
Abstract
Short indicator RNA sequences (<100 bp) persist after autoclaving and are recovered intact by molecular amplification. Primers targeting longer sequences are most likely to produce false positives due to amplification errors easily verified by melting curves analyses. If short indicator RNA sequences are used for virus identification and quantification then post autoclave RNA degradation methodology should be employed, which may include further autoclaving.
Similar content being viewed by others

Introduction
Quantitative PCR is a commonly used technique in microbiology as it employs a rapid, sensitive and specific detection of microbes that are difficult to culture or are non-culturable. One of the major limitations of this technique is the inability to differentiate between active and inactive viruses. Fully or partially intact genomes of inactivated viruses may produce positive PCR signals1,2,3,4. This can lead to overestimation of the quantity of active viruses in samples.
Autoclaving at 121.1°C induces cell and viral particle lysis and failure to recover viable organisms is considered as an evidence of its efficiency5. Degradation of genetic material is also assumed to occur and thus often PCR methods are used as verification in experimentation with autoclaved materials including autoclaved waters and wastewaters6. While many of these PCR methods target relatively larger indicator gene sequences, occasionally, most commonly for viruses, the target indicator genetic sequence may be a shorter sequence7,8,9,10,11,12. If degradation is incomplete, PCR signals due to non-degraded RNA sequences and false positive signals may confound subsequent experimentation and PCR based tests.
Previous reports have indicated that eukaryotic DNA13,14 or enveloped viruses DNA and RNA15,16 might survive standard autoclaving. These reports targeted sequences >100 bps and did not include sequencing verification of amplicons. However indicator viral RNA sequences, especially for non-enveloped viruses, are often short. If the longer sequences were more sensitive to degradation and fragmentation then it would be expected that shorter sequences might preferentially persist.
We have comprehensively verified the post-autoclave persistence of non-degraded RNA sequences of variable lengths for MS2 bacteriophage (ATCC® 15597-B1™) using quantitative reverse transcriptase PCR (qRT PCR) followed by verification of the amplicons melting behavior, their sequence size and identity. For this we employed 16 primer sets that were expected to produce amplicons in the 70 bp to 2953 bp range and a series of three autoclaving cycles. Following quantitation by qRT PCR putative sequence identity was verified by melting curve analysis and gel electrophoresis amplicon size visualization. Amplicons were cloned into plasmids and their identity further confirmed by sequencing. MS2 is an icosahedral positive sense-, single stranded, non-enveloped RNA coliphages17 and is a common viral surrogate used in environmental water quality research8.
Results
After the 1st autoclave cycle plaque assays were negative, confirming inactivation of MS2. However three commonly used indicator primer sets7 (listed here as A, B and C, Figure 1 and Supplementary materials), producing amplicons in the 70 to 77 bp range, permitted qRT PCR quantification at 0.18%, 0.015% and 0.009% respectively, of the original concentration. After the 2nd and 3rd autoclave cycles the genetic material degraded such that qRT PCR quantification could only be obtained after more than 35 quantification cycles (Cq) (Figure 1.i.). Primer set A targeted the RNA replicase β chain while primer sets B and C targeted assembly proteins on the RNA genome of MS2 bacteriophage7.

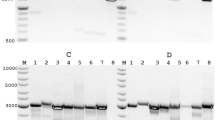
Quantification and quality control steps for three amplicons that were consistently recovered after repeated autoclaving.
(A), (B) and (C) indicate amplicons commonly employed as indicator sequences for the presence and quantification of MS2 coliphage7. Numeric subscripts indicate the number of autoclave cycles (e.g. A1, B1, C1). If no numeric subscript is present then the capital letter indicates results obtained from non-autoclaved positive controls. 1.i. The horizontal line represents the threshold Cq 40. The quantification cycles of A2 & A3, B2 & B3 and C2 & C3 are overlapping. 1.iv. Plasmid extracts are indicated by the subscript “p” (e.g. pA1, pB1, pC1). 1.v. is a paired comparison of A1, B1, C1 (bottom sequence in each pair) to A, B, C (top sequence in each pair). The first two pairs are sequenced in the 3′-5′ direction and the third pair in the 5′-3′ direction. The gels shown in this image have been cropped for convenience. The borders between separate gels have been marked with a black line.
