
Abstract
Alzheimer’s disease (AD) is characterized by a substantial loss of neurons and synapses throughout the brain. The exact mechanism behind the neurodegeneration is still unclear, but recent data suggests that spreading of amyloid-β (Aβ) pathology via extracellular vesicles (EVs) may contribute to disease progression. We have previously shown that an incomplete degradation of Aβ42 protofibrils by astrocytes results in the release of EVs containing neurotoxic Aβ. Here, we describe the cellular mechanisms behind EV-associated neurotoxicity in detail. EVs were isolated from untreated and Aβ42 protofibril exposed neuroglial co-cultures, consisting mainly of astrocytes. The EVs were added to cortical neurons for 2 or 4 days and the neurodegenerative processes were followed with immunocytochemistry, time-lapse imaging and transmission electron microscopy (TEM). Addition of EVs from Aβ42 protofibril exposed co-cultures resulted in synaptic loss, severe mitochondrial impairment and apoptosis. TEM analysis demonstrated that the EVs induced axonal swelling and vacuolization of the neuronal cell bodies. Interestingly, EV exposed neurons also displayed pathological lamellar bodies of cholesterol deposits in lysosomal compartments. Taken together, our data show that the secretion of EVs from Aβ exposed cells induces neuronal dysfunction in several ways, indicating a central role for EVs in the progression of Aβ-induced pathology.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) develops over decades and is the prevalent causal factor of dementia among the elderly1. The main characteristics of AD is the formation of amyloid-β (Aβ) plaques and neurofibrillary tangles, as well as pronounced inflammation. Based on the amyloid cascade hypothesis, accumulation of Aβ is the primary pathological event of AD, which in turn triggers inflammation and the appearance of neurofibrillary tangles, eventually leading to severe neuronal dysfunction2. Accumulating evidence indicate that the widespread neurodegeneration in the AD brain is a result of soluble Aβ aggregates, such as protofibrils, rather than insoluble Aβ fibrils3,4,5. However, the exact cellular and molecular mechanisms behind the Aβ-induced neuronal cell death remain elusive.
Since the majority of patients with sporadic AD do not display an increase in Aβ production, insufficient lysosomal degradation of Aβ has been suggested as a possible disease cause6,7. In line with such hypothesis, the cells’ ineffectiveness to clear protein aggregates is known to result in Aβ deposits as dense bodies, multivesicular bodies (MVBs) and autophagic vacuoles8,9,10. Moreover, intracellular Aβ accumulation may cause intercellular spreading of Aβ pathology via cell-to-cell contacts and secretion of extracellular vesicles (EVs)8,11. There are different forms of EVs, including larger microvesicles (MVs), that originate and bud directly from the plasma membrane and smaller exosomes, that are generated by invagination of the membrane of MVBs and released by exocytosis12. However, currently there are no consensus criteria to define different types of EVs, as they often overlap in size and share some surface markers13. EVs can be isolated by ultracentrifugation from various fluids, including cell culture medium, plasma, cerebrospinal fluid and urine, and constitute an important form of intercellular communication by delivering proteins, lipids, nucleic acids and sugar molecules to recipient cells through endocytosis14,15,16,17. Several studies have demonstrated that EVs can carry pathological proteins associated with neurodegenerative diseases, including aggregated forms of Aβ18, but their exact role in the disease process remain to be uncovered.
We have previously investigated the cellular responses of synthetic Aβ42 protofibrils in a mixed co-culture of astrocytes, neurons and oligodendrocytes. Interestingly, we found that the astrocytes rapidly engulf large amounts of Aβ42 protofibrils, followed by storage rather than degradation of the ingested material8. The ineffective degradation of Aβ results in astrocytic endosomal/lysosomal defects and secondary neuronal cell death, due to the secretion of neurotoxic EVs. Our previous investigations of the EV content demonstrated that EVs isolated from Aβ42 protofibril exposed cultures contain N-terminally truncated forms of Aβ42 and increased levels of apolipoprotein E (apoE)8,19. Truncated forms of Aβ are present in both intracellular and extracellular deposits in the AD brain20 and are known to be more resistant to degradation, more prone to aggregate and more toxic than full-length Aβ21. However, exactly how the toxic EVs affect the neurons was not examined. In this follow-up study, we have investigated the mechanisms behind EV-mediated neurotoxicity in detail.
Materials and methods
Synthetic Aβ42 protofibrils
Aβ42 protofibrils were produced according to a well-established protocol22,23,24. In short, synthetic Aβ42 peptides were dissolved in 10 mM NaOH, mixed with phosphate-buffered saline (PBS) to a concentration of 443 µM (2 mg/ml) and incubated at 37 °C for 30 min (unlabeled Aβ42, Innovagen or American Peptide Company Inc) or 45 min (Fluorescent HiLyte Fluor 555-labeled Aβ42, Anaspec Inc). The sample was then centrifuged for 5 min at 17,900×g at 4 °C to remove any insoluble aggregates, followed by 1:4 dilution of supernatant in sterile PBS to a final concentration of 0.5 mg/ml. The protofibril specific ELISA, based on mAb15822, was used to confirm the Aβ42 protofibril concentration. For transmission electron microscopy (TEM), Aβ42 protofibrils diluted 1:10 in PBS were dropped onto carbon coated 300-mesh copper grids, negatively stained with 1% Uranyl acetate for 5 min and air dried. The samples were analyzed using a Hitachi H-7100 transmission electron microscope. A careful characterization of the Aβ42 protofibrils has been performed previously, using HPLC-SEC, Thioflavin T staining, different ELISAs, in addition to TEM8,22,23,24.
Animals
All experiments were approved by the Uppsala County Animal Ethics Board (Ethical permit number: C75/13, valid until 2018-06-28 and Ethical permit number: 5.8.18-08472/18, valid until 2023-05-31), following the rules and regulations of the Swedish Animal Welfare Agency, in compliance with the European Communities Council Directive of 22 September 2010 (2010/63/EU). The mice were housed in a 12 h dark–light cycle in an enriched environment and had ad libitum access to food and water.
