
Abstract
Recent studies have demonstrated that human tau can be secreted by neurons and non-neuronal cells, an event linked to the propagation of tau pathology in the brain. In the present study, we confirmed that under physiological conditions, one tau-positive band was detected in the culture medium with an anti-tau antibody recognizing total tau and the Tau-1 antibody directed against unphosphorylated tau. We then examined whether tau secretion was modified upon insults. Tau secretion was increased by starvation [Earle's Balanced Salt Solution (EBSS)], inhibition of lysosomal function (leupeptin) and when both of these conditions were superimposed, this combined treatment having the most important effects on tau secretion. Interestingly, the pattern of tau secretion was distinct from that of control neurons when neurons were treated either with EBSS alone or EBSS + leupeptin. In these conditions, three tau-positive bands were detected in the culture medium. Two of these three bands were immunoreactive to Tau-1 antibody revealing that at least two tau species were released upon these treatments. Collectively, our results indicate that insults such as nutrient deprivation and lysosomal dysfunction observed in neurodegenerative diseases could result in an increase of tau secretion and propagation of tau pathology in the brain.
Similar content being viewed by others


Introduction
Neurofibrillary tangles (NFTs) composed of misfolded aggregated tau protein propagate in a predictable manner in Alzheimer's disease (AD)1,2,3,4. It is still unclear how NFTs spread in human brain. In recent years, a new concept has emerged that in several neurodegenerative diseases including AD, the spreading of misfolded protein aggregates in brain would result from cell-to-cell transmission5,6,7,8. This implies that NFTs would propagate in the brain by the release of misfolded tau aggregates from an affected neuron followed by its uptake in neighboring neurons. Consistent with this, recent studies, including our own demonstrated that tau can be secreted and endocytosed both in vitro and in vivo9,10,11,12,13,14,15,16,17,18,19,20,21. Most interestingly, in a transgenic mouse model where human tau overexpression was restricted to the entorhinal cortex, the first region to be affected in AD, the spreading of tau pathology was observed along synaptically connected circuits22,23. Although the mechanisms of the trans-synaptic spreading of tau pathology remain elusive, secretion of tau by pre-synaptic neurons and its uptake by post-synaptic neurons appear as a plausible cascade of events underlying the propagation of tau pathology in the brain.
Although it cannot be excluded with certainty that tau is not released in the extracellular space by dying neurons in AD brain, the presence of tau in the interstitial fluid in tau transgenic mice brain as well as the presence of tau in the cerebrospinal fluid (CSF) of tau transgenic mice before neurodegeneration indicate that extracellular tau can be released by an active process of secretion in vivo13,24. In vitro, human tau was shown to be secreted by several non-neuronal and neuronal cell lines when it was overexpressed8. More recently, endogenous tau was shown to be secreted by primary cortical neurons in normal conditions and this event increases upon neuronal activity25. In non-neuronal and neuronal cell lines, it was reported that tau secretion occurs through non-conventional secretory pathways12,14,18,26,27. This conclusion was mainly based on the fact that tau was found in exosomes and that the secretion of endogenous and overexpressed tau was insensitive to brefeldin A, a drug that blocks the conventional secretory pathway18,26,27,28. However, tau secretory pathways remain to be fully characterized.
Both full-length and cleaved tau were reported to be secreted in vitro. Secreted endogenous tau from primary cortical neurons, SH-SY5Y and iCell® neurons was full-length whereas overexpressed human tau secreted by M1C and Hela cells was cleaved at the C-terminal14,16,25. The overexpression of human tau in HEK293T resulted in the secretion of either full-length or cleaved species16,27. The phosphorylation state of secreted tau was also examined. Depending on the cell type, overexpressed human tau secreted by non-neuronal cells was either phosphorylated or importantly dephosphorylated at several sites12,14. Interestingly, the pool of tau found in exosomes produced by M1C cells overexpressing human tau was phosphorylated at several epitopes (AT180, AT100, AT270, AT8 and PHF-1) detected in AD brain, tau phosphorylated at T181 being enriched in exosomes18. In the case of tau secreted by primary cortical neurons, one study reported that it was phosphorylated at T181 and two studies showed that it was dephosphorylated at the epitope recognized by the Tau-1 antibody16,25.
An increase of tau in the CSF is noted in several neurodegenerative diseases including AD indicating that tau might be more secreted in pathological conditions29. In the present study, we examined whether tau secretion by primary cortical neurons would be modified upon insults. Both starvation by incubating neurons in EBSS and inhibition of lysosomal function by treatment with leupeptin significantly increased tau secretion. However, the most important increase of tau secretion was observed when both insults were superimposed. Upon EBSS and EBSS + leupeptin (E + L) but not leupeptin treatment, the pattern of tau secretion differed from that of control neurons. As a result of these treatments, three tau-positive bands were detected in the medium. Furthermore, two pools of tau were secreted by EBSS and E + L treated neurons based on their immunoreactivity to the Tau-1 antibody recognizing unphosphorylated tau. The present results revealed that starvation and lysosomal dysfunction exert positive effects on tau secretion and therefore these insults could contribute to the increased release of tau in pathological conditions.
Experimental Procedures
Preparation and transfection of mouse primary cortical neurons
The use of animals and all surgical procedures described in this article were carried out according to The guide to the Care and Use of Experimental Animals of the Canadian Council on Animal Care. Primary cortical cultures were prepared from E16 mouse embryos (C57BL6 mice). The cerebral cortices were treated with trypsin (0.025% at 37°C for 20 min). The reaction was stopped with trypsin inhibitor solution containing DNAse. Neurons were dissociated by several passages through a Pasteur pipette. The cells were then plated either on glass coverslips or on culture dishes coated with polylysine and laminin (Sigma, Oakville, ON, Canada). The neurons were maintained in Neurobasal medium (Invitrogen, Burlington, ON, Canada) supplemented with glutamax (Invitrogen) and B27 (Invitrogen). When specified, six days after plating, Lipofectamine 2000 (Invitrogen) was used to transfect neurons with pmRFP-LC330. Briefly, for 2 petris, 1 μg of plasmid DNA was mixed with 50 μl of Opti-MEM medium (Invitrogen) and 4 μl of Lipofectamine 2000 was mixed with 50 μl of Opti-MEM medium. Both mixtures were incubated for 5 min then combined and left standing for 20 min. The culture medium of each petri was removed, stored at 37°C for further use and replaced with 2 ml of fresh medium to which 40 μl of the DNA-Lipofectamine mix was added. After an incubation of 90 min at 37°C, cells were washed with a Hanks' balanced salts solution and the stored culture medium was put back in each petri.
