
Abstract
High-throughput AP-MS methods have allowed the identification of many protein complexes. However, most post-processing methods of this type of data have been focused on detection of protein complexes and not its subcomplexes. Here, we review the results of some existing methods that may allow subcomplex detection and propose alternative methods in order to detect subcomplexes from AP-MS data. We assessed and drew comparisons between the use of overlapping clustering methods, methods based in the core-attachment model and our own prediction strategy (TRIBAL). The hypothesis behind TRIBAL is that subcomplex-building information may be concealed in the multiple edges generated by an interaction repeated in different contexts in raw data. The CACHET method offered the best results when the evaluation of the predicted subcomplexes was carried out using both the hypergeometric and geometric scores. TRIBAL offered the best performance when using a strict meet-min score.
Similar content being viewed by others



Introduction
The discovery of protein complexes through high-throughput co-purification methods has increased the amount of available data to the extent that 43% of the reported protein complexes in interaction databases are estimated to be a result of this kind of experiments (see Supplementary Code). Traditionally, it has been argued that these methods produce high levels of noise1 although this claim has been contested2. Either way, complex detection from affinity-purification (AP) high-throughput (HT) data is not a straightforward process and to convert such data to a list of complexes demands the application of a series of post-processing steps that are still an open field of research3.
Raw data from an AP experiment is essentially a list of bait proteins mapped to all the prey proteins that they pulled out. Such a list is subject to false positives and false negatives (see Supplementary file, section 1, for a detailed review) and it is traditionally corrected by scoring the interactions according to different methods that measure the propensity of two proteins to interact given the background of interactions. Reliable interactions are integrated into a network which is then clustered to generate protein complexes3,4. These methods became very relevant as it was noticed that the differences between the conclusions of the first two main comprehensive maps of the yeast complexome were mainly a result of the pre-processing methods they employed3,5.
The way in which the scoring step is done has adopted a multiplicity of forms. The socio-affinity index (SA) scored the interaction between proteins i and j by including terms for how often i retrieves j and a term for how often pairs of proteins are seen together as preys. These were calculated as the log-odds of the number of times the proteins were observed together relative to what would be expected from their frequency in the data set6. Hart et al. postulated a scoring system based on the use of a hypergeometric distribution relative to a matrix model of interactions7. The Purification Enrichment score (PE) pointed out the limitants of the SA method, such as to include only positive evidence and not the inability of a protein to be identified by another and as being suitable mainly for cases where all proteins were both baits and preys. Alternatively, the authors used a naïve Bayes classifier, which estimates the probability of one hypothesis (interaction is reliable) relative to the probability of a second hypothesis (interaction is not reliable). The score was the log-ratio of these probabilities, computed using Bayes' theorem5. Finally, the Dice score was suggested as a simple alternative that focuses on comparing the co-purification patterns of two proteins across all different purification experiments; this is, constructing a pull-down matrix of proteins versus experiments and using a Dice index to compare each pair of protein profiles4. Additional scoring systems have been proposed in recent years8,9.
Regarding the clustering step, the options are even wider. The classical AP HT studies5,6 used methods such as Markov Clustering (MCL) and variations of Hierarchical Clustering3. However, many novel clustering methods have been proposed since then. We will review these methods below.
Finally, after scoring and clustering, the quality of the prediction strategy is commonly evaluated by comparison of the list of predicted complexes to a gold standard, that is, a manually curated database of protein complexes. A good agreement with this gold standard increases the confidence on the new complex predictions.
Protein subcomplex detection is an interesting special case of the more general complex prediction problem. A subcomplex can be defined as a functional (or predicted) complex which is a subset of a larger functional (or predicted) complex. In other words, the protein subunits of the subcomplex must be a subset of the protein subunits of the larger complex. Subcomplexes have been approached in different ways in the literature. One line of work depict them as clusters lying inside bigger network clusters, this is, the most connected region inside a bigger connected region, which is found using clustering strategies tailored for that purpose10. Other authors pay attention to the “cores” that repeat in several complexes and the “attachments” that make them different to each other6. Here the core of a core-attachment structure could be considered as a subcomplex. A similar approach focuses on studying multi-cluster and mono-cluster proteins after applying overlapping clustering algorithms to protein interaction networks11. Together with all these approaches, the “subcomplex” term can also be used to strictly define a biologically-relevant subcomplex, i.e., the subcomplexes that have been experimentally found to be functional and independent from the complex that contain them. Examples of subcomplexes are the TFIIK complex (related to the TFIIH complex), ADA-GCN5 complex (with SAGA complex), pre-replication complex (with replication complex) and ribonuclease MRP (with ribonuclease P). We will reserve the name “subcomplex” for these biologically meaningful macromolecular structures, while we will refer to “subcomplex predictions” to denote specific subcomplex representations.