Co-cultures of neurons and glia
Cerebral cortices were dissected from C57/BL6 E14 mouse embryos in Hank’s buffered salt solution (HBSS), supplemented with 100 U/ml penicillin and 100 µg/ml streptomycin and 8 mM Hepes buffer (all from Invitrogen). The cortices were centrifuged for 3 min at 150×g and the pellet was resuspended in HBSS. Any remaining blood vessels were let to sediment for 10 min at room temperature (RT) and the supernatant was centrifuged for 5 min at 150×g. The pellet was resuspended in proliferation medium; DMEM/F12 with added 1 × B27 supplement, 100 U/ml penicillin, 100 µg/ml streptomycin, 8 mM Hepes buffer, 10 ng/ml basic fibroblast growth factor (bFGF) (all from Invitrogen) and 20 ng/ml epidermal growth factor (EGF, Corning). The cells were expanded as neurospheres in non-treated cell culture flasks and passaged every second to third day, by dissociation in HBSS and resuspension in proliferation medium. Prior to experiments, the cells were plated on Poly-L-Ornithine (Sigma-Aldrich) and laminin (Invitrogen) coated coverslips or culture dishes in proliferation medium at a seeding density of 32,000 cells/cm2. The following day, the proliferation medium was replaced with differentiation medium (without bFGF and EGF) to initiate differentiation to a mixed population of 75 ± 8% astrocytes, 25 ± 8% neurons and 5 ± 3% oligodendrocytes8. During the 7-day differentiation period, the medium was changed every second to third day. Only neurospheres from passage 2–4 were used for experiments.
Neuronal cell cultures
Dissection and dissociation of C57/BL6 E14-E16 cerebral mouse cortices were performed as described above. The pellet was resuspended in neurobasal medium, supplemented with 1 × B27 supplement, 100 U/ml penicillin, 100 µg/ml streptomycin and 2 mM L-glutamine (all from Invitrogen). The neurons were cultured as a monolayer at a concentration of 25,000 cells/cm2 on Poly-l-Ornithine (Sigma-Aldrich) and laminin (Invitrogen) coated coverslips, electron microscopy dishes or cell culture dishes for 12 days in vitro (DIV12). The medium was fully replaced with fresh medium at DIV1, after which only half of the medium was replaced every second or third day.
Aβ42 protofibril exposure of co-cultures
Differentiated co-cultures were exposed to 0.1 µM Aβ42 protofibrils in differentiation medium for 24 h. Control cultures received fresh medium without Aβ42 protofibrils. No further aggregation of the protofibrils was noted in the medium during the 24 h exposure time8. After 24 h of exposure, cells were washed 3 times with medium and thereafter cultured in Aβ42 protofibril-free medium for 6 consecutive days (day 6), at which all medium was collected and stored at 4 °C. Fresh Aβ-free medium was added and the cultures were incubated for 6 additional days prior to the next medium collection at day 12.
Isolation and addition of EVs to neuronal cultures
The conditioned co-culture medium from day 6 was pooled with the equivalent medium from day 12 prior to ultracentrifugation. The pooled medium samples for each treatment and cell culture batch were centrifuged at 300×g for 5 min to remove any free-floating cells, followed by another centrifugation at 2,000×g for 10 min to remove any remaining cell debris. The supernatants were collected and transferred to ultracentrifuge tubes and centrifuged at 135,000×g at 4 °C for 1.5 h to isolate EVs, including larger MVs and exosomes. The vesicle pellets were resuspended in either PBS for TEM analysis or neurobasal medium, supplemented with penicillin, streptomycin, L-glutamine and B27 supplement, for treatment of neuronal cells. DIV12 neuronal cultures were incubated for 2 or 4 days with the EVs from untreated co-cultures or co-cultures that had been exposed to Aβ42 protofibrils. Parallel neuronal cultures received medium with or without 0.1 µM Aβ42 protofibrils, but no EVs.
Immunocytochemistry and labeling
Cells were fixed with 4% paraformaldehyde for 15 min at RT and washed 3 times with PBS. For the cholesterol staining, coverslips were incubated with filipin III (1:100 in cholesterol detection buffer, Cholesterol Assay kit, Abcam) for 1 h in the dark at RT. Prior to antibody incubation, the cells were permeabilized and blocked with 0.1% Triton X-100 with 5% normal goat serum (NGS) in PBS for 30 min at RT. Primary antibodies were diluted in 0.1% Triton X-100 with 0.5% NGS in PBS and the coverslips were incubated with the antibody solution for 1–4 h at RT. The primary antibodies used were: rabbit anti-Glial Fibrillary Acidic Protein (GFAP, 1:400, DAKO), mouse anti-βIII tubulin (1:200, Covance), mouse anti-2′,3′-Cyclic-nucleotide 3′-phosphodiesterase (CNPase, 1:500, Sigma-Aldrich), rabbit anti-synaptophysin (1:50, Abcam), rabbit anti-lysosomal-associated membrane protein 1 (LAMP1, 1:100, Abcam), mouse anti-Aβ 4G8 (1:200, Biolegend). The coverslips were washed 3 times with 1 × PBS and then incubated with secondary antibodies, also diluted in 0.1% Triton X-100 with 0.5% NGS in PBS for 45 min at 37 °C. The secondary antibodies used were AlexaFluor 488, 555 or 350 against mouse or rabbit (1:200, Molecular Probes). The coverslips were washed 3 times with PBS and mounted on microscope slides using EverBrite hard-set medium with or without DAPI (Biotium) or Vectashield hard-set mounting medium with DAPI (DAKO). For the cholesterol stainings, Triton X-100 was not included in the blocking solution or antibody dilution. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, Roche biochemicals) assay was performed according to the manufacturer’s instructions (protocol 10, to quantify apoptotic cells). For visualization of mitochondria, the neuronal cultures were transfected with CellLight Mitochondria-GFP (Bacman 2.0, ThermoFisher Scientific). The reagent was added in the neurobasal medium 3 days after the addition of EVs according to the manufacturer’s instructions and the neuronal cultures were incubated for 24 h prior to fixation.