Treatment with EBSS and leupeptin
Seven days after plating, neurons were incubated either in EBSS alone (Invitrogen), 20 μM leupeptin (Sigma) diluted in water or EBSS and 20 μM leupeptin for 3–9 hrs. Control cells were incubated in complete Neurobasal medium for 3–9 hrs. Then, the medium was harvested and the cells lysed as described below.
Quantification of cell death and cell membrane integrity
Cell death was evaluated by trypan blue exclusion method. Briefly, neurons were cultured on glass coverslips. Cells were washed twice with phosphate buffered saline (PBS) and then incubated in 0.2% trypan blue (Sigma) diluted in PBS for 4 min at room temperature (RT). Cells were then washed once with PBS and fixed in 4% paraformaldehyde for 5 min at 4°C and then 10 min at RT. The number of blue cells (dead cells) and total cells were counted in ten different fields and the cell death percentage was evaluated by the ratio of blue cells on the total number of cells.
Cell membrane integrity was assessed by the measurement of the LDH activity in the culture medium using the LDH cytotoxicity assay kit from Cayman Chemical Company (Ann Arbor, MI, USA), according to the manufacturer's instructions. Triplicates were performed for each sample. The LDH content in the samples was measured with a BIO-TEK Elx800 plate reader.
Preparation of cell lysates
After the culture medium was collected, the cells were immediately washed twice with PBS and once with PBS containing 0.5 M NaCl to detach proteins non-specifically attached to the cell surface31. The cells were then lysed in modified RIPA buffer [Tris HCl pH 7.5 50 mM, NaCl 150 mM, Triton X-100 0.5%, sodium deoxycholate (SDC) 0.5%, sodium dodecyl sulfate (SDS) 1%, dithiothreitol (DTT) 1 mM, NaF 50 mM, Na3VO4 5 mM], protease inhibitor cocktail 1× (Complete EDTA-free from Roche Diagnostics, Indianapolis, IN), phosphatase inhibitor cocktail 2× (PhosSTOP from Roche Diagnostics). Protein assay was performed with Bio-Rad DC Protein assay (Bio-Rad Laboratories Ltd., Mississauga, ON, Canada).
Immunoprecipitation of tau from culture medium
After treatment, the culture medium was collected from control and treated cells and centrifuged at 3000 RPM for 10 min at RT to remove cell debris. To analyze the pattern of secreted tau isoforms, tau was immunoprecipitated from the culture medium. For each condition, 60 μl of magnetic beads coupled with anti-mouse antibodies (Invitrogen, Dynabeads® M-280 Sheep anti-Mouse IgG) or with anti-rabbit antibodies (Invitrogen, Dynabeads® M-280 Sheep anti-Rabbit IgG) were washed in PBS containing 0.1% BSA and incubated O/N at 4°C with one of the following antibodies: 0.4 μg Tau-5 (Invitrogen) or 2 μg A0024 (DAKO) respectively. The beads were then washed and incubated for 2 hrs at 4°C with 1.5 ml of the culture medium. The complex bead-antibody-antigen was then washed in PBS, resuspended in 30 μl of sample buffer 1× and boiled for 5 min. Immunoblotting was performed as described below.
Immunoblotting
Equal amounts of proteins were loaded in each lane and electrophoresed on a 10% polyacrylamide gel. Following SDS-PAGE separation, proteins were electrophoretically transferred to a nitrocellulose membrane. Then the membranes were incubated in 5% milk diluted in 0.2% Tween-20 Tris buffered saline (TBST) for 1 hr. The nitrocellulose stripes were incubated with the primary antibodies O/N at 4°C. They were then washed in TBST and incubated with the peroxidase-conjugated secondary antibodies. Membranes were again washed and revealed by chemiluminescence (Amersham Pharmacia Biotech, Quebec, Quebec, Canada). The image acquisition and densitometry were performed with a ChemiDoc™ MP System. After incubation with tau antibodies, membranes were stripped to visualize the level of γ-actin (Santa-Cruz Biotechnology Inc, Santa-Cruz, CA) as a loading control for the cell lysates. Membranes were also incubated with the mouse or rabbit peroxidase-conjugated secondary antibodies to reveal IgG as a loading control for immunoprecipitates. The quantification of LC3-II was done according to the procedure recommanded in the last version of Guidelines for the use and interpretation of assays for monitoring autophagy published in 201232. The changes of LC3-II levels were quantified in comparison to those of actin and not to those of LC3-I.
Dephosphorylation of tau by lambda-protein phosphatase
Cells were lysed in Ripa buffer (1 mM PMSF, protease inhibitors cocktail 1×, 1 μM okadaic acid, 0.25% Na-deoxycholate, 50 mM Tris-HCl ph7.4, 1% Nonidet P-40, 0.9% NaCl, 1 mM EDTA) with a syringe and centrifuged at 20000× G for 20 min at 4°C. Forty μl of supernatant was mix with 200 units of lambda protein phosphatase (BioLabs) and incubated at 30°C for 3 hrs. The reaction was stopped by adding 1× sample buffer and boiled for 5 min at 95°C.
Immunofluorescence on primary cortical neurons
Treated neurons grown on coverslips were fixed in 4% paraformaldehyde prepared in PBS for 30 min. Neurons were then permeabilized with 0.2% Triton X-100 in PBS for 5 min. Cultures were kept in PBS until they were processed for immunofluorescence. For immunofluorescence, coverslips were blocked with 5% normal goat serum (NGS) (Invitrogen) in PBS. Then coverslips were stained with the antibody A0024 (1:500) (DAKO). After 3 washes in PBS, coverslips were incubated with anti-rabbit antibody coupled to FITC (1:500, Jackson ImmunoResearch). The antibody was diluted in the blocking solution. Incubations were carried out at RT for 1 hr. Coverslips were then washed in PBS and mounted in mowiol. Labeled cells were visualized with an axioplant Zeiss fluorescence microscope using either ×63 or ×100 objective.
Formation of autophagosomes was quantified in control and neurons treated with either EBSS alone or EBSS + leupeptin that were transfected with RFP-LC3. The formation of autophagosomes was confirmed by the presence of RFP-LC3 positive dots.
Electron microscopy
Primary cortical neurons were fixed in 0.1 M cacodylate buffer (pH 7.4; 37°C) containing 1.0% glutaraldehyde (MECALAB, Québec, Canada) and 1.5% tannic acid (Fisher, Québec, Canada). Cells were postfixed with 1% osmium tetroxide (EMS, PA, USA) in cacodylate buffer at 4°C. After several washes in buffer, neurons were dehydrated in graded ethanol, infiltrated and embedded in Epon 812 (MECALAB), according to standard technique33. Ultrathin sections were obtained using a Reichert Ultracut S ultramicrotome and mounted on formvar-carbon coated nickel grids (MECALAB). Sections were stained with uranyl acetate and lead citrate and examination was performed with a Philips CM 100 electron microscope.