The problem of subcomplex detection from an AP experiment is interesting due to the fact that we can find a high number of complex-subcomplex pairs in protein interaction databases. For example, the iRefIndex, which is a consolidation of 13 of the most popular protein interaction databases, contains 8145 cases of complex-subcomplex pairs12. But, at the same time, some of the algorithms used in the AP complex prediction pipeline, such as MCL, are non-overlapping clustering algorithms, making impossible any subcomplex detection. New methods that face the subcomplex detection problem have recently appeared but they need to be assessed and compared. The first line of research would be the incorporation of overlapping clustering algorithms to the complex detection pipeline. The second type of methods is based on the core-attachment model. Further on, we will introduce a third type of strategy in this paper and compare it to the other two.
Regarding the first type of methods employed to detect subcomplexes in AP data, there are many available clustering algorithms that could be applied to the networks resulting from the scoring step. Most have been proposed following the assumption that complexes can be reconstructed from highly densely connected regions in the network13: RNSC and MCL are some important examples14. A review of twelve of these algorithms15 claim that the best prediction method is “Infomap”, followed by “Fast modularity” and “Potts model approach”. However, it has also been argued that densely connected regions do not reflect functional units; hence, alternative ways to look at the complex prediction problem have been proposed13,16,17. In addition, complex prediction algorithms have also evolved from algorithms that generate non-intersecting or non-overlapping clusters to algorithms that take into account the fact that protein complexes share subunits with other complexes, i.e., overlapping community detection algorithms. Some of these methods are MCODE18, CFinder19, Link-communities10,20, OCG11, ClusterONE21 and RSRGM17.
Regarding the second type of methods that allow subcomplex detection, an early strategy to take into account the overlapping nature of complexes and apply it to complex identification from AP experiments was introduced by Gavin et al.6, who, after applying the SA score to AP data, performed repeated hierarchical clustering using different parameters to generate overlapping complexes that they described as composed of common “cores” and “attachments”. Two recent papers,22 and23, present a review of current clustering methods applied to protein complex identification. A group of methods, including CORE24, COACH25 and CACHET26, deserve a special mention for explicitly incorporating the above-mentioned “core-attachment” model (other methods include: Markov random fields27 and CODEC28).
In this paper, we will explore the detection of biologically relevant subcomplexes from AP data through some of the above-mentioned techniques and suggest a new strategy which might be able to improve them. We will evaluate the use of some recent methods that are able to detect “nested communities” or “hierarchies”, in order to prove whether or not these nested communities can detect biologically meaningful subcomplexes, while we introduce a new method to identify subcomplexes from AP data. We start from the premise that most of the previous procedures remove the multi-edge nature of pull-down data in an interaction network (i.e., bait-prey co-occurrences), which is the network signature of a subcomplex. For example, some scoring methods reduce all co-appearances of a bait and a prey protein to one single weighted interaction, whereas some clustering methods explicitly remove any subcomplex candidates. An example of this is the Leu4–Leu9 interaction from Gavin's dataset6: This interaction does not end up in any complexes when using the PE score, whilst the SA scores produce three complexes and the Dice score produces 2 complexes containing Leu4, Leu9 and a few more subunits. A Leu4–Leu9 complex does not appear in the results with any scoring method and, in fact, Leu4 and Leu9 are the subunits of the alpha-isopropylmalate synthase. This demonstrates how a highly scored copurification, which happens to be a complex, may get filtered out by the clustering method. Here we introduce TRIBAL (TRIad-Based ALgorithm), a novel method to identify subcomplexes from AP data that preserves and exploits this co-occurrence information.
Results
TRIBAL algorithm
In order to predict subcomplexes, we have designed a simple strategy that keeps the multi-edge information after the scoring and clustering steps, assuming that such information could include subcomplex information to some yet undetermined extent.
The first step of our algorithm is the generation of a pull-down matrix in order to compare purification patterns; this is similar to what is done in Zhang et al., but, instead of recording the purification or non-purification of a protein per experiment, we recorded the co-purification of pairs of proteins per experiment. This way, the Dice score is not used to compare patterns of purification between proteins but patterns of co-purification between pairs of proteins.
Each interaction between a bait and a pair of co-purifying preys will be scored using a Dice index and the ones above the cutoff (we will explain cutoff selection below) will be considered reliable.
Each reliable interaction (formed by three proteins) will be converted to a graph, using a spoke model representation, i.e., the bait will have an edge to each of the two prey proteins. All of these triads are integrated into a purification network. Unlike other methods, the conversion from triads of proteins to a network will generate multiple (repeated) edges. We have kept the multi-edge nature of this network, as this was our initial goal.
We then take one set of known or predicted complexes as a template and we insert the multiple edges inside the complexes. The proteins with multiple edges inside a complex will be predicted to be a subcomplex. Figure 1 summarizes these four steps.