Time-lapse microscopy
Time-lapse experiments were performed at 37 °C in humidified 5% CO2 in air, using a Nikon Biostation IM Live Cell Recorder (Nikon). The cells were cultured at a concentration of 25,000 cells/cm2, in time-lapse culture dishes (VWR) and pictures were taken every 10th min for up to 48 h8.
Transmission electron microscopy
Transmission electron microscopy was performed as described previously8,19.
EVs
EV samples were added onto a formvar-coated 200-mesh grid (Oxford 11 Instruments) and incubated for 45 min. The grid was dried, and 1% uranyl acetate was added to the grid and incubated for 10 s. The grid was left to dry for at least 15 min before visualization in a Tecnai G2 transmission electron microscope (TEM, FEI company) with an ORIUS SC200 CCD camera and Gatan Digital Micrograph software (both from Gatan Inc.).
Cells
The neuronal cultures were fixed in 2.5% glutaraldehyde and 1% paraformaldehyde. The cells were then rinsed with 0.15 M sodium cacodylate (pH 7.2–7.4) for 10 min and incubated in fresh 1% osmium tetraoxide in 0.1 M sodium cacodylate for 1 h at RT. After incubation, the sodium cacodylate was rinsed away to dehydrate the dishes with 70% ethanol for 30 min, 95% ethanol for 30 min and > 99% ethanol for 1 h. A thin plastic layer (Agar 100 resin kit, Agar Scientific Ltd) was added to the dishes and incubated for 1 h. The plastic was poured off and a new plastic layer was added onto the dishes for incubation overnight in a desiccator. Next, the plastic was heated to enable its removal after which a new thicker plastic layer was added before another incubation for 1 h in a desiccator. Cells were covered with 3 mm plastic and polymerized in the oven at 60 °C for 48 h. Embedded cells were sectioned by using a Leica ultracut UTC ultrotome (Rowaco AB) and visualized with a Tecnai G2 transmission electron microscope (FEI company) with an ORIUS SC200 CCD camera and Gatan Digital Micrograph software (both from Gatan Inc.).
Cell lysis
Cell lysis was performed as described previously19. In short, cell culture medium was thoroughly removed from the dishes and the cells were lysed in ice-cold lysis buffer (20 mM Tris pH 7.5, 0.5% Triton X-100, 0.5% deoxycholic acid, 150 mM NaCl, 10 mM EDTA, 30 mM NaPyro), supplemented with a protease inhibitor cocktail (ThermoScientific). The lysates were transferred to protein LoBind tubes (Eppendorf) and incubated for 30 min on ice prior to centrifugation at 10,000×g for 10 min at 4 °C. The supernatants were transferred to new tubes and stored at − 70 °C until analysis.
Western blot analysis
Western blot analysis was performed as described previously19. In short, protein concentrations of the total cell lysates were measured with Pierce BCA protein kit (ThermoFisher Scientific), according to the manufacturer’s instructions. A total of 18 µg protein was mixed with Bolt LDS Sample buffer and Sample Reducing agent (both from ThermoFisher Scientific) and incubated for 5 min at 95 °C to denature the proteins. Samples were loaded on a Bolt 4–12% Bis–Tris plus gel and run in Bolt MES sodium dodecyl sulfate (SDS) running buffer (both from ThermoFisher Scientific) for 22 min at 200 V. Chameleon duo pre-stained protein ladder (Li-Cor) was used for visualization of gel migration, protein size and orientation. Transfer to a PVDF membrane was performed for 1 h at 20 V in Bolt transfer buffer containing 10% methanol, 0.1% Bolt antioxidant (ThermoFisher Scientific) and 0.01% SDS. Blocking of the membrane was performed in 5% bovine serum albumin (BSA, Sigma-Aldrich) in 0.1% tris-buffered saline-Tween (TBS-T) for 1 h on shake at RT, prior to incubation with primary antibody in 0.5% BSA in 0.1% TBS-T O/N at 4 °C. Primary antibodies used in the study were rabbit anti-Synaptophysin (1:20,000, Abcam) and mouse anti-β-actin (β-actin, 1:1,000, Cell Signaling). Following extensive washes in TBS-T, the membrane was incubated with horseradish peroxidase-conjugated (HRP) secondary goat anti-rabbit or goat anti-mouse antibody (1:20,000, Pierce) in 0.5% BSA in 0.1% TBS-T for 1 h on shake at RT. Development of membrane was performed with enhanced chemiluminescence (ECL, GE Healthcare) by using a ChemiDoc XRS with Image Lab Software to visualize and measure the intensity of the immunoreactive bands (Bio-Rad Laboratories).
Analyses and statistics
All experiments were performed on 3 independent cell cultures, derived from mouse embryos of different pregnant females. A wide-field microscope (Zeiss AxioImager Z1) and Zen 2012 software was used for image acquisition and analysis. Confocal micrographs were taken with a Zeiss LSM 700 confocal microscope. The number of apoptotic, TUNEL + cells and living cells with non-condensed nuclei was manually counted in totally 30 captured images for each treatment. The number of cells with disrupted mitochondrial networks in CellLight Mitochondria-GFP transfected cultures was manually counted and normalized against the total number of transfected, GFP-expressing cells. A minimum of 135 transfected cells per treatment were analyzed. Since the immunocytochemistry data did not meet the assumption of normal distribution with the Shapiro–Wilk’s W-test, Mann–Whitney U-test was used for the mitochondrial data (when only two groups were compared, n = 135) and Kruskal Wallis test followed by Dunn’s multiple comparison test was used for the TUNEL data (when 4 groups were compared, n = 30). The Western blot data was analyzed using Mann–Whitney U-test (n = 3). All statistical tests were two-tailed. All results are presented in bar charts with mean ± standard deviation. Levels of significance were set to *p < 0.05, **p < 0.01 and ***p < 0.001.
Ethics approval
All experiments involving animals were approved by the Uppsala County Animal Ethics Board (ethical permit number: C17/13 and 5.8.18-08472/18), following the rules and regulations of the Swedish Animal Welfare Agency, in compliance with the European Communities Council Directive of 22 September 2010 (2010/63/EU).