Statistical analysis
The Gaussian distribution of the data was analyzed using the Kolmogorov and Smirnov method (p > 0.05). Statistical significance was evaluated with a two-tailed Paired t-test for the quantification of the percentage of neurons presenting the formation of LC3 positive structures, the increase of LC3-II analyzed by western blotting, the analysis of cell viability and the amount of LDH released in the culture medium. A one-way analysis of variance (ANOVA) Kruskal-Wallis test followed by a Dunn's multiple comparison test was used to compare the increase of LDH between the treatments. The ratio LDH in the medium of treated neurons/LDH in the medium of control neurons was used for this analysis since for leupeptin treated neurons, LDH was measured in complete medium whereas for EBSS and EBSS + leupeptin treated neurons, LDH was measured in EBSS. A two-tailed Paired t-test was used for the analysis of the amount of tau secretion when the data presented a Gaussian distribution and a Wilcoxon signed-rank test if the data did not present a Gaussian distribution. A Wilcoxon signed-rank test was used to analyze the changes of tau phosphorylation in treated neurons since these data did not present a Gaussian distribution. The statistical analysis was performed using the GraphPad InStat 3 software and p < 0.05 was considered significant. To compare the effects of the three treatments on tau secretion, statistical significance was evaluated with a two-way ANOVA with match values followed by Tukey's multiple comparison test. The ANOVA analysis was performed using Prism 6.0c software.
Results
A significant increase of tau secretion by primary cortical neurons upon starvation
In two recent studies, it was shown that endogenous full-length tau could be secreted by primary cortical neurons16,25. We examined tau secretion by these neurons after 7 days of plating and confirmed that tau was released in the medium (Figure 1a). In the cell lysate, two main bands at 52 and 57 kDa were detected with the anti-tau antibody A0024 from DAKO, a marker of total tau. As previously reported, a single band located at 52 kDa was detected in the medium by the antibody recognizing total tau when tau was enriched from the culture medium by immunoprecipitation25. To demonstrate that tau found in the medium was not released by neuronal cell death, the trypan blue exclusion method was used to monitor cell death. The percentage of the cell viability was 99.7 ± 0.108 in 6 independent cultures (p < 0.0001). This low percentage of cell death indicated that tau found in the medium was most likely released by a specific process of secretion as previously shown25.

EBSS, leupeptin and EBSS + leupeptin increased the secretion of endogenous tau by primary cortical neurons.
(a) In normal conditions (CTL), one tau-positive band at 52 kDa was detected in the culture medium by an antibody revealing total tau (A0024). A similar pattern was observed when neurons were treated with leupeptin (L). When neurons were treated either with EBSS (E) or EBSS + leupeptin (E + L), three tau-positive bands at 47, 52 and 57 kDa were detected in the culture medium. In the cell lysate of control neurons, two tau-positive bands at 52 and 57 kDa were revealed by the anti-tau antibody A0024 (n = 6). (b) Quantification of total tau in culture medium by densitometry upon EBSS, leupeptin and E + L treatments (n = 6). The intensities of the bands were expressed in arbitrary units. Values represent mean ± s.e.m.; *p < 0.05; **p < 0.01; ***p < 0.001. (c) Cell damage was evaluated by measuring the amount of LDH released in the culture medium (n = 6). Since control and treated neurons were not incubated in the same medium, the amount of LDH in the EBSS treatment was compared to that of EBSS without cell. The amount of LDH in the leupeptin treatment was compared to that of neurobasal medium without cell. The increase of LDH in the medium was not statistically different (n.s) between the treatments. Values represent mean ± s.e.m.; *p < 0.05; **p < 0.01. Blots used in this figure were cropped. Full-length blots were included in supplementary figure 1.
We then investigated whether tau secretion would be altered upon insults. Neurons are very sensitive to nutrient deprivation, a condition that can occur in neurodegenerative diseases including AD34. In the present study, nutrient deprivation was induced by incubating neurons in EBSS, a medium containing glucose but devoid of amino acids and growth factors. EBSS is known to result in an extreme induction of autophagy characterized by the accumulation of autophagic vacuoles (AVs) in neurons35. Two approaches were used to monitor the formation of AVs. Firstly, the production of LC3-II, a phosphatidylethanolamine-modified isoform of LC3-I generated and translocated to nascent autophagosomes when macroautophagy is induced was examined in the cell lysate of treated neurons by western blotting (Figure 2a)36. In EBSS treated neurons, the increase of LC3-II was not statistically significant. A similar observation was reported in a previous study performed in primary cortical neurons under extreme induction of autophagy where the high turnover of LC3-II prevented its accumulation35. Secondly, the accumulation of AVs by EBSS was confirmed by using RFP-LC3, a marker of autophagosomes, on fixed neurons30. As expected, the number of neurons presenting AVs was significantly higher for neurons incubated in EBSS than for control neurons (Figure 2b).

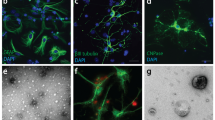
EBSS and E + L treatments induced AVs in primary cortical neurons.
(a) LC3-II was analyzed by western blotting in the cell lysate prepared from EBSS (E), leupeptin (L) and EBSS + leupeptin (E + L) treated neurons. The LC3-II levels were compared to those of actin. For the analysis of LC3-II, 10 μg of cell lysate were loaded (n = 6). Values represent mean ± s.e.m.; *p < 0.05. (b) Control (CTL) and EBSS treated neurons were transfected with RFP-LC3 to monitor the formation of AVs (arrows) (n = 3). Scale bar = 10 μm. The number of cells containing AVs in treated neurons was significantly higher than in control neurons (n = 3). Values represent mean ± s.e.m.; *p < 0.05. (c) Control and E + L treated neurons were transfected with RFP-LC3 to monitor the formation of AVs (arrows) (n = 4). Scale bar = 10 μm. The number of cells containing AVs in treated neurons was significantly higher than in control neurons (n = 4). Values represent mean ± s.e.m.; **p < 0.01. (d) The formation of AVs in the cell body and neurites of neurons was confirmed by electron microscopy (arrows). AVs contained electron-dense material. Scale bar = 2 μm for left panel and 500 nm for middle and right panels. Magnification = 5800 × for left panel and 25 000 × for middle and right panels.
