the UCSC Genome Browser (http://genome.ucsc.edu). Mapping of the heterogeneous sequences to these mini-genomes was performed with Bowtie v.1.0.069. Using in-house Perl scripts, the solution closest to the TSS (and its associated gene identification number; i.e. GeneID) was selected and if more than one solution resulted from this filtering step, one was selected randomly. For the GO terms analysis, the frequency of the reads corresponding to each GeneID was scaled from 0 to 100 for each viral transcripts. The GeneID were then multiplied by the rescaled frequency to better reflect their enrichment and the analysis of GO terms corresponding to biological processes enriched was performed in R with GoProfiles 1.20.0 (available at http://bioconductor.org/biocLite.R). To account for bias due to the gene set we used, mRNA expression profile of both uninfected A549 and M-1 cells, as determined previously by microarray70, was used and GO terms enrichment analysis was performed as above. All further data analysis was performed using in-house R scripts. Pearson's χ2 tests were performed in R using pair-wise comparisons between the populations of the heterogeneous sequences located at the 5′ end of the eight IAV mRNAs, as well as the populations of genes that the heterogeneous sequences mapped to. Pearson's χ2 tests were also used in pair-wise comparisons of the lists of GO terms associated with each IAV mRNA.
References
Fodor, E. The RNA polymerase of influenza a virus: mechanisms of viral transcription and replication. Acta Virol 57, 113–122 (2013).
Taylor, J. M. et al. Use of specific radioactive probes to study transcription and replication of the influenza virus genome. J Virol 21, 530–540 (1977).
Mark, G. E., Taylor, J. M., Broni, B. & Krug, R. M. Nuclear accumulation of influenza viral RNA transcripts and the effects of cycloheximide, actinomycin D and alpha-amanitin. J Virol 29, 744–752 (1979).
Scholtissek, C. & Rott, R. Synthesis in vivo of influenza virus plus and minus strand RNA and its preferential inhibition by antibiotics. Virology 40, 989–996 (1970).
Caton, A. J. & Robertson, J. S. Structure of the host-derived sequences present at the 5′ ends of influenza virus mRNA. Nucleic Acids Res 8, 2591–2603 (1980).
Dhar, R., Chanock, R. M. & Lai, C. J. Nonviral oligonucleotides at the 5′ terminus of cytoplasmic influenza viral mRNA deduced from cloned complete genomic sequences. Cell 21, 495–500 (1980).
Beaton, A. R. & Krug, R. M. Selected host cell capped RNA fragments prime influenza viral RNA transcription in vivo. Nucleic Acids Res 9, 4423–4436 (1981).
Engelhardt, O. G., Smith, M. & Fodor, E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 79, 5812–5818 (2005).
Bouloy, M., Plotch, S. J. & Krug, R. M. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro. Proc Natl Acad Sci U S A 75, 4886–4890 (1978).
Plotch, S. J., Bouloy, M., Ulmanen, I. & Krug, R. M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23, 847–858 (1981).
Plotch, S. J., Bouloy, M. & Krug, R. M. Transfer of 5′-terminal cap of globin mRNA to influenza viral complementary RNA during transcription in vitro. Proc Natl Acad Sci U S A 76, 1618–1622 (1979).
Krug, R. M., Broni, B. A. & Bouloy, M. Are the 5′ ends of influenza viral mRNAs synthesized in vivo donated by host mRNAs? Cell 18, 329–334 (1979).
Bouloy, M., Morgan, M. A., Shatkin, A. J. & Krug, R. M. Cap and internal nucleotides of reovirus mRNA primers are incorporated into influenza viral complementary RNA during transcription in vitro. J Virol 32, 895–904 (1979).
Bouloy, M., Plotch, S. J. & Krug, R. M. Both the 7-methyl and the 2′-O-methyl groups in the cap of mRNA strongly influence its ability to act as primer for influenza virus RNA transcription. Proc Natl Acad Sci U S A 77, 3952–3956 (1980).
Robertson, H. D., Dickson, E., Plotch, S. J. & Krug, R. M. Identification of the RNA region transferred from a representative primer, beta-globin mRNA, to influenza mRNA during in vitro transcription. Nucleic Acids Res 8, 925–942 (1980).
Li, M. L., Rao, P. & Krug, R. M. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J 20, 2078–2086 (2001).
Vreede, F. T. & Fodor, E. The role of the influenza virus RNA polymerase in host shut-off. Virulence 1, 436–439 (2010).
Braam, J., Ulmanen, I. & Krug, R. M. Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell 34, 609–618 (1983).
Biswas, S. K. & Nayak, D. P. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J Virol 68, 1819–1826 (1994).
Argos, P. A sequence motif in many polymerases. Nucleic Acids Res 16, 9909–9916 (1988).
Poch, O., Sauvaget, I., Delarue, M. & Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J 8, 3867–3874 (1989).
Fechter, P. et al. Two aromatic residues in the PB2 subunit of influenza A RNA polymerase are crucial for cap binding. J Biol Chem 278, 20381–20388 (2003).
Fechter, P. & Brownlee, G. G. Recognition of mRNA cap structures by viral and cellular proteins. J Gen Virol 86, 1239–1249 (2005).