Results
Endocytosis of EVs results in intracellular Aβ deposits in cortical neurons
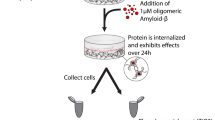
In order to study EV-mediated Aβ neurotoxicity, we isolated EVs from cells with intracellular Aβ deposits and added these to cortical neurons (Fig. 1a). More specifically, co-cultures, containing mainly astrocytes (Fig. 1b), but also neurons (Fig. 1c) and a few oligodendrocytes (Fig. 1d) were exposed to Aβ42 protofibrils for 24 h (Fig. 1e) or were left untreated. Similar to our previous findings8,19,25, the astrocytes in the co-culture ingested large amounts of Aβ42 protofibrils, which accumulated intracellularly in lysosomal compartments (Fig. 1f). Following Aβ42 protofibril exposure, the co-cultures were cultured without Aβ for 12 days and EVs were isolated from the conditioned medium. TEM analysis of the EV preparation confirmed the presence of exosomes and larger MVs (Fig. 1g). Cortical neurons (Fig. 1h) were exposed to EVs for 2 or 4 days, prior to analysis. TEM analysis and time-lapse microscopy indicated that the added EVs attached to and were endocytosed by the neurons (Fig. 2a,b). Immunocytochemistry of neuronal cultures followed by confocal microscopy confirmed that intracellular Aβ deposits were present in the Aβ42 protofibril-EV exposed neurons (Fig. 2c).

Isolation of EVs from cultures with glial Aβ deposits.. Schematic experimental set-up for studying EV-mediated neurodegeneration (a). Co-cultures of astrocytes (b), neurons (c) and a few oligodendrocytes (d) were exposed to 0.1 µM Aβ42 protofibrils (e) or left untreated. During the exposure time the astrocytes in the culture ingested and accumulated large amounts of aggregated Aβ (f). After 24 h incubation, the cells were washed and incubated in Aβ-free medium for 12 days. Conditioned medium from day 6 and 12 was pooled and ultracentrifuged and successful isolation of EVs was verified by TEM. The EV samples were shown to contain a mixture of exosomes and larger MVs (g). Following isolation, the EVs were resuspended in neurobasal medium and added to cortical neuronal cultures (h) for 2 or 4 days. Scale bars: (b), (c), (d), (f) and (h) = 20 µm.

Endocytosis of EVs results in intracellular Aβ deposits in cortical neurons. TEM analysis indicated that EVs attached to and were endocytosed by cortical neurons (asterisks, a). The process of EV uptake by the neurons could also be monitored with time-lapse microscopy (asterisk, b). Immunocytochemistry followed by confocal microscopy, confirmed intracellular Aβ deposits in Aβ42 protofibril-EV exposed neurons (c). Scale bars: (b) = 10 µm and (c) = 20 µm.
EVs isolated from Aβ42 protofibril exposed co-cultures induce apoptosis of cortical neurons
To investigate if EVs secreted from Aβ42 protofibril exposed neuroglial co-cultures affected the survival of cortical neurons, we labeled the neurons with the apoptotic marker TUNEL and quantified the number of apoptotic cells. The relative number of TUNEL + neurons was analyzed in cell cultures exposed to Aβ42 protofibril-EVs, control-EVs or Aβ42 protofibrils (no EVs) and in untreated control cultures. There were no differences in the relative number of TUNEL + neurons in cultures exposed to Aβ42 protofibrils only and untreated controls. Moreover, control-EVs did not affect neuronal survival, compared to the untreated control cultures (Fig. 3a). However, Aβ42 protofibril-EVs significantly increased the number of TUNEL + neurons, compared to control-EVs at both day 2 (p < 0.001, Fig. 3a) and day 4 (p < 0.001, Fig. 3b). Representative images of the TUNEL labeling in cultures exposed to control-EVs and Aβ42 protofibril-EVs are shown in Fig. 3c. Taken together, the TUNEL analysis indicated a decreased neuronal survival upon exposure to Aβ42 protofibril-EVs.

Aβ42 protofibril-EVs induce apoptosis of cortical neurons. The relative number of TUNEL + neurons was analyzed 2 and 4 days after EV addition to verify the number of apoptotic cells. Parallel neuronal cultures received medium only (control) or Aβ42 protofibrils. There was a significant increase in apoptotic neurons in cultures exposed to Aβ42 protofibril-EVs at day 2 (a) and day 4 (b), compared to neurons that received control-EVs, control medium or medium with Aβ42 protofibrils only (*p < 0.05, **p < 0.01 and ***p < 0.001). Representative images of the TUNEL labeling of neuronal cultures exposed to control-EVs and Aβ42 protofibril-EVs (c). Scale bar: (c) = 20 µm.
Aβ42 protofibril-EVs cause severe mitochondrial stress in cortical neurons
Since we observed an increased cell death in neuronal cultures following exposure to Aβ42 protofibril-EVs, we next sought to investigate the health status of living neurons in the cultures. To examine whether Aβ42 protofibril-EV exposed neurons have a disrupted energy metabolism, neurons were transfected with CellLight Mitochondria-GFP 24 h prior to fixation. Neurons that received control-EVs displayed elongated, branched, healthy mitochondrial networks throughout the cells (Fig. 4a). However, in Aβ42 protofibril-EV exposed cultures, many neurons displayed a disrupted, unhealthy mitochondrial network and mitochondrial swelling (Fig. 4a). The percentage of neurons with healthy mitochondria, was quantified in relation to the total number of transfected cells. There was a significant decrease in the percentage of healthy mitochondria in cultures exposed to Aβ42 protofibril-EVs, compared to control-EVs (p < 0.001, Fig. 4b). The morphology of mitochondria was further visualized by using TEM. The results verified that neurons exposed to control-EVs exhibited healthy mitochondria with clear inner cristae (Fig. 4c), while mitochondria in neurons exposed to Aβ42 protofibril-EVs were clearly affected, showing loss of structure of the outer membrane as well as the inner cristae, and abnormal size compared to the control-EV exposed neurons (Fig. 4d,e).