Interestingly, when neurons were incubated in EBSS, the pattern of tau secretion differed from that of control neurons (Figure 1a). In the culture medium of control neurons, only one tau-positive band was detected with the antibody A0024 whereas in the medium of treated neurons, three tau-positive bands were found, one at 52 kDa also present in control neurons and two additional bands, one at 57 kDa noted in the cell lysate and one at 47 kDa. The band at 52 kDa was the most abundant tau-positive band in the medium of treated neurons. The band at 47 kDa indicated that secreted tau was cleaved and/or dephosphorylated (see below). Furthermore, tau secretion was increased ~5 times (p = 0.014) by EBSS treatment (Figure 1b). To eliminate the possibility that tau was released by cell death in the medium of treated neurons, cell viability was evaluated using the trypan blue exclusion method. The percentage of neurons taking up trypan blue was evaluated to be less than 1% for both control and treated neurons and was not significantly different between these two groups (p = 0.8733). The above observations indicated that the increased release of tau could occur by a specific process of secretion upon EBSS treatment.
A significant increase of tau secretion by primary cortical neurons is observed when lysosomal function is inhibited
In several neurodegenerative diseases including AD, lysosomal function has been shown to be deficient. We verified if such a condition would affect tau secretion by primary cortical neurons. Lysosomal function was inhibited by leupeptin, an inhibitor of cysteine and aspartyl proteases such as cathepsins found in lysosomes. The pattern of tau secretion upon leupeptin treatment was identical to that noted in normal conditions as a single tau-positive band located at 52 kDa was detected in the culture medium (Figure 1a). However, tau secretion was increased ~2 times (p = 0.03) by leupeptin (Figure 1b). Neuronal cell death could not be responsible for the increase of tau secretion as indicated by the percentage of cell viability that was 99.77 ± 0.04 and 99.52 ± 0.08 for control and treated neurons respectively (n = 6).
Highest levels of tau secretion is observed when EBSS and leupeptin treatments are superimposed
In neurodegenerative diseases, more than one insult can act on neurons. In the present study, starvation and inhibition of the lysosomal function were superimposed by incubating neurons in EBSS and leupeptin (E + L) for at least 3 hrs. In a previous study, this treatment was shown to result in an accumulation of AVs35. In normal conditions, after their formation, autophagosomes are rapidly eliminated by their fusion with lysosomes32,37. Leupeptin prevents the elimination of autophagosomes by inhibiting lysosomal function35. To monitor the efficacy of our treatment, three approaches were used to visualize the accumulation of AVs. Firstly, we analyzed the amount of LC3-II by western blotting (Figure 2a). LC3-II was 2.81 ± 0.51 times higher in E + L treated neurons than in control neurons (p = 0.017). Secondly, the accumulation of AVs was monitored by RFP-LC3 (Figure 2c). The number of cells containing AVs was significantly higher for treated neurons than control neurons. Thirdly, the presence of AVs was confirmed by electron microscopy (Figure 2d). The morphology of AVs was similar to that described by Boland et al'0835.
Interestingly, the pattern of tau secretion by E + L treated neurons was identical to that observed with EBSS treatment. Indeed, three tau-positive bands at 47, 52 and 57 kDa were found in the medium (Figure 1a). E + L treatment induced the most significant increase of tau secretion by cortical neurons in comparison to either EBSS or leupeptin alone as the amount of total tau was ~8 times (p = 0.0002) higher in the medium of E + L treated neurons than in the medium of control neurons (Figure 1b). The amount of tau secreted by E + L treated neurons was not significantly different from that of EBSS treated neurons although it had the tendency to be higher. Starvation and inhibition of lysosomal function seem to have additive effects on tau secretion since the amount of tau released upon combination of these insults was very close to the sum of tau secretion generated by these individual treatments. The percentage of neurons taking up trypan blue was evaluated to be less than 1% for both control and treated neurons and was not significantly different between these two groups (p = 0.083) indicating that tau was not released by dying neurons in the medium of E + L treated neurons. To eliminate the possibility that the increase of tau in the culture medium was caused by cell damage induced by the treatments, we measured the amount of lactate dehydrogenase (LDH) activity, a marker of cell membrane integrity, in the culture medium38. The LDH was increased ~1.22 (p = 0.0124), 1.08 (p = 0.0172) and 1.19 (p = 0.0095) times in the medium of EBSS, leupeptin and E + L treated neurons, respectively, compared to the amount in the medium of control neurons revealing that cell membrane integrity was changed by our treatments (Figure 1c). This indicated that changes of membrane permeabilization could be responsible for the increased release of tau in the medium. However, the increase of LDH in the culture medium was not statistically different between the treatments (p = 0.1453) whereas tau secretion was indicating that changes in cell membrane integrity could not be the sole factor contributing to the increased release of tau (Figure 1c). Collectively, the above observations indicated that one pool of tau could be released by changes of the cell membrane integrity and another pool by a specific process of secretion.
In primary cortical neurons, we could confirm that E + L treatment induced an accumulation of AVs. Since we observed an important increase of tau secretion upon the accumulation of AVs by E + L treatment, one could speculate that these structures contributed to the release of tau. Indeed, recent studies reported that autophagosomes were involved in unconventional secretion39. To verify this possibility, we examined whether tau was enriched in AVs in E + L treated neurons (Figure 3). AVs were visualized by transfecting neurons with RFP-LC3 as described in Figure 2. In most control neurons, RFP-LC3 presented a diffuse staining in the cell body and neurites whereas tau was mainly enriched in the neurites as revealed by the anti-tau antibody A0024 (Figure 3a). In E + L treated neurons, several RFP-LC3 positive AVs were observed in the cell body and neurites but no enrichment of tau was noted in these structures. As illustrated in Figure 3b, tau staining was not concentrated in AVs in the cell body. Tau staining was observed in axonal swellings containing AVs (Figure 3b, arrow). However, tau staining was not enriched in the AVs. Similarly, no enrichment of tau staining was noted in AVs formed in neurites (Figure 3c).

Tau was not enriched in AVs found in E + L treated neurons.
Control and E + L treated neurons transfected with RFP-LC3 were fixed and stained with an antibody directed against total tau (Green). (a) Control neurons transfected with RFP-LC3. (b) Presence of several AVs in the cell body of E + L treated neurons as revealed by RFP-LC3. No enrichment of tau was noted in RFP-LC3-positive structures in the cell body and neurites (arrow) of E + L treated neurons (n = 7). (c) Tau staining was not enriched in a RFP-LC3-positive structure (arrow) found in a neurite of E + L treated neurons. Scale bar = 10 μm.