After one autoclaving, amplicons in the 100 through 900 bps range (nine primer sets, D, E, F, G, H, I, J, L and O) could either not be quantified via qRT PCR or required over 35 Cq's (Supplementary information, Table 1). Moreover the quantifiable amplicons were shorter than expected (<100 bps and as short as ~50 bps; see Supplementary Figure 3) and generally melted at lower temperatures. While for the non-autoclaved controls these primers produced amplicons with melting peaks (Tm) in the range of 80–90°C, after autoclaving they produced amplicons exhibiting melting peaks in the range of 65–80°C (see Supplementary Materials). The positive controls for the amplicons obtained with these nine primer sets were 100% identical to their expected sequence whereas all the autoclaved controls were unable to be sequenced because of their extremely small size. An exception was a medium sized amplicon (I, 335 bps) that, while barely recoverable after autoclaving (>35 Cq's) could still be recovered as intact sequence after autoclaving. Another four targeted sequences (primer sets, K, M, N and P), aimed at producing amplicons longer than 900 bps, while amplifiable in-vitro, could not be amplified in-vivo even from the positive control.
No non-specific amplification was observed after the 1st autoclave cycle for any of the A, B and C amplicons as described by the peak melting temperature alignment. The Tm of the positive controls and the quantified amplicons from these 3 primer sets were in the same range of 75–85°C (Figure 1. ii.). The amplicon sizes for these three primer sets were confirmed at <100 bps by agarose gel electrophoresis (Figure 1. iii.). After the 2nd and 3rd autoclave cycles recovered amplicons A produced only very diffuse wide bands on the electrophoresis gels. Diffuse electrophoretic bands were not observed for amplicons obtained from the non-autoclaved control samples.
The three short amplicons A, B and C obtained after the 1st autoclave cycle as well as their respective positive controls were cloned and sequenced. Restriction digests of plasmids confirmed inserts shorter than 100 bp (Figure 1. iv.). Presence of the inserts in the plasmids was also verified by PCR (Figure 1. iv.). Subsequent sequencing confirmed the identity of the amplified sequences (Figure 1. v.). This indicates that short RNA regions were not completely degraded after the 1st cycle of autoclaving and could still be quantified by qRT PCR.
Discussion
These results suggests that short MS2 indicator gene sequences, of less than 100 bp, commonly used to quantify MS2 abundance7,8,9,10,12, may lead to false abundance estimates. A combination of culture based plaque assay and possibly targeting of larger indicator sequences in qRT PCR would therefore be recommended for an accurate quantitation of virions in environmental samples. Pecson et al.3 have shown that short heating events, 72°C for 3 min, do not eliminate MS2 amplifiable genomic fragments of 240–340 bp range. However simple PCR amplification does not guarantee an amplicon's identity. Susceptibility to degradation through heat of different genomic regions varies due to both the particularities of the secondary structure and the level of genomic association with proteins3. The genome of MS2 has a complex secondary structure18. Melting temperatures (Tm) and the level of energy required to degrade secondary structures, expressed as changes in the Gibbs free energy (ΔG), provide a qualitative thermodynamic understanding of the structure of the amplicons. As expected amplicons'ΔG values had a significant positive correlation with their Tm (adjusted R2 = 66.8%, p = 0.001) and length (adjusted R2 = 95.4%, p < 0.001). Our results suggest that longer amplicon sequences have a tendency to produce non-specific amplifications evident on electrophoresis gels and through melting curve analyses (see Supplementary materials) but that shorter RNA fragments are not completely degraded by autoclaving and may be recovered intact by molecular amplification techniques. However while most of the tested > 100 bps amplicons were likely fragmented (see gel sizing in Supplementary Figure 3) at least one of the medium sized amplicon (I) was still recoverable at the expected size, supporting some of the earlier reports obtained for eukaryotic DNA13,14 or enveloped viruses DNA and RNA15,16 and with its sequence intact as confirmed by Sanger sequencing (data not shown). Nevertheless all > 100 bps amplicons required > 35 Cq's to be quantified.