Aβ42 protofibril-EV exposure induces mitochondrial stress in cortical neurons. Neurons were transfected with CellLight Mitochondria-GFP 24 h prior to fixation (at day 3), targeting cellular mitochondria. In cultures exposed to control-EVs the neurons displayed an elongated, branched mitochondrial network (healthy), while neurons exposed to Aβ42 protofibril-EVs frequently displayed a disrupted mitochondrial network and mitochondrial swelling (unhealthy) (a). Quantification revealed a significant decrease in the percentage of healthy mitochondria in neuronal cultures exposed to Aβ42 protofibril-EVs, compared to control-EVs (***p < 0.001) (b). TEM analysis of neurons exposed to control-EVs verified that their mitochondria appeared healthy (white arrow head) (c), whereas Aβ42 protofibril-EV exposed neurons had abnormally large, swollen and disrupted mitochondria (white arrow heads) (d,e). Scale bar: (a) = 20 µm.
Impaired synaptic organization in Aβ42 protofibril-EV exposed cortical neurons
Next, we aimed to study if Aβ42 protofibril-EVs induce synaptic alterations. For this purpose, neurons were stained with the neuronal-specific marker βIII tubulin and the synaptic vesicle marker synaptophysin. Neurons that received control-EVs displayed long axonal and dendritic networks at day 2 (Fig. 5a). In contrast, addition of Aβ42 protofibril-EVs resulted in severe synaptic loss and disrupted dendrites (Fig. 5a). Western blot analysis of the lysed neurons revealed that intracellular synaptophysin levels were unaffected in all neuronal cultures at day 2 (Fig. 5b; Supplementary Fig. 1). This was confirmed by intensity measurements of synaptophysin immunoreactive bands, analyzed relative to the loading control β-actin (Fig. 5c). To further investigate the effect of Aβ42 protofibril-EVs on the neuronal network, we performed TEM. The Aβ42 protofibril-EV exposed neurons displayed distinct pathology, including accumulation of filled vesicles and vacuoles (Fig. 5d), and abnormal swelling of processes (Fig. 5e). Taken together, our data suggest that the synaptophysin levels were not altered in response to Aβ42 protofibril-EVs, but its structural organization within the neurons was markedly affected.

Aβ42 protofibril-EV exposure results in synaptic loss in cortical neurons. Neuronal cultures were co-immunostained with the neuronal-specific marker βIII tubulin and the synaptic vesicle marker synaptophysin. In cultures exposed to control-EVs, the neurons displayed a large network of synaptophysin-positive processes at day 2. In contrast, neurons exposed to Aβ42 protofibril-EVs displayed a severely disrupted network (a). Intracellular synaptophysin levels, measured by Western blot analysis for 3 independent neuronal cell culture batches, showed no differences between Aβ42 protofibril-EV exposed neurons (+) and neurons that received control-EVs (−), control medium (−) Aβ42 protofibrils (+), respectively (b,c). TEM analysis of neuronal processes in cultures exposed to Aβ42 protofibril-EVs exhibited pathological signs of abnormal swelling and accumulation of filled vesicles (d,e). Scale bar: (a) = 20 µm.
Aβ42 protofibril-EVs induce enlarged vacuoles and waste accumulation in cortical neurons
In order to investigate subcellular changes in the neurons following exposure to Aβ42 protofibril-EVs, we performed additional TEM analysis. In comparison to cortical neurons treated with control-EVs (Fig. 6a) or free-floating Aβ42 protofibrils (Fig. 6b), Aβ42 protofibril-EV exposed neurons displayed multiple enlarged vacuoles. The vacuoles appeared to be empty or filled with diverse waste material (Fig. 6c,d). For example, the neurons contained many dense vacuoles (Fig. 6c) and lysosomal or endosomal compartments containing condensed mitochondria (Fig. 6e). The pathological vacuolization in combination with the high load of waste indicates that the neurons’ degradation capacity was severely affected by the exposure to Aβ42 protofibril-EVs.

Addition of Aβ42 protofibril-EVs induces vacuole formation in neuronal cell bodies. TEM analysis demonstrated that the cell bodies of neurons exposed to control-EVs (a) or free-floating Aβ42 protofibrils (b) were compact and healthy, while Aβ42 protofibril-EVs induced massive vacuole formation in the neuronal cell bodies (c). A higher magnification of the dashed area in c display a group of vesicles filled with debris (c’). Also, some of the larger vacuoles were filled with various forms of waste (d), including condensed mitochondria (e). A higher magnification of the dashed area is shown in (e’).
Lamellar body formation is frequently present in EV exposed cortical neurons
Interestingly, neurons that received either Aβ42 protofibril-EVs or control-EVs displayed multilamellar bodies (Fig. 7a,b). Multilamellar bodies are structures with multiple membranes, generated in lysosomal compartments, primarily consisting of undegraded phospholipids and cholesterol. In various disease conditions, the appearance of multilamellar bodies has been shown to be closely related to excessive cholesterol accumulation26,27. Hence, we next stained the neuronal cultures with the fluorescent cholesterol-binding dye filipin III. Co-stainings using specific antibodies against the lysosomal protein LAMP1, followed by fluorescence and confocal microscopy, confirmed that multilamellar bodies in Aβ42 protofibril-EV exposed cortical neurons consisted of cholesterol deposits in lysosomal compartments (Fig. 7c,d). A zoomed out image of the stained cell culture shown is shown in Supplementary Fig. 2.

Addition of EVs induces the formation of lamellar inclusions. Vesicles filled with lipid membranes were observed in TEM analysis of cortical neurons exposed to both control-EVs and Aβ42 protofibril-EVs (arrowheads, a, b). Immunocytochemical stainings, followed by fluorescence microscopy (c) and confocal microscopy (d) demonstrated that cholesterol deposits in neurons exposed to Aβ42 protofibril-EVs were situated in LAMP-1 positive compartments, verifying the presence of lamellar inclusions. Scale bar: (c) = 5 µm and (d) = 20 µm.