Dephosphorylated tau species were secreted by EBSS and E + L treated neurons
The presence of a tau-positive band migrating at 47 kDa in the culture medium of EBSS and E + L treated neurons indicated that these treatments might have induced dephosphorylation of secreted tau. To verify this point, we examined the phosphorylation state of tau secreted by control and treated neurons (Figure 4a). As previously reported, tau found in the medium of control neurons was detected by the antibody Tau-1, an antibody directed against unphosphorylated tau25. In the medium of neurons treated with EBSS and E + L, the bands at 52 kDa and 47 kDa were immunoreactive to the antibody Tau-1 but not the band at 57 kDa. The low amount of this band could not account for its absence of detection by Tau-1. Indeed, this antibody could detect the low amount of tau present in the medium in normal conditions. The amount of Tau-1-positive tau in the medium of E + L treated neurons was ~9 times (p = 0.0016) higher than in the medium of control neurons (Figure 4b). The above results revealed that EBSS and E + L treated neurons secreted two pools of tau, one Tau-1 positive and one Tau-1 negative. No increase of Tau-1 staining was noted for tau secreted by neurons treated with leupeptin. Secreted tau was not detectable with the phospho-tau antibodies (pS214, pT217 and pS422) indicating that its phosphorylation was very low (data not shown).

Significant increased secretion of dephosphorylated tau by EBSS and E + L treatments.
(a) In the culture medium of control neurons (CTL) and leupeptin treated neurons (L), only one tau-positive band at 52 kDa was detected with the Tau-1 antibody. In the medium of EBSS (E) and E + L treated neurons, the bands at 47 and 52 kDa but not the one at 57 kDa were detected with this antibody (n = 6). (b) Quantification by densitometry of tau dephosphorylated at the Tau-1 antibody epitope in culture medium of control and E + L treated neurons (n = 11). The intensities of the bands were expressed in arbitrary units. Values represent mean ± s.e.m.; **p < 0.01 (c) In the cell lysate of EBSS and E + L treated neurons, an important decrease of the band at 57 kDa and the apparition of a band at 47 kDa were revealed by the antibody A0024 (n = 13). An additional weak band was also observed between 52 and 57 kDa. In the cell lysate of EBSS and E + L treated neurons, the bands at 52 and 47 kDa were highly immunoreactive to the Tau-1 antibody whereas only the band at 52 kDa was detected in the cell lysate of control and leupeptin treated neurons (n = 11). Blots used in this figure were cropped. Full-length blots were included in supplementary figure 2. A higher exposure time of panel (a) was included in supplementary figure 3 for better detection of tau signal in the medium of control neurons.
Dephosphorylation of tau by EBSS and E + L treatment
We then verified whether tau dephosphorylation by E + L treatment occurred before it was secreted by examining its phosphorylation in the cell lysate. Total tau staining revealed an important decrease of the band at 57 kDa and the presence of an additional band at 47 kDa in the cell lysate of treated neurons (Figure 4c). An additional weak band located between 52 and 57 kDa was also noted. To verify if tau was dephosphorylated in E + L treated neurons, the cell lysate was stained with the Tau-1 antibody (Figure 4c). In control neurons, this antibody mainly detected the band at 52 kDa whereas in treated neurons, it strongly revealed the bands located at 52 kDa and 47 kDa showing that tau found in these bands was importantly dephosphorylated. We then examined whether tau dephosphorylation was induced by either EBSS or leupeptin. In neurons incubated in EBSS, the levels of tau dephosphorylation were very similar to those noted in E + L treated neurons whereas no tau dephosphorylation was observed upon leupeptin treatment as revealed by the Tau-1 antibody staining (Figure 4c). It is interesting to note that the band at 57 kDa that was barely detectable in cell lysate was present in the culture medium of treated neurons indicating that tau present in this band was importantly released by treated neurons (Figures 1a and 4c).
We further characterized the pattern of tau dephosphorylation in the lysate of E + L treated neurons since this treatment had the most important effects on tau secretion. The bands positive to the Tau-1 antibody were less immunoreactive to the phospho-specific anti-tau antibodies in treated neurons than in control neurons (Figure 5a). Interestingly, the antibody detecting tau phosphorylated at T181 was the sole phospho-specific anti-tau antibody that could detect the band at 47 kDa (Figure 5a). No phospho-specific anti-tau antibody could detect the band at 57 kDa in the cell lysate of treated neurons. This could be explained by its low levels in the cell lysate. The signal of the Tau-1 antibody was 3.75 ± 0.54 times higher in the cell lysate of E + L treated neurons than in the lysate of control neurons (p = 0.0010) but the decrease of the phospho-tau antibodies was not statistically different (Figure 5b).

Dephosphorylation of tau by E + L treatment.
(a) The bands positive to tau-1 antibody were less immunoreactive to the phospho-tau antibodies in the cell lysate of E + L treated neurons (n = 3) (b) Quantification of tau detected by Tau-1 and phospho-specific tau antibodies in the cell lysate of E + L treated and control neurons. Values represent mean ± s.e.m.; ***p < 0.001. (c) Incubation of the cell lysates from control neurons with lambda (λ) protein phosphatase resulted in an increase of tau gel mobility reminiscent to that observed in E + L treated neurons. No further increase of tau gel mobility was observed when the cell lysate of E + L treated neurons was incubated with λ protein phosphatase (n = 3). Blots used in this figure were cropped. Full-length blots were included in supplementary figure 2.
The above results showed that E + L treatment resulted in tau dephosphorylation. To further demonstrate that tau gel mobility shift observed in E + L treated neurons was mainly caused by its dephosphorylation, an in vitro dephosphorylation assay was used. Cell lysates from control neurons were incubated with lambda protein phosphatase. Then, the pattern of tau-positive bands was analyzed by western blotting. As expected, when tau was dephosphorylated in the lysate of control cells, the pattern of tau-positive bands was reminiscent to that observed in neurons treated with E + L indicating that dephosphorylation contributed to the increased gel mobility of tau (Figure 5c). In contrast, when the cell lysate of treated neurons was dephosphorylated in vitro, no further tau gel mobility shift was noted indicating that tau was already dephosphorylated by the E + L treatment (Figure 5c). From the above results, it appeared that tau secretion was enhanced in conditions where its dephosphorylation was increased. It remains to be determined whether dephosphorylation favors tau secretion by neurons.
Discussion
In the present study, we demonstrated that tau secretion by primary cortical neurons is increased upon insults. Both starvation and inhibition of lysosomal function resulted in an increased secretion of tau. However, the most dramatic effect was noted when both insults were superimposed. Interestingly, the pattern of tau secretion varied depending on the insult. Upon leupeptin treatment only one tau-positive band was released as noted for control neurons whereas neurons treated with either EBSS or E + L secreted three tau-positive bands. Furthermore, in all conditions tested, a pool of secreted tau was dephosphorylated at the Tau-1 antibody epitope. All together, our observations indicate that insults such as nutrient deprivation and lysosomal dysfunction known to take place in neurodegenerative diseases could increase tau secretion and thereby the propagation of tau pathology in the brain.