Methods
Study design
Ten mL of the MS2 virus stock of 1010 pfu mL−1 was autoclaved three times with 3 mL of the sample removed after each autoclave cycle. Each autoclave cycle was verified to maintain a temperature of 121°C and pressure of 21 psi (144.8 kPa) for 20 minutes. Each autoclave cycle took a total of 50 minutes to complete. Non-autoclaved virus culture was used as positive control. Detection and enumeration of the virus was carried out by plaque assay and qRT PCR. The latter employed multiple primer sets that targeted 16 different RNA regions of the MS2 genome. The identity of the amplified PCR products was verified by cloning and subsequent sequencing. All experiments were carried out in triplicate.
MS2 bacteriophage
MS2 bacteriophage stock was obtained from the laboratory of Dr. C. Gerba at the University of Arizona and propagated using the double layer agar USEPA method 1601 (2001). Escherichia coliHS (pFamp)R host strain was purchased from American Type Culture Collection (ATCC-700891).
Primer design
A total of 16 different primer sets were selected based on amplicon length, GC% and melting temperature to produce amplicons in the 69 to 2953 base pairs range (Methods Table 1). MS2 bacteriophage has a genome size of 3569 bp18. Primers were synthesized by Eurofins MWG Operon (Huntsville, AL).
Quantitative reverse transcriptase polymerase chain reaction (qRT PCR)
MS2 target copies were estimated by qRT PCR. RNA was extracted with the Invitrogen PureLink RNA/DNA viral extraction kit (Invitrogen, Carlsbad, CA) without the use of carrier RNA according to the manufacturer instructions. Extracts were stored at −80°C and qRT PCR was performed within 48 h of sampling. The iScript TM One-Step qRT PCR kit with SYBR Green (Bio-Rad, Hercules, CA) was used for the assay. Primers were added to a final concentration of 400 nM. The reaction was set up for 25 μL with 5 μL of template RNA. Non-template controls (NTC's) were included, where RNA template was replaced by nuclease free water. Assays were carried out on a Bio-Rad CFX Connect™ detection system (Biorad, Hercules CA). Thermocycling was carried out with an initial cycle of 50°C for 10 min to synthesize cDNA and 95° C for 5 min followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. This was followed by development of melting curves from 65°C to 95°C with 0.05 seconds incremental steps of 0.5°C. All tests were run in triplicate.
RNA standards were prepared from total genomic RNA extracted from a high titer of purified MS2. The RNA extracted was quantified as 40 ng μL−1 with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). This was diluted into four 10-fold dilution standards ranging between 1 ng μL−1 and 1 pg μL−1. A genome size of 3,569 nucleotides with an average molecular mass of 330 Da per nucleotide7 was assumed. Thus a 1 ng μL−1 RNA was calculated to contain 5 × 108 copies μL−1. The quantification cycle (Cq) values of all 15 sets (4 set of standards and 1 NTC with 3 replicates each) were averaged using the Bio-Rad's CFX Connect software. The standards were run using primer sets A, B and C with efficiencies of 91.7%, 99.7% and 98.2% and R2 of 0.9928, 0.9991 and 0.9951 respectively. A linear regression master curve was created to correlate the Cq values to copy numbers.
Plaque assay
Infective MS2 virions were quantified by standard double agar layer plaque assay (USEPA Method 1601). Samples were vortexed for 60 s. An aliquot of 500 μL was mixed into 5 mL of 0.7% Tryptic Soy Agar, 0.015% Ampicillin sodium salt and 0.015% Streptomycin sulphate. To this 100 μL of log-phase growth E. coli ATCC 700891 host were added. The mix, maintained at 46°C, was vortexed for 5 s and then overlayed onto a 1.5% Tryptic Soy Agar layer with the same composition of antibiotics. After the agar solidified, it was incubated at 37°C for 24 h. Infective MS2 was calculated in terms of plaque forming units (pfu) per mL. Four replicates were tested for each sample.