TRIBAL algorithm.
In order to select the best cutoff value for the modified Dice score and the best template for the final step, the performance of the method was assessed by applying it to different templates and 11 different cutoff values of the modified Dice reliability score, ranging from 0.0001 to 0.2. This range was selected due to the size of the resulting PIN: Below 0.001, the size of the PINs does not increase, whereas, after 0.2, PINs then contain a small amount of edges (less than 1000). The best setup for TRIBAL (the one that maximizes the precision and the number of validated subcomplex predictions, while minimizes the size of the network) is the set of predicted complexes using Link communities clustering plus PE scoring, with a cutoff value of the reliability score of 0.05. In this case, TRIBAL predicts 18 subcomplexes with a precision of 100%. Details regarding template and cutoff selection can be found in the Supplementary file (section 3).
Complex and subcomplex predictions
Protein complex predictions using each combination of scoring and overlapping clustering methods were performed. First, we scored Gavin's raw data using four of the most popular scoring systems: The SA score6, the Hart score7, the PE score5 and the “Dice” score4 (see Methods). We used the cutoff values defined in the original papers (SA < 4, Hart > 0.01, PE < = 1.5, Dice < = 0.15) and we obtained four lists of reliable edges. The size of these lists varied from 6528 edges for Hart to 18278 for PE, including 14004 for SA and 16447 for Dice.
The edge lists were used to generate four Protein Interaction Networks (PINs). PIN-Dice contains 2192 nodes and 16447 edges, PIN-Hart contains 639 nodes and 6528 edges, PIN-PE contains 2344 nodes and 18278 edges and PIN-SA contains 2005 nodes and 14004 edges.
The four PINs were clustered using two different overlap-detecting clustering methods: “Link-communities”10,20 and “OCG”11. A description of the results can be observed in the Supplementary file (Supplementary Table 1).
For comparison purposes, we also constructed clusters by using traditional hierarchical clustering. Dice-H (Dice scoring and hierarchical clustering) produces 2293 communities, Hart-H produces 544 and SA-H produces 608.
Subcomplex predictions using the above-mentioned overlapping clustering strategies were generated by extracting all predicted complexes which were contained (meet-min = 1.0) by at least one other predicted complex. The result was a prediction of 102 subcomplexes by using Dice score and Link communities, 35 subcomplexes by PE score and Link communities, 67 subcomplexes by SA score and Link communities, 29 subcomplexes by Dice score and OCG, 7 subcomplexes by Hart score and OCG and 34 subcomplexes by PE score and OCG.
Subcomplexes predictions using the CACHET method were generated by removing all complexes without attachments and then selecting the cores of the remaining complexes, as explained in Methods. The result was 309 subcomplex predictions.
TRIBAL is a subcomplex prediction strategy, so all its predictions are subcomplexes. TRIBAL was applied to Gavin's dataset as explained in Methods, producing 18 subcomplex predictions. It is important to notice that the edges of the Dice-scored network are a subset of the edges of the PE-network, and, even more, TRIBAL's edges are a subset of the Dice-score (and, therefore, PE) methods. Therefore, TRIBAL seems to be generating a more strict network, with edges already validated for other methods, but with the difference that we keep the multiple edges that the other methods have removed.
Analysis of predictions
As explained in Methods, protein complex predictions were compared to a MIPS gold standard. For comparison purposes, we added examples using traditional hierarchical clustering. Table 1 summarizes the percentage of predicted complexes that can be found in the MIPS reference set (i.e., the Precision or PPV) and the percentage of MIPS complexes that can be found at the predicted set (i.e., the Recall or Sensitivity), when using a p-value smaller than 0.05 (after a hypergeometric test) to determine a match. Some researchers might be more interested in a few high precision results (despite having a high number of false negatives or missing hits) and some others might be interested in a high recall (many results, despite having a high number of false positives). However, it is a common practice to find a balance between false positives and false negatives, or between precision and recall. There are several computational methods to do that and two of the most popular are the F-measure and the area under the ROC curve. In the following tables, we show the first one. Results suggest that PE schemes offer the best precision while OCG outperforms Link communities in terms of recall. Therefore, it is the PE score plus OCG clustering which offers the best F-measure.
Other studies use different methods to determine what a match is. For example, Wu et al.26 use a “geometric index” (also called NA-score) smaller than 0.2. The results under this alternative method can be found in Table 2. In this case, results partially contradict the previous analysis and now Link communities has the best performance, with the best F-measure belonging to a combination of Dice score and Link communities clustering. We hypothesize that the reason of this difference might be the fact that the hypergeometric score is more sensitive to the size of the complexes and OCG is predicting complexes of a size larger than expected, as it can be observed in the Supplementary file (section 4).
However, our main interest is not to evaluate the ability to identify complexes but the ability to identify subcomplexes. We begin by verifying that all datasets under study contain subcomplexes: The MIPS dataset (our reference set) includes 16 subcomplexes. Raw data includes 348 groups of proteins contained in others. None of the sets produced by hierarchical clustering display subcomplexes, as these are non-overlapping methods. The predicted sets (Dice-lcomm, PE-lcomm, SA-lcomm, Dice-OCG, Hart-OCG, PE-OCG, CACHET and TRIBAL) display between 7 and 309 subcomplexes.
In order to evaluate the subcomplex detection abilities of these methods, we performed two different analyses. Table 3 summarizes the precision and recall analysis for predicted subcomplexes when compared to the full MIPS dataset, using a hyper-geometric score as a criterion for a match. Table 4 does the same when using the geometric score. The results show that CACHET has the best F-measure in both cases. Despite the fact that TRIBAL has the best precision in both cases, CACHET has a far better recall which ultimately leads to a better F-measure.
Meet-min index evaluation
The hyper-geometric index, likewise the Jaccard index, the geometric index and the Dice score, is designed to measure similarity between two sets. However, we are not only interested in the similarity; we are also interested in the fact that one set contains the other. In this case, the meet-min index is the best validation criterion, as discussed in Figure 3. A meet-min value of 1.0 indicates that the smaller complex is a subset of the larger and (1 – meetmin) gives the proportion of proteins in the subcomplex left out of the complex.