The fact that the percentage of cell viability between control and treated neurons was not statistically different indicated that tau was not released in the culture medium by dying neurons upon insults. However, we noted an increase of LDH in the culture medium revealing changes of cell membrane integrity in treated neurons. This indicated that tau could be released by an increase of membrane permeabilization38. Although we cannot rule out this possibility, our data indicate that it would not be the sole mechanism responsible for the increase of tau in the culture medium. Indeed, the increase of LDH in the culture medium was not statistically different between the treatments indicating that changes of cell membrane integrity were similar in all the treatments. On the other hand, the increase of tau secretion between E + L and leupeptin and EBSS and leupeptin was statistically different. Based on the above data, we concluded that tau could be released by both changes of membrane permeabilization and a specific process of secretion when neurons were treated with EBSS and E + L. According to a recent study, the release of LDH could be secondary to the extracellular accumulation of tau38. In this study, it was shown that extracellular tau oligomers can form pores allowing LDH to escape from the cytoplasm to the medium. If LDH was released in the medium by tau-induced pores in our experimental conditions, it would signify that tau was primarily released in the medium by a specific secretory process. This possibility remains to be demonstrated since we were not successful in demonstrating that tau released by treated neurons was oligomeric (data not shown).
Starvation exerted a dramatic effect on tau secretion by primary cortical neurons. This could signify that tau is secreted by membranous structures that are present and accumulate in this condition. During starvation, there is an extreme induction of autophagy characterized by the accumulation of AVs32. Autophagosomes were shown to participate in unconventional secretion39. In the present experimental conditions, AVs might not contribute to the increase of tau secretion since tau was not enriched in these structures. The increase of tau secretion by E + L treatment was higher than that induced by starvation alone although the difference between these two treatments was not statistically significant. The main difference between EBSS and E + L treatments is that in EBSS but not in E + L treated neurons, the autophagic flux was complete. Indeed, in a previous study, it was reported that when neurons were treated with EBSS and leupeptin, AVs were filled with material whereas upon treatment with EBSS alone, neurons presented empty AVs as expected after their fusion with functional lysosomes35. The present results indicate that complete autophagic flux is not necessary to induce a significant increase of tau secretion by starved neurons. The fact that tau was not enriched in AVs and that the inhibition of autophagic flux did have a major impact of tau secretion could indicate that the increase of tau release by EBSS and E + L treated neurons was not linked to the induction of autophagy but rather to starvation per se. For example, important alterations of signaling and membrane trafficking are observed in starved cells. These changes could enhance the interaction of tau with proteins and/or membranous structures involved in its secretion.
The inhibition of lysosomal function by leupeptin resulted in a modest increase of tau secretion. Lysosomes were shown to be able to fuse with the plasma membrane to release their content in the extracellular space40. It is known that tau can be degraded in lysosomes41,42. Therefore, it is possible that inhibition of cathepsins by leupeptin resulted in less degradation of tau and this would in turn increase the amount of tau that could be released by lysosome exocytosis. An endo-lysosomal secretory pathway has also been reported where lysosomes fuse with endosomes before their fusion with the plasma membrane43. In such a case, extracellular tau that has been endocytosed could be found in lysosomes after their fusion with tau-containing endosomes. The highest levels of tau secretion were observed when both starvation and inhibition of lysosomal function were superimposed. These two conditions seem to act in parallel to increase tau secretion since the amount of tau released by E + L treated neurons was very close to that obtained when the amounts of each treatment were added. All together, our results indicate that tau can be released by several pathways, which by acting in parallel, can result in an important increase of tau secretion in pathological conditions.
Upon starvation, three tau-positive bands were noted in the culture medium. The release of different tau species by this treatment could indicate that more than one secretory pathways are involved in tau secretion in this condition. Tau seems to be secreted by unconventional secretory pathways as noted for other proteins involved in neurodegenerative diseases12,14,18,26,27. Several unconventional secretory pathways exist and therefore different tau species might have access to distinct pathways8. Furthermore, several studies including ours have reported that two pools of tau, a cytosolic and membranous one are present in neurons44,45,46,47. Each of these pools could have access to different secretory pathways in starved neurons. The tau species released upon starvation differed by their phosphorylation state. Phosphorylation of tau was shown to regulate its association with the membranes. In a recent study, it was reported that dephosphorylated tau was associated with the plasma membrane46. In our previous study, we showed that hyperphosphorylated tau and tau dephosphorylated at the epitope of Tau-1 were not found in the same membranous subfractions in AD brain45. Hyperphosphorylated and dephosphorylated tau at the Tau-1 epitope were found in the subfraction enriched in ER membranes and in the Golgi membranes respectively. The differential association of hyperphosphorylated and hypophosphorylated tau with membranes could give them access to distinct secretory pathways more or less efficient in releasing tau.
Upon starvation, tau dephosphorylation most likely occurred before secretion since in the cell lysate, we noted an important decrease of tau phosphorylation at several sites accompanied by an increased immunoreactivity of tau to the Tau-1 antibody. The dephosphorylation of tau by starvation was unexpected. However, this phenomenon is not unique to this pathology since other insults were reported to induce a decrease of tau phosphorylation. Oxidative stress induced by hydrogen peroxide treatment caused a dephosphorylation of tau in primary cortical neurons48. Excitotoxicity induced by homocysteine that activates both glutamate receptors and phosphatase 2A resulted in tau dephosphorylation in cerebellar granule cells49. Other insults such as hypoxia and glucose deprivation were also reported to induce a decrease of tau phosphorylation in both rat and human brain slices50. Dephosphorylation of tau was also reported in vivo during ischemia in both brain and spinal cord51,52,53. Interestingly, our previous study revealed that at an advanced stage of AD, both hyperphosphorylated and hypophosphorylated tau are present45. A pool of dephosphorylated tau at the epitope recognized by the Tau-1 antibody was also observed by another group in AD brain54. Collectively the above observations indicate that some of the above insults could be responsible for the generation of hypophosphorylated tau whereas other insults such as the accumulation of Aβ could be responsible for the increase of hyperphosphorylated tau in AD brain55,56,57,58. In normal conditions, it is still unclear whether tau was dephosphorylated before secretion. A dephosphorylated pool of tau at the Tau-1 antibody epitope was detected in the cell lysate prepared from control neurons and therefore it is possible that this pool was secreted. On the other hand, phosphatases located along tau secretory pathway could contribute to tau dephosphorylation. For example, the phosphatase tissue non-specific alkaline phosphatases (TNAP) present in the plasma membrane were shown to be responsible for the dephosphorylation of extracellular tau59. Upon starvation, a tau-positive band located at 57 kDa was observed in the medium. Interestingly, this band was not immunoreactive to the Tau-1 antibody showing that tau contained in this band was not significantly dephosphorylated at this epitope. Our results demonstrated that both phosphorylated and hypophosphorylated tau can be released by primary cortical neurons but the amount of each of these pools of tau varied depending on the insult and thereby could be used as an indication of neuronal injury.