Cloning and sequencing
The amplified products from qRT PCR were analyzed on 3% agarose gel (50 V for 3 hrs), purified using QIAquick PCR purification kit (Qiagen), as per manufacturer's instructions, followed by elution in 100 μL of sterilized distilled water. Eluate concentrations were measured with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Purified PCR products were ligated into pGEM T Easy vector (Promega). The ligation reaction mix contained 7.5 μL of 2X Rapid Ligation buffer, 1 μL of pGEM T easy vector, 5.5 μL of purified PCR product and 1 μL of T4 DNA Ligase with a final volume of 15 μL. Ligations were carried out at 16°C in a water bath, overnight. Transformation was carried out with 15 μL ligation mix added to 200 μL of freshly prepared E. coli XL1 Blue competent cells and 3.4 μL of 10% β-Mercaptoethanol solution. Transformation mixes were kept on ice for 30 min followed by heat shock at 42°C for 90 sec and then placed immediately on ice for 5 min. Upon transformation 1000 μL of pre-warmed Luria Bertani (LB) broth was added and the mix was incubated at 37°C for 1½ hrs on a horizontal shaker set at 180 rpm. The transformed cells were centrifuged at 4000 rpm for 10 min. The pellet was resuspended in 100 μL of the supernatant and plated on LB agar amended with carbenicillin (100 μg mL−1), 20% Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 2% X-gal. Plates were incubated at 37°C for 14–18 h. Colonies picked from plates were used for plasmid extraction (QIAprep Spin Miniprep Kit, Qiagen). Plasmids were quantified on a NanoDrop ND-1000 spectrophotometer. Positive clones were confirmed by visualization of EcoR1 restriction digests of the extracted plasmids on a 3% agarose gel (50 V run for 3 h). Plasmids with a confirmed insert were sequenced at Functional Biosciences (Madison, WI).
References
Katayama, H., Nakamura, M. & Ohgaki, S. [PCR determination of inactivated RNA coliphage Qβ] Advances in Water and Wastewater Technology [Matsuo, T., Hanaki, K., Takizawa, S. & Satoh, H. (eds)] [101–108] (Elsevier, Amsterdam, 2001).
Blackmer, F., Reynolds, K. A., Gerba, C. P. & Pepper, I. L. Use of integrated cell culture-PCR to evaluate the effectiveness of poliovirus inactivation by chlorine. Appl Environ Microb 66, 2267–2268 (2000).
Pecson, B. M., Martin, L. V. & Kohn, T. Quantitative PCR for determining the infectivity of bacteriophage MS2 upon inactivation by heat, UV-B radiation and singlet oxygen: advantages and limitations of an enzymatic treatment to reduce false-positive results. Appl Environ Microb 75, 5544–5554 (2009).
Ma, J. F., Straub, T. M., Pepper, I. L. & Gerba, C. P. Cell culture and PCR determination of poliovirus inactivation by disinfectants. Appl Environ Microb 60, 4203–4206 (1994).
Joslyn, L. J. [Sterilization by heat] Disinfection, Sterilisation and Preservation [Block, S. S. (ed)] [695–728] (Lippincott Williams & Wilkins, Philadelphia, 2001).
Limsawat, S. & Ohgaki, S. Fate of liberated viral RNA in wastewater determined by PCR. Appl Environ Microb 63, 2932–2933 (1997).
O'Connell, K. P. et al. Real-time fluorogenic reverse transcription-PCR assays for detection of bacteriophage MS2. Appl Environ Microb 72, 478–483 (2006).
Dreier, J., Störmer, M. & Kleesiek, K. Use of bacteriophage MS2 as an internal control in viral reverse transcription-PCR assays. J Clin Microbiol 43, 4551–4557 (2005).