Guilligay, D. et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 15, 500–506 (2008).
Blaas, D., Patzelt, E. & Kuechler, E. Cap-recognizing protein of influenza virus. Virology 116, 339–348 (1982).
Blaas, D., Patzelt, E. & Kuechler, E. Identification of the cap binding protein of influenza virus. Nucleic Acids Res 10, 4803–4812 (1982).
Penn, C. R., Blaas, D., Kuechler, E. & Mahy, B. W. Identification of the cap-binding protein of two strains of influenza A/FPV. J Gen Virol 62 (Pt 1), 177–180 (1982).
Ulmanen, I., Broni, B. A. & Krug, R. M. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc Natl Acad Sci U S A 78, 7355–7359 (1981).
Dias, A. et al. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458, 914–918 (2009).
Hara, K., Schmidt, F. I., Crow, M. & Brownlee, G. G. Amino acid residues in the N-terminal region of the PA subunit of influenza A virus RNA polymerase play a critical role in protein stability, endonuclease activity, cap binding and virion RNA promoter binding. J Virol 80, 7789–7798 (2006).
Yuan, P. et al. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 458, 909–913 (2009).
Yamanaka, K., Ishihama, A. & Nagata, K. Reconstitution of influenza virus RNA-nucleoprotein complexes structurally resembling native viral ribonucleoprotein cores. J Biol Chem 265, 11151–11155 (1990).
Biswas, S. K., Boutz, P. L. & Nayak, D. P. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J Virol 72, 5493–5501 (1998).
Poole, E., Elton, D., Medcalf, L. & Digard, P. Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology 321, 120–133 (2004).
Hagen, M., Chung, T. D., Butcher, J. A. & Krystal, M. Recombinant influenza virus polymerase: requirement of both 5′ and 3′ viral ends for endonuclease activity. J Virol 68, 1509–1515 (1994).
Desselberger, U., Racaniello, V. R., Zazra, J. J. & Palese, P. The 3′ and 5′-terminal sequences of influenza A, B and C virus RNA segments are highly conserved and show partial inverted complementarity. Gene 8, 315–328 (1980).
Flick, R., Neumann, G., Hoffmann, E., Neumeier, E. & Hobom, G. Promoter elements in the influenza vRNA terminal structure. RNA 2, 1046–1057 (1996).
Datta, K., Wolkerstorfer, A., Szolar, O. H., Cusack, S. & Klumpp, K. Characterization of PA-N terminal domain of Influenza A polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res 41, 8289–8299 (2013).
Zhang, S., Wang, J., Wang, Q. & Toyoda, T. Internal initiation of influenza virus replication of viral RNA and complementary RNA in vitro. J Biol Chem 285, 41194–41201 (2010).
Pritlove, D. C., Fodor, E., Seong, B. L. & Brownlee, G. G. In vitro transcription and polymerase binding studies of the termini of influenza A virus cRNA: evidence for a cRNA panhandle. J Gen Virol 76 (Pt 9), 2205–2213 (1995).
Deng, T., Vreede, F. T. & Brownlee, G. G. Different de novo initiation strategies are used by influenza virus RNA polymerase on its cRNA and viral RNA promoters during viral RNA replication. J Virol 80, 2337–2348 (2006).
Shaw, M. W. & Lamb, R. A. A specific sub-set of host-cell mRNAs prime influenza virus mRNA synthesis. Virus Res 1, 455–467 (1984).
Rao, P., Yuan, W. & Krug, R. M. Crucial role of CA cleavage sites in the cap-snatching mechanism for initiating viral mRNA synthesis. EMBO J 22, 1188–1198 (2003).
Fodor, E., Pritlove, D. C. & Brownlee, G. G. Characterization of the RNA-fork model of virion RNA in the initiation of transcription in influenza A virus. J Virol 69, 4012–4019 (1995).
Hagen, M., Tiley, L., Chung, T. D. & Krystal, M. The role of template-primer interactions in cleavage and initiation by the influenza virus polymerase. J Gen Virol 76 (Pt 3), 603–611 (1995).
Baumann, M., Pontiller, J. & Ernst, W. Structure and basal transcription complex of RNA polymerase II core promoters in the mammalian genome: an overview. Mol Biotechnol 45, 241–247 (2010).
Shi, L., Summers, D. F., Peng, Q. & Galarz, J. M. Influenza A virus RNA polymerase subunit PB2 is the endonuclease which cleaves host cell mRNA and functions only as the trimeric enzyme. Virology 208, 38–47 (1995).
Geerts-Dimitriadou, C., Zwart, M. P., Goldbach, R. & Kormelink, R. Base-pairing promotes leader selection to prime in vitro influenza genome transcription. Virology 409, 17–26 (2011).
Klumpp, K., Hooker, L. & Handa, B. Influenza virus endoribonuclease. Methods Enzymol 342, 451–466 (2001).
Zhang, L., Kasif, S., Cantor, C. R. & Broude, N. E. GC/AT-content spikes as genomic punctuation marks. Proc Natl Acad Sci U S A 101, 16855–16860 (2004).