In two recent studies, it was reported that in physiological conditions, tau secreted by primary cortical neurons was dephosphorylated at the Tau-1 epitope16,25. In all conditions that were tested in the present study, a pool of secreted tau was dephosphorylated at the Tau-1 antibody epitope. All Tau-1 positive bands detected in the cell lysate were found in the medium in both control and treated neurons. It remains to be determined whether dephosphorylated tau was preferentially secreted by primary cortical neurons. Upon starvation, the proportion of the bands at 52 and 47 kDa was similar in both the cell lysate and medium indicating that the most dephosphorylated tau species were not more secreted than the less dephosphorylated ones. The fact that hypophosphorylated tau was secreted by control and treated neurons could indicate that mechanisms are present by these cells to eliminate toxic proteins, their secretion being one of them. Indeed, intracellular hypophosphorylated tau was reported to induce apoptosis when it was overexpressed in non-neuronal cells60. Hypophosphorylated tau was also shown to be toxic in Drosophila61. In this model, the toxicity of hypophosphorylated tau was correlated to the impairment of axonal transport. Interestingly, extracellular hypophosphorylated tau was also shown to be toxic. In a previous study, extracellular hypophosphoryated tau was reported to induce a sustained increase of intracellular calcium by the activation of the muscarinic M1 and M3 receptors leading to cell death59,62. The present study indicates that upon insults, the increased release of hypophosphorylated tau could benefit to neurons to prevent apoptosis. On the other hand, this release could compromise neuronal survival by its extracellular toxicity.
It remains unclear whether phosphorylation regulates tau secretion since both phosphorylated and unphosphorylated tau species were detected extracellularly. In Hela cells, we reported that a tau mutant mimicking phosphorylation was more secreted than one mimicking dephosphorylation12. Tau secreted by exosomes was shown to be phosphorylated at several sites found in AD18. In normal conditions, primary cortical neurons were shown to release unphosphorylated tau25. However, in the present study, we report that several tau species were secreted some phosphorylated and other unphosphorylated by cortical neurons upon insults. Collectively, the above observations indicate that phosphorylation per se might not be the determinant factor regulating tau secretion but rather the cellular context which influences both tau phosphorylation and tau access to secretory pathways.
Tau secretory pathways in primary cortical neurons remain to be identified. However, our data showed that this cellular system is suitable to examine these pathways in physiological conditions and upon insults. The distinct pattern of secreted tau species by insults and the additive effects of these insults on tau secretion when they are combined indicate that tau might be secreted by several pathways as noted for other proteins involved in neurodegenerative diseases63,64,65,66. It is still unknown whether the different tau species that are released exert distinct toxic effects and/or whether they contribute distinctly to the propagation of tau pathology in the brain. It is crucial to unravel the distinct contribution of tau species to these events to develop therapeutic strategies to prevent neuronal dysfunction and to abrogate the spreading of tau pathology in AD brain.
References
Grundke-Iqbal, I. et al. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J biol chem 261, 6084–6089 (1986).
Grundke-Iqbal, I. et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83, 4913–4917 (1986).
Braak, H. & Braak, E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991).
Morris, M., Maeda, S., Vossel, K. & Mucke, L. The many faces of tau. Neuron 70, 410–426 (2011).
Brundin, P., Melki, R. & Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11, 301–307 (2010).
Frost, B. & Diamond, M. I. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 11, 155–159 (2010).
Walker, L. C., Diamond, M. I., Duff, K. E. & Hyman, B. T. Mechanisms of Protein Seeding in Neurodegenerative Diseases. Arch Neurol 1–7 (2012).
Mohamed, N. V., Herrou, T., Plouffe, V., Piperno, N. & Leclerc, N. Spreading of tau pathology in Alzheimer's disease by cell-to-cell transmission. Eur J Neurosci 37, 1939–1948 (2013).
Kim, W., Lee, S. & Hall, G. F. Secretion of human tau fragments resembling CSF-tau in Alzheimer's disease is modulated by the presence of the exon 2 insert. FEBS Lett 584, 3085–3088 (2010).
Kim, W. et al. Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis 19, 647–664 (2010).
Avila, J. et al. Looking for novel functions of tau. Biochem Soc trans 40, 653–655 (2012).
Plouffe, V. et al. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One 7, e36873 (2012).
Yamada, K. et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31, 13110–13117 (2011).
Chai, X., Dage, J. L. & Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol Dis 48, 356–366 (2012).
Wu, J. W. et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J biol chem 288, 1856–1870 (2012).
Karch, C. M., Jeng, A. T. & Goate, A. M. Extracellular tau levels are influenced by variability in tau that is associated with tauopathies. J biol chem 287, 42751–42762 (2012).
Kfoury, N., Holmes, B. B., Jiang, H., Holtzman, D. M. & Diamond, M. I. Trans-cellular propagation of Tau aggregation by fibrillar species. J biol chem 287, 19440–19451 (2012).
Saman, S. et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J biol chem 287, 3842–3849 (2012).
Clavaguera, F. et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11, 909–913 (2009).
Frost, B., Jacks, R. L. & Diamond, M. I. Propagation of tau misfolding from the outside to the inside of a cell. J biol chem 284, 12845–12852 (2009).
Guo, J. L. & Lee, V. M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J biol chem 286, 15317–15331 (2011).
de Calignon, A. et al. Propagation of Tau Pathology in a Model of Early Alzheimer's Disease. Neuron 73, 685–697 (2012).
Liu, L. et al. Trans-synaptic spread of tau pathology in vivo. PLoS One 7, e31302 (2012).
Barten, D. M. et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis 24 Suppl 2, 127–141 (2011).
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W. & Hanger, D. P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO reports 14, 389–394 (2013).
Simon, D. et al. Tau overexpression results in its secretion via membrane vesicles. Neurodegener Dis 10, 73–75 (2012).
Simon, D., Garcia-Garcia, E., Royo, F., Falcon-Perez, J. M. & Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett 586, 47–54 (2012).
Saraste, J., Palade, G. E. & Farquhar, M. G. Temperature-sensitive steps in the transport of secretory proteins through the Golgi complex in exocrine pancreatic cells. Proc Natl Acad Sci USA 83, 6425–6429 (1986).