Discussion
Knowledge of the exact mechanisms behind neuronal atrophy in AD, as well as information about other cell types’ contribution to the neurodegeneration process is still very limited. To enable the development of effective therapeutic interventions for AD patients, a better understanding of the basic cellular mechanisms underlying the disease progression is crucial. In the present study we are clarifying the impact of EVs in Aβ-induced neurotoxicity. Taken together, our results show that EVs from Aβ42 protofibril exposed neuroglial co-cultures induce mitochondrial disturbances, synaptic loss and neuronal cell death in cortical neurons, indicating that EVs could play a critical role in the neurodegeneration process of AD.
Many lines of evidence suggest that Aβ pathology and inflammation are closely associated. Both reactive astrocytes and microglia are frequently found in the AD brain, especially around the plaques28, but their impact on AD progression remains elusive. While the formation of a glial capsule around the Aβ deposits may have a protective effect on the brain tissue by isolating the toxic Aβ species, the glial cells may also spread neurotoxic products to surrounding cells, for example via EVs8,29,30.
The aim of the present study was to elucidate in which ways EVs, released from the major brain cell types, are involved in Aβ-mediated neurodegeneration. For this purpose, EVs isolated from Aβ42 protofibril exposed neuroglial co-cultures were added to cortical neurons. Subsequently the viability of the neurons was analyzed, as well as their health status with respect to morphology, synapses and mitochondria. Astrocytes are the most abundant glial cell type in the human brain and they are crucial for maintaining tissue homeostasis31. Astrocytes have a wide variety of functions, including metabolic support of neurons, modification of synapse signaling, recycling of neurotransmitters, blood–brain barrier regulation and glymphatic clearance31,32,33. In the pathological brain, astrocytes are converted to a reactive, inflammatory state34. The impact of reactive astrocytes on AD progression is probably largely due to their uptake and release of substances from the microenvironment that they share with neurons. Astrocytes effectively engulf dead cells, damaged synapses and protein aggregates8,35,36,37,38,39. Interestingly, reactive astrocytes have been shown to be more efficient than microglia in taking up Aβ40. The fact that reactive astrocytes with high Aβ load are frequently found in the human AD brain, further confirms the importance of astrocytes in Aβ pathology35,38.
EVs are a heterogeneous group of secretory vesicles that are known to be crucial for cell-to-cell communication41. It has been shown that EVs can promote neuronal progression and regeneration, but also contribute to neurodegenerative diseases42. In the present investigation, endocytosis of EVs by cortical neurons was verified by time-lapse microscopy and TEM. Moreover, EV-mediated spreading of Aβ deposits was demonstrated, using immunocytochemistry and confocal imaging. The majority of the cells in the co-cultures, from which the EVs were isolated, were astrocytes that displayed large intracellular Aβ deposits. We have previously shown that astrocytes ingest very large amounts of Aβ42 protofibrils that are accumulated intracellularly in lysosomal compartments, rather than degraded8. Interestingly, the Aβ42 deposits co-localize with LAMP-1, but not with LysoTracker labelling, indicating that the Aβ42 containing lysosomes are not fully mature8. Importantly, the inability of astrocytes to clear Aβ leads to increased release of neurotoxic N-terminally truncated forms of Aβ in EVs8. It is likely that the secretion of EVs may serve as a clearing mechanism, in which the astrocytes try to get rid of toxic material, such as aggregated proteins that they have failed to degrade. Our previous analyses have further demonstrated that apoE is highly elevated in EVs isolated from Aβ42 protofibril exposed co-cultures19. In the present study, we show that Aβ42 protofibril exposure of neuroglial co-cultures results in the release of neurotoxic EVs. TUNEL assays demonstrated that neuronal cultures exposed to Aβ42 protofibril-EVs contained a significantly increased number of apoptotic nuclei at both 2 and 4 days, compared to cultures exposed to control-EVs. Interestingly, free-floating Aβ42 protofibrils (without EVs) were much less toxic than the Aβ42 protofibril-EVs. This can be explained by the fact that astrocytes in the co-culture only partly degrade the ingested Aβ42 protofibrils and instead secrete a more toxic, truncated form of Aβ aggregates8. Since EVs can be internalized by target cells through different pathways, including endocytosis, phagocytosis and macropinocytosis, it is possible that Aβ aggregates that are situated within EVs are more harmful to the neurons, because of their entering route17. Neuronal uptake of Aβ packed in EVs is probably more effective than uptake of free-floating aggregates and the ingested Aβ may also end up in different intracellular compartments and thereby induce more damage.
The function of mitochondria has been suggested to be severely compromised in AD. Mitochondria from AD brains have reduced membrane potential, increased permeability and express excessive levels of reactive oxygen species (ROS), the latter known to culminate in oxidative stress and cell damage43,44,45. Mitochondria also have an important role in the regulation of cell death. Upon apoptotic stimuli, mitochondria release several death factors, which may trigger neurodegeneration in AD46. Here, we show that Aβ42 protofibril-EVs induce mitochondrial disruption in neurons, suggesting that mitochondria are unable to maintain the energy homeostasis of the cell and are thus likely to undergo apoptosis. Quantification of neurons with healthy and unhealthy mitochondria showed that there was a significant decrease in the percentage of healthy mitochondria in neuronal cultures exposed to Aβ42 protofibril-EVs, compared to control-EVs. Moreover, TEM analysis verified that neurons exposed to control-EVs exhibited healthy mitochondria, while neurons exposed to Aβ42 protofibril-EVs did not. The mitochondria in these neurons were abnormal in size and displayed loss of structure of the outer membrane as well as the inner cristae.