Number and percentage of validated predicted subcomplexes using TRIBAL and six other methods.
TRIBAL outperforms CACHET and all combinations of scoring strategies and overlapping clustering methods, for a meet-min equal to 1.0, that is, in terms of perfect containment of a subcomplex by a reference complex. This aplies to both (a) the number of validated subcomplexes and (b) the precision or percentage of validated subcomplexes.

An example of similarity and containment metrics.
The Jaccard and Geometric indexes are able to measure the similarity between two sets. The higher Jaccard and Geometric indexes indicate that the two sets in (a) are more “similar” to each other than the two sets in (b). In opposition, the Meet-min is a better measure of containment. The higher meet-min index shows that the two sets in (b) are a perfect set and subset, while the two sets in (a) are only overlapping. The scores show that case (a) is an example of a good similarity with a not so good containment, while (b) is an example of a good containment with a poor similarity.
For this reason, we use the meet-min index to verify that predicted subcomplexes are not only similar but contained by MIPS complexes.
Figure 2 depicts the number and percentage of validated predicted subcomplexes for TRIBAL, CACHET and six other methods which combine different scoring and clustering systems, using the meet-min index as comparison criterion. The results show that TRIBAL outperforms all other methods for meet-min equal to 1.0. CACHET shows a good performance to low meet-min values, but it decreases strongly when meet-min increases.
Analysis of subcomplexes
In order to understand the reasons of the inferior performance of the overlapping clustering methods, we studied the structure and identity of their predicted subcomplexes.
Initially, we identified some real biological complex-subcomplex pairs among the complexes in MIPS, including:
-
1
TIM22 complex with TIM9-TIM10 complex
-
2
TFIIH complex with TFIIK complex
-
3
SAGA complex with ADA-GCN5 complex
-
4
Replication complex with: pre-replication complex, replication initiation complex, post-replication complex, DNA polymerase deltaIII, DNA polymerase epsilonII and DNA polymerase zeta.
-
5
Ribonuclease P with ribonuclease MRP
-
6
Cytoskeleton with: microtubules and tubulin-associated proteins
However, when reviewing the complexes predicted by link-communities and PE, we found real subcomplexes that the method fails to predict and false subcomplexes that the method is predicting. For instance:
-
1
NUP84-NPC subcomplex: This is a subcomplex of the nuclear pore complex (NPC). The algorithm does not predict this; instead, it predicts three other subcomplexes which are truncated versions of NUP84-NPC.
-
2
GLE2-NUP116-NUP82 subcomplex: This is also a subcomplex of the NPC and has a biologically relevant subcomplex which is NUP82. The algorithm does not detect these two but two truncated versions of GLE2-NUP116-NUP82 instead.
-
3
NUP57 subcomplex: Similar to NUP84-NPC, the algorithm detects one truncated version of NUP57 instead of the relationship to the NPC.
-
4
Polymerase-alpha-primase complex: This one contains two real subcomplexes: DNA primase and DNA polymerase alpha. The algorithm identifies two truncated versions of Polymerase-alpha-primase instead.
-
5
TREX complex has a subcomplex called THO complex and in turn, there is an extended version of TREX that contains it. However, the algorithm shows three truncated versions of TREX.
-
6
alpha-alpha-trehalose-phosphate-synthase complex: This one contains one subcomplex called TPS1-TPS2 complex (trehalose biosynthetic pathway). The algorithm instead detects two truncated versions of the larger complex.
-
7
The NuA4-HAT complex contains a few real subcomplexes: EAF3-EAF5-EAF7 subcomplex, ACT1-ARP4-SWC4-YAF9 subcomplex and ESA1-EPL1-YNG2-EAF6 subcomplex. The algorithm instead detects two versions of NuA4-HAT.
Details of the previous comparisons can be found in Supplementary Code.
At the same time, OCG produced complexes and subcomplexes of a very large size. While the average size of a complex in MIPS is 6.3 subunits, the predicted subcomplexes display the following average sizes: TRIBAL = 5.61, CACHET = 5.58, Dice + lcomm = 4.71, PE + lcomm = 4.69, SA + lcomm = 4.25, raw data = 4.00, Dice + OCG = 24.93 and PE + OCG = 24.00. The large size of the OCG complexes allows them to intersect many subcomplexes. Such complexes do not have a biological meaning but our first comparison method (hyper-geometric) seems to be favouring this case. A complete description of the size distribution of complexes and subcomplexes can be found in the Supplementary file (section 4).
In summary, results suggest that Link-communities's predicted subcomplexes are mainly truncated versions of a well-predicted complex, while OCG's subcomplexes lack biological meaning.
Discussion
We have unveiled the limitants of AP-MS processing methods regarding their ability to predict subcomplexes and we have also suggested TRIBAL as an alternative solution to this.