Hampel, H. et al. Total and phosphorylated tau protein as biological markers of Alzheimer's disease. Exp Gerontol 45, 30–40 (2010).
Kimura, S., Noda, T. & Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 (2007).
Madeira, A., Pommet, J. M., Prochiantz, A. & Allinquant, B. SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J 19, 1905–1907 (2005).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 (2012).
Luft, J. H. Improvements in epoxy resin embedding methods. J Biophys Biochem Cytol 9, 409–414 (1961).
Allen, S. J., Watson, J. J. & Dawbarn, D. The neurotrophins and their role in Alzheimer's disease. Curr Neuropharmacol 9, 559–573 (2011).
Boland, B. et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 28, 6926–6937 (2008).
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T. & Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15, 1101–1111 (2004).
Nixon, R. A. et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J neuropathol exp neurol 64, 113–122 (2005).
Flach, K. et al. Tau oligomers impair artificial membrane integrity and cellular viability. J biol chem 287, 43223–43233 (2012).
Duran, J. M., Anjard, C., Stefan, C., Loomis, W. F. & Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J cell biol 188, 527–536 (2010).
Andrei, C. et al. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell 10, 1463–1475 (1999).
Wang, Y. et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet 18, 4153–4170 (2009).
Kruger, U., Wang, Y., Kumar, S. & Mandelkow, E. M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 33, 2291–2305 (2012).
Laulagnier, K. et al. Role of AP1 and Gadkin in the traffic of secretory endo-lysosomes. Mol Biol Cell 22, 2068–2082 (2011).
Farah, C. et al. Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton 63, 710–724 (2006).
Perreault, S., Bousquet, O., Lauzon, M., Paiement, J. & Leclerc, N. Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J neuropathol exp neurol 68, 503–514 (2009).
Pooler, A. M. et al. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging 33, 431 e427–438 (2012).
Brandt, R., Léger, J. & Lee, G. Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J cell biol 131, 1327–1340 (1995).
Davis, D. R., Anderton, B. H., Brion, J. P., Reynolds, C. H. & Hanger, D. P. Oxidative stress induces dephosphorylation of tau in rat brain primary neuronal cultures. J neurochem 68, 1590–1597 (1997).
Kuszczyk, M., Gordon-Krajcer, W. & Lazarewicz, J. W. Homocysteine-induced acute excitotoxicity in cerebellar granule cells in vitro is accompanied by PP2A-mediated dephosphorylation of tau. Neurochem Int 55, 174–180 (2009).
Burkhart, K. K., Beard, D. C., Lehman, R. A. & Billingsley, M. L. Alterations in tau phosphorylation in rat and human neocortical brain slices following hypoxia and glucose deprivation. Exp Neurol 154, 464–472 (1998).
Mailliot, C. et al. Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. J Cereb Blood Flow Metab 20, 543–549 (2000).
Shackelford, D. A. & Nelson, K. E. Changes in phosphorylation of tau during ischemia and reperfusion in the rabbit spinal cord. J neurochem 66, 286–295 (1996).
Shackelford, D. A. & Yeh, R. Y. Dephosphorylation of tau during transient forebrain ischemia in the rat. Mol Chem Neuropathol 34, 103–120 (1998).
Kopke, E. et al. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J biol chem 268, 24374–24384 (1993).
Götz, J. Tau and transgenic animal models. Brain Res Brain Res Rev 35, 266–286 (2001).
Zheng, W. H., Bastianetto, S., Mennicken, F., Ma, W. & Kar, S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience 115, 201–211 (2002).
Zempel, H., Thies, E., Mandelkow, E. & Mandelkow, E. M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation and destruction of microtubules and spines. J Neurosci 30, 11938–11950 (2010).
Jin, M. et al. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA 108, 5819–5824 (2011).
Diaz-Hernandez, M. et al. Tissue non-specific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J biol chem 285, 32539–32548 (2010).
Liu, X. A. et al. Tau dephosphorylation potentiates apoptosis by mechanisms involving a failed dephosphorylation/activation of Bcl-2. J Alzheimers Dis 19, 953–962 (2010).
Talmat-Amar, Y. et al. Important neuronal toxicity of microtubule-bound Tau in vivo in Drosophila. Hum Mol Genet 20, 3738–3745 (2011).
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T. & Avila, J. Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol Cell Neurosci 37, 673–681 (2008).
Urushitani, M. et al. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 9, 108–118 (2006).
Gomes, C., Keller, S., Altevogt, P. & Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci lett 428, 43–46 (2007).
Gousset, K. et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11, 328–336 (2009).
Fevrier, B. et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA 101, 9683–9688 (2004).
Acknowledgements
The authors would like to thank Michel Lauzon for his valuable technical assistance for the electron microscopy and Miguel Chagnon for his help for the statistical analysis. We would like to thank Diane Gingras for helpful discussion. The authors would also like to thank Dr. T. Yoshimori for the construct RFP-LC3 obtained from Addgene. This work was supported by the Canadian Institute of Health Research (MOP-114874) (NL) and an FRSQ infrastructure grant to the Groupe de Recherche sur le sytème nerveux central (GRSNC) (NL). N-V M has a studentship from the GRSNC. G. R.-L. has a post-doctoral fellowship from the GRSNC.
Author information
Authors and Affiliations
Contributions
N.-V.M. was involved in all the experiments included in the present study and prepared figures 1, 2, 3, 4 and 5. V.P. performed tau immunoprecipitation for figures 1 and 4 and analyzed the results presented in figure 5A and B. G.R.-L. performed the morphological analysis for figure 2C and participated to the analysis of the results included in figure 5A and B.N.L. wrote the paper. E.P. was involved in the in vitro dephosphorylation assay. All the authors reviewed the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mohamed, NV., Plouffe, V., Rémillard-Labrosse, G. et al. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep 4, 5715 (2014). https://doi.org/10.1038/srep05715
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05715
This article is cited by
-
LncRNA PVT1 Promotes Neuronal Cell Apoptosis and Neuroinflammation by Regulating miR-488-3p/FOXD3/SCN2A Axis in Epilepsy
Neurochemical Research (2023)
-
Synaptic tau: A pathological or physiological phenomenon?
Acta Neuropathologica Communications (2021)
-
Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy
Molecular Neurodegeneration (2020)
-
Unconventional secretion factor GRASP55 is increased by pharmacological unfolded protein response inducers in neurons
Scientific Reports (2019)
-
Endolysosome and Autolysosome Dysfunction in Alzheimer’s Disease: Where Intracellular and Extracellular Meet
CNS Drugs (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