Extensive research has been investigating the synaptic dysfunction in AD. In post-mortem human AD brains, a significant loss of important synaptic vesicle proteins, including synaptophysin, SV2 and p65 has been observed, particularly in the neocortex and hippocampus47,48. Furthermore, it has been shown that increased levels of Aβ oligomers correlate with the loss of synaptic markers in post-mortem frontal cortex samples from AD patients49. In the present study, we demonstrate that Aβ42 protofibril-EV exposure induces synaptic defects in cortical neurons. Analysis with TEM demonstrated that the Aβ42 protofibril-EVs induce axonal swelling and deposition of waste-filled vesicles in the neuronal processes. In addition, immunocytochemistry demonstrated that Aβ42 protofibril-EVs caused severe disruption of the neuronal network.
Interestingly, Aβ42 protofibril-EV exposed neurons displayed multiple enlarged vacuoles that appeared either empty or filled with diverse waste material, indicating that their degradation capacity was severely affected. Some of the vacuoles had the typical appearance of lipid droplets, but EV exposed neurons also showed the distinct feature of multilamellar bodies. Multilamellar bodies, also called lamellar inclusions, are multi-layered lysosomal organelles that store and secrete lipids as their main function. They can be found in various types of cells in the body and are for example frequent in the lung and epidermis. Interestingly, lamellar bodies also exist in the brain, but only under pathological conditions, such as Niemann-Pick disease, where lysosomal storage is severely affected26,27. Immunocytochemistry of the cortical neurons with the cholesterol-binding dye filipin III indicated that the lamellar bodies consisted of cholesterol deposits in lysosomal compartments.
Taken together, our data show that the secretion of EVs from Aβ42 protofibril exposed cells induces neuronal dysfunction in several ways, indicating a critical role for EVs in the progression of Aβ-induced pathology. A pathogenic role for EVs in spreading of aggregation-prone proteins has been implicated in various neurodegenerative diseases, including AD50,51. However, many questions remain to be answered, including the involvement of various cell types and how to interfere with the spreading. In conclusion, our analysis of the neurotoxic effects of Aβ42 protofibril-EVs in terms of synapse alterations, mitochondrial impairment, neuronal swelling, vacuolization, degradation defects and increased apoptosis strongly supports the hypothesis that EVs contribute to Aβ-induced pathology and we encourage further studies aimed to reverse or prevent the negative actions of EVs in order to limit disease propagation.
Data availability
All data is included in the article. Raw data for the mitochondria, TUNEL and Western blot graphs are available from the corresponding author upon request. All data generated or analyzed during this study are included in this article.
References
Prince, M. et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9, 63–75. https://doi.org/10.1016/j.jalz.2012.11.007 (2013).
Hardy, J. A. & Higgins, G. A. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992).
Walsh, D. M. et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002).
Lee, S. J., Nam, E., Lee, H. J., Savelieff, M. G. & Lim, M. H. Towards an understanding of amyloid-beta oligomers: characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 46, 310–323. https://doi.org/10.1039/c6cs00731g (2017).
Williams, T. L. & Serpell, L. C. Membrane and surface interactions of Alzheimer’s Abeta peptide–insights into the mechanism of cytotoxicity. FEBS J. 278, 3905–3917. https://doi.org/10.1111/j.1742-4658.2011.08228.x (2011).
Nixon, R. A., Yang, D. S. & Lee, J. H. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4, 590–599 (2008).
Mawuenyega, K. G. et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774. https://doi.org/10.1126/science.1197623 (2010).
Sollvander, S. et al. Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 11, 38. https://doi.org/10.1186/s13024-016-0098-z (2016).
Cataldo, A. M., Hamilton, D. J., Barnett, J. L., Paskevich, P. A. & Nixon, R. A. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J. Neurosci. 16, 186–199 (1996).
Steele, J. W. et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 18, 889–897. https://doi.org/10.1038/mp.2012.106 (2013).
Wang, Y., Cui, J., Sun, X. & Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 18, 732–742. https://doi.org/10.1038/cdd.2010.147 (2011).
Levy, E. Exosomes in the diseased brain: first insights from in vivo studies. Front. Neurosci. 11, 142. https://doi.org/10.3389/fnins.2017.00142 (2017).
Yanez-Mo, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesic. 4, 27066. https://doi.org/10.3402/jev.v4.27066 (2015).
Keller, S., Ridinger, J., Rupp, A. K., Janssen, J. W. & Altevogt, P. Body fluid derived exosomes as a novel template for clinical diagnostics. J. Transl. Med. 9, 86. https://doi.org/10.1186/1479-5876-9-86 (2011).
Thery, C., Amigorena, S., Raposo, G. & Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 22, 1. https://doi.org/10.1002/0471143030.cb0322s30 (2006).
Lasser, C. et al. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J. Transl. Med. 9, 9. https://doi.org/10.1186/1479-5876-9-9 (2011).
Mulcahy, L. A., Pink, R. C. & Carter, D. R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 3, 1. https://doi.org/10.3402/jev.v3.24641 (2014).
Hill, A. F. Extracellular vesicles and neurodegenerative diseases. J. Neurosci. 39, 9269–9273. https://doi.org/10.1523/JNEUROSCI.0147-18.2019 (2019).
Nikitidou, E. et al. Increased release of apolipoprotein E in extracellular vesicles following amyloid-beta protofibril exposure of neuroglial co-cultures. J. Alzheimers Dis. https://doi.org/10.3233/JAD-170278 (2017).
Perez-Garmendia, R. et al. Identification of N-terminally truncated pyroglutamate amyloid-beta in cholesterol-enriched diet-fed rabbit and AD brain. J. Alzheimers Dis. 39, 441–455. https://doi.org/10.3233/JAD-130590 (2014).
De Kimpe, L. et al. Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Abeta pathology at the lysosome. Age (Dordr) 35, 673–687. https://doi.org/10.1007/s11357-012-9403-0 (2013).
Englund, H. et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J. Neurochem. 103, 334–345. https://doi.org/10.1111/j.1471-4159.2007.04759.x (2007).
Magnusson, K. et al. Specific uptake of an amyloid-beta protofibril-binding antibody-tracer in AbetaPP transgenic mouse brain. J. Alzheimers Dis. 37, 29–40. https://doi.org/10.3233/JAD-130029 (2013).