We have traditionally assumed that the fact that a pair of proteins shows multiple evidence of co-purification and few evidences of independent purification is a demonstration of the confidence that we can have in the quality of such an interaction. However, here we start from the assumption that it could also mean that the interaction can be repeated in multiple overlapping complexes. The current complex detection pipeline discards the second type of information because the scoring step converts all co-purifications to one interaction record. For this reason, a method to score interactions without removing the information of proteins co-purifying in different contexts (i.e. different complexes), could be useful.
The scoring of triads of proteins instead of interactions has been our first attempt to design such a method. This is, instead of studying the pattern of co-purification and independent purification of a bait and a prey, we study the bait and pairs of preys. This way, we apply the Dice score to a modified pulldown matrix which contains the baits versus the pairs of preys that copurify with them. After removing low quality data and generating a PIN with the high quality triads, we also get a PIN with multiple edges.
We tested and compared the performance of different combinations of scoring methods (Dice, PE, Hart, SA) and overlapping clustering algorithms (Link-communities, OCG), with one method based on the core-attachment model (CACHET) and our own method (TRIBAL). All these three types of methods are based on different assumptions; for example, that subcomplexes can be extracted from high-degree (highly connected) regions of the network or, in our case, that co-purification information with different baits is not only useful as a data quality measure but can also include co-complex information in different biological contexts. These assumptions can be partially validated through the performances of the methods. The results show that CACHET gives the best results in terms of similarity (geometric or hypergeometric indexes) to the MIPS reference set (this is mainly due to its better recall), while TRIBAL offers the best precision in terms of similarity and the best performance in terms of containment metrics (meet-min index). From the group of overlapping clustering methods, Link-communities shows better results than OCG.
Regarding limitations, the nature of the TRIBAL method leads to a small number of predictions. New strategies to solve this limitation and address the subcomplex problem will be needed. The possibility of replacing the use of templates with clustering algorithms that do not find dense modules but functional subunits, or the possibility of using clustering algorithms designed for multiple edges, should be considered. Another important limitation is the lack of a gold standard or reference set specifically designed for subcomplexes.
We have made a thorough analysis of the traditional complex prediction pipeline for AP-MS experiments. Besides problems due to coverage, false positives and inconsistencies regarding mutual pull-down (see Supplementary File), most traditional methods are not able to detect subcomplexes due to decisions made in the scoring and clustering steps. Based on this knowledge, we have identified the best strategies to detect subcomplexes, including the development of TRIBAL, a simple strategy that improves the precision of subcomplex prediction compared to previous methods. These strategies are the initial attempts to specifically address the subcomplex detection problem in co-purification data. This paper suggests that overlapping clustering methods fail to detect subcomplexes from AP data while the core-attachment model used by CACHET and similar software seems to be the best option to this date. However, it also suggests that alternatives such as the co-purification matrix introduced by TRIBAL deserve more attention, as they show potential thanks to a high precision, especially when we evaluate the meet-min index, i.e., subcomplexes absolutely contained by larger complexes. Finally, research in subcomplex identification demands the generation of curated gold standards for subcomplexes, which will be an important step to achieve a better validation of predictions.
Methods
All analyses in this paper were performed using R v.2.15.2 and some of its packages, including “iRefR” v.1.0012, “igraph” v.0.64 and “org.Sc.sgd.db” v.2.9.1. All code needed to reproduce the following analyses can be found as Supplementary code.
Purification data
The raw purification dataset used in this study was taken from the Tandem Affinity Purification study of Gavin et al.6 on S.cerevisiae, available as supplementary material of their paper. It consists of 2166 experiments, using 1849 baits in one experiment each and 143 baits in 317 experiments repeated two or more times. A degree distribution of the purified proteins shows a power-law-like distribution, with a few high-degree nodes (Apa1, Cic1, Hhf1, Mak21, Psa1 and Pwp2) and a greater number of low-degree nodes, including 254 baits only purifying themselves. 912,333 out of 2,344,695 pairs of purified groups display some overlap of one or more proteins, while 601 out of 2,344,695 pairs display a meet-min index of 1, indicating that one is a subset of the other one.
The “org.Sc.sgd.db” R library was used to convert SGD IDs to gene names.
Scoring indexes
After data is collected, it is scored using the four above-mentioned popular scoring methods. The SA score is defined as follows:

where

and

Here A denotes the socio-affinity index, S is a spoke model-related term and M is a matrix model-related term.
-
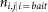 is the number of times that protein i retrieves protein j when i is tagged.
is the number of times that protein i retrieves protein j when i is tagged. -
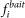 is the fraction of purifications where i was a bait.
is the fraction of purifications where i was a bait. -
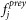 is the fraction of all retrieved preys that were j.
is the fraction of all retrieved preys that were j. -
nbait is the total number of purifications (i.e., of baits).
-
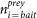 is the number of preys retrieved with protein i as bait.
is the number of preys retrieved with protein i as bait. -
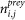 is the number of times that i and j co-purify with baits different to i or j.
is the number of times that i and j co-purify with baits different to i or j. -
nprey is the number of preys observed with a specific bait (excluding itself).
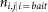 is the number of times that protein i retrieves protein j when i is tagged.
is the number of times that protein i retrieves protein j when i is tagged.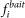 is the fraction of purifications where i was a bait.
is the fraction of purifications where i was a bait.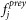 is the fraction of all retrieved preys that were j.
is the fraction of all retrieved preys that were j.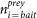 is the number of preys retrieved with protein i as bait.
is the number of preys retrieved with protein i as bait.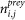 is the number of times that i and j co-purify with baits different to i or j.
is the number of times that i and j co-purify with baits different to i or j.The Hart score makes use of a hypergeometric test to compute the probability of an interaction being observed at random:

where

Here k is the number of times the interaction is observed (success sample), n is the total number of interactions for the first protein (sample), m is the total number of interactions for the second protein (success population) and N is the total number of interactions in the full data set (population). Here, the interactions assume a matrix model.
The PE score is computed as follows:

where




Here S is a spoke-term, M is a matrix-term, i is a bait, j is a prey and k is a purification. r is the probability of a true association to be detected, while pijk is the probability of i and j being observed together for non specific reasons. For details regarding the estimation of these parameters, we refer the reader to Collins et al.5.
The Dice score is defined as follows:

Where i and j are two proteins vectors, with values of 1 for each experiment where that protein was pulled down as a prey and a value of zero otherwise. q is the number of elements (experiments) where both i and j have 1's (appear in the same purification), r is the number of elements where i has 1 and j has 0 and s is the number of elements where i has 0 and j has 1, this is, (r + s) is the number of cases where the i-j pair does not co-purify.
SA, Hart, PE and Dice scores applied to the raw data and to the clustering methods below specified, were computed using the ProCope software29.
Our modified Dice score for the TRIBAL algorithm is explained in Results.
Clustering methods
Link-communities is a method that uses communities of edges and not of nodes. OCG hierarchically merges edges into modules, checking the value of a special modularity function Q. Both OCG and Link-communities were performed using the “linkcomm” R package20.
CACHET
The 369 complexes generated by CACHET from Gavin's dataset, were retrieved from their web page26.
In order to generate subcomplexes, we selected all complexes containing both core and attachments (i.e., we removed all only-core complexes). Subcomplexes were defined as the cores of those core-attachment sets.
Evaluation methods
In order to evaluate the quality of the complex predictions, we used the S.cerevisiae complex MIPS dataset as a gold standard and compared it to every predicted complexes dataset. We highlight that alternative gold standard sets have been proposed30; however, the MIPS data set is still used as a validation instrument for the complex detection tools above mentioned17,21,29.
The comparison is done through three strategies: Firstly, through a hypergeometric test, as defined in eq. (5). In this test, the population is the total number of proteins, the population success is the size of the MIPS complex, the sample is the predicted complex (predicted either by any combination of the scores and clustering algorithms here employed, CACHET or TRIBAL) and the success sample is the size of the intersection between MIPS and the predicted complex. The overlap is considered significant when p-value < 0.05.
Secondly, the geometric index, defined as follows:

Where one complex is predicted and the other one belongs to the reference set. The overlap is considered significant when Geom > 0.2.
Thirdly, the meet-min index, defined as follows:

Where one complex is predicted and the other one belongs to the reference set. A meet-min index of 1.0 indicates that one complex perfectly contains the other one.
The relationship and distinction between the meet-min and the other metrics can be observed in figure 3.
The performance of each method was assessed by computing their precision, recall and F-measure, defined as follows:



Where TP = True positives (matches), FP = False positives (mispredictions) and FN = False negatives.
TRIBAL algorithm
Both TRIBAL and its evaluations were coded using R 2.15.2 and its packages “iRefR” and “igraph” and they are available as Supplementary code.
References
von Mering, C. et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 399–403 (2002).
Yu, H. et al. High-quality binary protein interaction map of the yeast interactome network. Science 322, 104–10 (2008).
Wodak, S. J., Pu, S., Vlasblom, J. & Seraphin, B. Challenges and rewards of interaction proteomics. Mol Cell Proteomics 8, 3–18 (2009).
Zhang, B., Park, B. H., Karpinets, T. & Samatova, N. F. From pull-down data to protein interaction networks and complexes with biological relevance. Bioinformatics 24, 979–86 (2008).
Collins, S. R. et al. Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Mol Cell Proteomics 6, 439–50 (2007).
Gavin, A. C. et al. Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–6 (2006).
Hart, G. T., Lee, I. & Marcotte, E. R. A high-accuracy consensus map of yeast protein complexes reveals modular nature of gene essentiality. BMC Bioinformatics 8, 236 (2007).
Xie, Z., Kwoh, C. K., Li, X. L. & Wu, M. Construction of co-complex score matrix for protein complex prediction from AP-MS data. Bioinformatics 27, i159–66 (2011).
Yu, X., Ivanic, J., Wallqvist, A. & Reifman, J. A novel scoring approach for protein co-purification data reveals high interaction specificity. PLoS Comput Biol 5, e1000515 (2009).
Ahn, Y. Y., Bagrow, J. P. & Lehmann, S. Link communities reveal multiscale complexity in networks. Nature 466, 761–4 (2010).
Becker, E., Robisson, B., Chapple, C. E., Guenoche, A. & Brun, C. Multifunctional proteins revealed by overlapping clustering in protein interaction network. Bioinformatics 28, 84–90 (2012).
Mora, A. & Donaldson, I. M. iRefR: an R package to manipulate the iRefIndex consolidated protein interaction database. BMC Bioinformatics 12, 455 (2011).
Wang, Z. & Zhang, J. In search of the biological significance of modular structures in protein networks. PLoS Comput Biol 3, e107 (2007).
Brohee, S. & van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7, 488 (2006).
Lancichinetti, A. & Fortunato, S. Community detection algorithms: a comparative analysis. Phys Rev E Stat Nonlin Soft Matter Phys 80, 056117 (2009).
Pinkert, S., Schultz, J. & Reichardt, J. Protein interaction networks--more than mere modules. PLoS Comput Biol 6, e1000659 (2010).
Zhang, X. F., Dai, D. Q., Ou-Yang, L. & Wu, M. Y. Exploring overlapping functional units with various structure in protein interaction networks. PLoS One 7, e43092 (2012).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2 (2003).
Adamcsek, B., Palla, G., Farkas, I. J., Derenyi, I. & Vicsek, T. CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics 22, 1021–3 (2006).
Kalinka, A. T. & Tomancak, P. linkcomm: an R package for the generation, visualization and analysis of link communities in networks of arbitrary size and type. Bioinformatics 27, 2011–2 (2011).
Nepusz, T., Yu, H. & Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods 9, 471–2 (2012).
Li, X., Wu, M., Kwoh, C. K. & Ng, S. K. Computational approaches for detecting protein complexes from protein interaction networks: a survey. BMC Genomics 11 Suppl 1, S3 (2010).
Srihari, S. & Leong, H. W. A survey of computational methods for protein complex prediction from protein interaction networks. J Bioinform Comput Biol 11, 1230002 (2013).
Leung, H. C., Xiang, Q., Yiu, S. M. & Chin, F. Y. Predicting protein complexes from PPI data: a core-attachment approach. J Comput Biol 16, 133–44 (2009).
Wu, M., Li, X., Kwoh, C. K. & Ng, S. K. A core-attachment based method to detect protein complexes in PPI networks. BMC Bioinformatics 10, 169 (2009).
Wu, M., Li, X. L., Kwoh, C. K., Ng, S. K. & Wong, L. Discovery of protein complexes with core-attachment structures from Tandem Affinity Purification (TAP) data. J Comput Biol 19, 1027–42 (2012).
Rungsarityotin, W., Krause, R., Schodl, A. & Schliep, A. Identifying protein complexes directly from high-throughput TAP data with Markov random fields. BMC Bioinformatics 8, 482 (2007).
Geva, G. & Sharan, R. Identification of protein complexes from co-immunoprecipitation data. Bioinformatics 27, 111–7 (2011).
Krumsiek, J., Friedel, C. C. & Zimmer, R. ProCope--protein complex prediction and evaluation. Bioinformatics 24, 2115–6 (2008).
Pu, S., Wong, J., Turner, B., Cho, E. & Wodak, S. J. Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res 37, 825–31 (2009).
Acknowledgements
The authors would like to acknowledge the assistance provided by the Office of the Deputy Vice Chancellor for Research and Graduate Studies (NRF Grant Ref. No. 21T021 and 31T046) at the United Arab Emirates University (UAEU). AM also thanks the Institute of Clinical Physiology (CNR, Pisa, Italy) for hosting him during the end of this project.
Author information
Authors and Affiliations
Contributions
N.Z. and A.M. contributed to the conceptual idea of the study and directed the writing of the manuscript. A.M. and N.Z. conceived and designed the experiments. A.M. performed the experimental work. A.M. and N.Z. analyzed the results. N.Z. and A.M. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary file
Supplementary Information
Supplementary Code
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zaki, N., Mora, A. A comparative analysis of computational approaches and algorithms for protein subcomplex identification. Sci Rep 4, 4262 (2014). https://doi.org/10.1038/srep04262
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04262
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