Lamb, R. A. & Lai, C. J. Sequence of interrupted and uninterrupted mRNAs and cloned DNA coding for the two overlapping nonstructural proteins of influenza virus. Cell 21, 475–485 (1980).
Markoff, L. & Lai, C. J. Sequence of the influenza A/Udorn/72 (H3N2) virus neuraminidase gene as determined from cloned full-length DNA. Virology 119, 288–297 (1982).
Vreede, F. T., Gifford, H. & Brownlee, G. G. Role of initiating nucleoside triphosphate concentrations in the regulation of influenza virus replication and transcription. J Virol 82, 6902–6910 (2008).
Lamb, R. A., Lai, C. J. & Choppin, P. W. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: colinear and interrupted mRNAs code for overlapping proteins. Proc Natl Acad Sci U S A 78, 4170–4174 (1981).
Bishop, D. H., Gay, M. E. & Matsuoko, Y. Nonviral heterogeneous sequences are present at the 5′ ends of one species of snowshoe hare bunyavirus S complementary RNA. Nucleic Acids Res 11, 6409–6418 (1983).
Doan, L., Handa, B., Roberts, N. A. & Klumpp, K. Metal ion catalysis of RNA cleavage by the influenza virus endonuclease. Biochemistry 38, 5612–5619 (1999).
Chung, T. D. et al. Biochemical studies on capped RNA primers identify a class of oligonucleotide inhibitors of the influenza virus RNA polymerase. Proc Natl Acad Sci U S A 91, 2372–2376 (1994).
Kawakami, K., Ishihama, A. & Hamaguchi, M. RNA polymerase of influenza virus. I. Comparison of the virion-associated RNA polymerase activity of various strains of influenza virus. J Biochem 89, 1751–1757 (1981).
Plotch, S. J. & Krug, R. M. Influenza virion transcriptase: synthesis in vitro of large, polyadenylic acid-containing complementary RNA. J Virol 21, 24–34 (1977).
Plotch, S. J. & Krug, R. M. Segments of influenza virus complementary RNA synthesized in vitro. J Virol 25, 579–586 (1978).
Garcin, D. et al. The 5′ ends of Hantaan virus (Bunyaviridae) RNAs suggest a prime-and-realign mechanism for the initiation of RNA synthesis. J Virol 69, 5754–5762 (1995).
van Knippenberg, I., Lamine, M., Goldbach, R. & Kormelink, R. Tomato spotted wilt virus transcriptase in vitro displays a preference for cap donors with multiple base complementarity to the viral template. Virology 335, 122–130 (2005).
Simons, J. F. & Pettersson, R. F. Host-derived 5′ ends and overlapping complementary 3′ ends of the two mRNAs transcribed from the ambisense S segment of Uukuniemi virus. J Virol 65, 4741–4748 (1991).
Duijsings, D., Kormelink, R. & Goldbach, R. In vivo analysis of the TSWV cap-snatching mechanism: single base complementarity and primer length requirements. EMBO J 20, 2545–2552 (2001).
Bouloy, M., Pardigon, N., Vialat, P., Gerbaud, S. & Girard, M. Characterization of the 5′ and 3′ ends of viral messenger RNAs isolated from BHK21 cells infected with Germiston virus (Bunyavirus). Virology 175, 50–58 (1990).
Chou, Y. Y. et al. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog 9, e1003358 (2013).
Ping, J. et al. Genomic and protein structural maps of adaptive evolution of human influenza A virus to increased virulence in the mouse. PLoS ONE 6, e21740 (2011).
Dankar, S. K. et al. Influenza A virus NS1 gene mutations F103L and M106I increase replication and virulence. Virol J 8, 13 (2011).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009).
Dankar, S. K. et al. Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol J 10, 243 (2013).
Acknowledgements
This work was funded by a grant from the Natural Science and Engineering Research Council of Canada (NSERC Canada) awarded to M. Pelchat. The authors wish to acknowledge the genotyping platform of the McGill University and Genome Quebec Innovation Centre for technical assistance.
Author information
Authors and Affiliations
Contributions
M.P. and E.G.B. designed the project. D.S. performed the library preparation for high-throughput sequencing. L.R. and M.P. wrote the required in-house Perl and R scripts. M.P., L.R. and D.S. conducted data analyses. D.S., E.G.B. and M.P. contributed to writing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Raw sequencing data have been deposited on the Sequence Read Archive of NCBI [SRA: SRR1301043].
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Sikora, D., Rocheleau, L., Brown, E. et al. Deep sequencing reveals the eight facets of the influenza A/HongKong/1/1968 (H3N2) virus cap-snatching process. Sci Rep 4, 6181 (2014). https://doi.org/10.1038/srep06181
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06181
This article is cited by
-
Cut site preference allows influenza A virus PA-X to discriminate between host and viral mRNAs
Nature Microbiology (2023)
-
Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis
Nature Reviews Microbiology (2016)
-
siRNAs targeting PB2 and NP genes potentially inhibit replication of Highly Pathogenic H5N1 Avian Influenza Virus
Journal of Biosciences (2015)
-
Structural insight into cap-snatching and RNA synthesis by influenza polymerase
Nature (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