Julian, T. R., Tamayo, F. J., Leckie, J. O. & Boehm, A. B. Comparison of surface sampling methods for virus recovery from fomites. Appl Environ Microb 77, 6918–6925 (2011).
Wolf, S., Hewitt, J., Rivera-Aban, M. & Greening, G. E. Detection and characterization of F+ RNA bacteriophages in water and shellfish: application of a multiplex real-time reverse transcription PCR. J Virol Methods 149, 123–128 (2008).
Porter-Jordan, K. et al. Nested polymerase chain reaction assay for the detection of cytomegalovirus overcomes false positives caused by contamination with fragmented DNA. J Med Virol 30, 85–91 (1990).
Venkatesan, D. Isolation and Detection of Indicator MS2 Coliphage in different environment and sea foods by PEG Precipitation and GAC-UAPB-RT-PCR method. Advanced BioTech 7, 26–32 (2008).
Gefrides, L. A., Powell, M. C., Donley, M. A. & Kahn, R. UV irradiation and autoclave treatment for elimination of contaminating DNA from laboratory consumables. Forensic Sci Int-Gen 4, 89–94 (2010).
Dwyer, D. E. & Saksena, N. Failure of ultra-violet irradiation and autoclaving to eliminate PCR contamination. Mol Cell Probe 6, 87–88 (1992).
Espy, M. J., Uhl, J. R., Sloan, L. M. & Rosenblatt, J. E. Detection of vaccinia virus, herpes simplex virus, varicella-zoster virus and Bacillus anthracis DNA by LightCyclerNested polymerase chain reaction after autoclaving: Implications for biosafety of bioterrorism agents in. Mayo Clin Proc 77, 624–628 (2002).
Elhafi, G., Naylor, C., Savage, C. & Jones, R. Microwave or autoclave treatments destroy the infectivity of infectious bronchitis virus and avian pneumovirus but allow detection by reverse transcriptase-polymerase chain reaction. Avian Pathol 33, 303–306 (2004).
Van Duin, J. & Tsareva, N. [Single-stranded RNA phages] The Bacteriophages [Calendar, R. L. (ed)] [175–196] (Oxford University Press, Oxford, 2006)
Fiers, W. R. et al. Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene. Nature 260, 500–507 (1976).
Ye, J. et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC bioinformatics 13, 134 (2012).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic acids res 31, 3406–3415 (2003).
SantaLucia, J. A unified view of polymer, dumbbell and oligonucleotide DNA nearest-neighbor thermodynamics. P Natl Acad Sci USA 95.4, 1460–1465 (1998).
Acknowledgements
This work was supported by the New Mexico State University Agricultural Experiment Station, by the U.S. Department of Energy under contract DE-EE0003046 awarded to the National Alliance for Advanced Biofuels and Bioproducts and by NIH through grant R25GM061222.
Author information
Authors and Affiliations
Contributions
V.V.U. and A.U. designed the experiment, V.V.U. and V.J. carried out the experiment, V.V.U. and A.U. analyzed the data, V.V.U., A.U. and G.S. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Info File
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Unnithan, V., Unc, A., Joe, V. et al. Short RNA indicator sequences are not completely degraded by autoclaving. Sci Rep 4, 4070 (2014). https://doi.org/10.1038/srep04070
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04070
This article is cited by
-
An Environmental DNA Primer for Microbial and Restoration Ecology
Microbial Ecology (2023)
-
Investigation of potential safety hazards during medical waste disposal in SARS-CoV-2 testing laboratory
Environmental Science and Pollution Research (2021)
-
Applications of environmental DNA (eDNA) in ecology and conservation: opportunities, challenges and prospects
Biodiversity and Conservation (2020)
-
Enzymatic Pre-treatment of Wastewater to Minimize Recovery by Reverse Transcriptase PCR of RNA from Inactive Bacteriophages
Current Microbiology (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