Sehlin, D. et al. Large aggregates are the major soluble Abeta species in AD brain fractionated with density gradient ultracentrifugation. PLoS ONE 7, e32014. https://doi.org/10.1371/journal.pone.0032014 (2012).
Sollvander, S. et al. The Abeta protofibril selective antibody mAb158 prevents accumulation of Abeta in astrocytes and rescues neurons from Abeta-induced cell death. J. Neuroinflamm. 15, 98. https://doi.org/10.1186/s12974-018-1134-4 (2018).
Lajoie, P., Guay, G., Dennis, J. W. & Nabi, I. R. The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies. J. Cell Sci. 118, 1991–2003. https://doi.org/10.1242/jcs.02324 (2005).
Garcia-Sanz, P., Orgaz, L., Fuentes, J. M., Vicario, C. & Moratalla, R. Cholesterol and multilamellar bodies: Lysosomal dysfunction in GBA-Parkinson disease. Autophagy 14, 717–718. https://doi.org/10.1080/15548627.2018.1427396 (2018).
Orre, M. et al. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 35, 1–14. https://doi.org/10.1016/j.neurobiolaging.2013.07.008 (2014).
Sastre, M., Klockgether, T. & Heneka, M. T. Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int. J. Dev. Neurosci. 24, 167–176. https://doi.org/10.1016/j.ijdevneu.2005.11.014 (2006).
Joshi, P. et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 21, 582–593. https://doi.org/10.1038/cdd.2013.180 (2014).
Sofroniew, M. V. & Vinters, H. V. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. https://doi.org/10.1007/s00401-009-0619-8 (2010).
Eroglu, C. & Barres, B. A. Regulation of synaptic connectivity by glia. Nature 468, 223–231. https://doi.org/10.1038/nature09612 (2010).
Verkhratsky, A., Nedergaard, M. & Hertz, L. Why are astrocytes important?. Neurochem Res 40, 389–401. https://doi.org/10.1007/s11064-014-1403-2 (2015).
Pekny, M. et al. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 131, 323–345. https://doi.org/10.1007/s00401-015-1513-1 (2016).
Gomez-Arboledas, A. et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66, 637–653. https://doi.org/10.1002/glia.23270 (2018).
Loov, C., Mitchell, C. H., Simonsson, M. & Erlandsson, A. Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. Glia https://doi.org/10.1002/glia.22873 (2015).
Loov, C., Hillered, L., Ebendal, T. & Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 7, e33090. https://doi.org/10.1371/journal.pone.0033090 (2012).
Nagele, R. G., D’Andrea, M. R., Lee, H., Venkataraman, V. & Wang, H. Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 971, 197–209 (2003).
Lindstrom, V. et al. Extensive uptake of alpha-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell Neurosci. 82, 143–156. https://doi.org/10.1016/j.mcn.2017.04.009 (2017).
Nielsen, H. M. et al. Astrocytic A beta 1–42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia 58, 1235–1246. https://doi.org/10.1002/glia.21004 (2010).
Lee, S., Mankhong, S. & Kang, J. H. Extracellular vesicle as a source of alzheimer’s biomarkers: opportunities and challenges. Int. J. Mol. Sci. 20, 1. https://doi.org/10.3390/ijms20071728 (2019).
Janas, A. M., Sapon, K., Janas, T., Stowell, M. H. & Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta 1139–1151, 2016. https://doi.org/10.1016/j.bbamem.2016.02.011 (1858).
Parker, W. D. Jr. & Parks, J. K. Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology 45, 482–486 (1995).
Islam, M. T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 39, 73–82. https://doi.org/10.1080/01616412.2016.1251711 (2017).
Hamanaka, R. B. & Chandel, N. S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513. https://doi.org/10.1016/j.tibs.2010.04.002 (2010).
Moreira, P. I., Cardoso, S. M., Santos, M. S. & Oliveira, C. R. The key role of mitochondria in Alzheimer’s disease. J. Alzheimers Dis. 9, 101–110 (2006).
Masliah, E., Terry, R. D., DeTeresa, R. M. & Hansen, L. A. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 103, 234–239 (1989).
Scheff, S. W. & Price, D. A. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199. https://doi.org/10.1002/ana.410330209 (1993).
Pham, E. et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 277, 3051–3067. https://doi.org/10.1111/j.1742-4658.2010.07719.x (2010).
Rajendran, L. et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 103, 11172–11177. https://doi.org/10.1073/pnas.0603838103 (2006).
Saman, S. et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. https://doi.org/10.1074/jbc.M111.277061 (2012).
Acknowledgements
This study was supported by grants from the Swedish Research Council, the Swedish Alzheimer Foundation, the Swedish Brain Foundation, Åhlén Foundation, Stohnes Foundation, Gamla Tjänarinnor Foundation and Uppsala Berzelii Technology Centre for Neurodiagnostics. Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
C.B. optimized and performed experiments, interpreted data and wrote the manuscript; E.N. optimized and performed experiments, interpreted data and wrote the manuscript; L.S-G. performed experiments, interpreted data and revised the manuscript; M.I. interpreted data and revised the manuscript; D.S. interpreted data and revised the manuscript; A.E. designed and coordinated the study, interpreted data and wrote the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beretta, C., Nikitidou, E., Streubel-Gallasch, L. et al. Extracellular vesicles from amyloid-β exposed cell cultures induce severe dysfunction in cortical neurons. Sci Rep 10, 19656 (2020). https://doi.org/10.1038/s41598-020-72355-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72355-2
This article is cited by
-
Amyloid-β accumulation in human astrocytes induces mitochondrial disruption and changed energy metabolism
Journal of Neuroinflammation (2023)
-
Intracellular deposits of amyloid-beta influence the ability of human iPSC-derived astrocytes to support neuronal function
Journal of Neuroinflammation (2023)
-
Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology
Acta Neuropathologica Communications (2023)
-
Altered Distribution of SNARE Proteins in Primary Neurons Exposed to Different Alpha-Synuclein Proteoforms
Cellular and Molecular Neurobiology (2023)
-
ISGylation is induced in neurons by demyelination driving ISG15-dependent microglial activation
Journal of Neuroinflammation (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
