
Abstract
Technological improvements have resulted in increased discovery of new microRNAs (miRNAs) and refinement and enrichment of existing miRNA families. miRNA families are important because they suggest a common sequence or structure configuration in sets of genes that hint to a shared function. Exploratory tools to enhance investigation of characteristics of miRNA families and the functions of family-specific miRNA genes are lacking. We have developed, miRNAVISA, a user-friendly web-based tool that allows customized interrogation and comparisons of miRNA families for hypotheses generation and comparison of per-species chromosomal distribution of miRNA genes in different families. This study illustrates hypothesis generation using miRNAVISA in seven species. Our results unveil a subclass of miRNAs that may be regulated by genomic imprinting and also suggest that some miRNA families may be species-specific, as well as chromosome- and/or strand-specific.
Similar content being viewed by others



Introduction
microRNA (miRNA) are a class of evolutionarily conserved endogenous non-coding RNAs that may act as gene regulators in both plants and animals1,2,3,4,5. Mature miRNA are excised from longer single-stranded precursor (pre-miRNA) transcripts that fold into hairpin-like RNA secondary structures6,7. Each pre-miRNA originates from a cistronic or polycistronic transcript (pri-miRNA) containing several hairpins that are thought to collaborate in providing a common function8. Without loss of generality, a pre-miRNA is considered as an independent miRNA gene9,10,11,12. The latest miRBase13,14 release 19 (R19) contains 21,264 experimentally validated miRNA genes (green bars in Fig. 1a) expressing 25,141 mature miRNA (red bars in Fig. 1a) in 193 species (Fig. 1c). About 50% of the species are from the animal kingdom whereas plant, viral and protist (chromalveolata and mycetozoa) kingdoms approximately represent 35%, 26% and 3% of the database entries, respectively. One miRNA gene can yield more than one mature miRNA.

Growth of the miRBase database over time.
(a) The green, red and blue bars show the number of miRNA genes (hairpins), classified miRNA genes and mature miRNA in each miRBase release, respectively. One miRNA gene can yield more than one mature miRNA and therefore the total number of the latter can exceed that of the former. The increase in number of validated mature miRNA and their genes is mainly a consequence of improved high throughput sequencing. (b) Increasing number of miRNA families with time. (c) Number of species where gene regulation by miRNA has been reported in different miRBase releases; also see Supplementary Fig. S1 online. The increase in number of miRNA families and coverage of species has benefited from increased miRNA gene numbers, community annotation and improved computational algorithms12,15,19.
For almost a decade, some of the miRNA genes have been categorized into different groups, named miRNA families, based on the mature miRNA, sequence and/or structure of pre-miRNAs14,15. About 73% (15,554) of the miRNA genes in miRBase R19 are assigned to (1,543) miRNA families. miRNA families are important because they suggest a common sequence or structure configuration in sets of genes and hence function6. In fact, miRNA genes in a family can exhibit full conservation of the mature miRNA or partial conservation of only the seed sequences at positions 2–8 of the functional mature miRNA8. Interestingly, it has been observed that miRNA genes in the same miRNA family are non-randomly co-localized and well organized around genes involved in infectious, immune system, sensory system and neurodegenerative diseases, development and cancer8.
As the number reported mature miRNA and their genes (Fig. 1a), miRNA families (Fig. 1b) and coverage of species (Fig. 1c) continues to grow almost exponentially, attention is now shifting to elucidating the function of these miRNAs and their influence in biochemical pathways and diseases. However, tools for exploration analysis of high-dimension categorical data that characterizes miRNA family categories are still lacking to enhance hypothesis driven investigations of their constituent genes, their functional roles and general properties. This study attempts to address this issue by providing a tool that can be used to evaluate the following questions. What attributes and characteristics are encoded in miRNA families? Which characteristics can be used to examine potent inter-miRNA family relationships and/or define clan(s) of miRNA families? How can the diversity of miRNA families across species, lineages and/or kingdoms be interrogated? How can the genomic distribution of family-annotated miRNA genes be summarized and compared in different genomes? What information can be inferred when jointly interrogating the genomic distribution of miRNA genes, organization (spatial co-location on specific chromosomes) and characteristics of miRNA families? Do family-specific miRNA genes exist as clusters? And if so, what are the general functions of clustered miRNA genes belonging to different miRNA families and what are the links between family-specific mature miRNAs and biochemical pathways and/or diseases?
Insights into intra- and inter-miRNA-family relationships are still scarce. At the gene level, several machine learning algorithms exist to find new members of miRNAs families based on sequence and/or structure conservation and clustered genes based on intergenic distance6,8,16,17,18,19. However, there is a need to jointly interrogate and integrate information about the genomic distribution of miRNA genes and their sequence and/or structure organization implied by miRNA family categories. Such interrogation is useful to better understand both the properties and function of family-annotated miRNA genes and to establish the characteristics that may define inter-miRNA family relationships.
Results from sequence-based or intergenic-based clustering methods imply common ancestry that is defined by weak miRNA sequence similarity and/or localization on a single spatially unique genomic region6. Lu et al. (2008)20 found evidence that miRNA derived from clustered miRNA genes tend to have similar functional roles and disease associations. Clustered genomic arrangements can however involve miRNA genes from different miRNA families (see Supplementary Table S1 online). Nonetheless, given that structural evolution is thought to be slower than sequence evolution6, the curation of miRNA families based on structural clustering can suggest more general functional commonalities in sets of genes than those hinted by sequence-/intergenic-based methods. That is, existence of miRNA family groupings (clans of families i.e. sub-classes) that have shared general characteristics. Currently, there are no tools in existence that can be used to explore the existence of such miRNA family sub-classes.
Only a handful of software tools exploit the information availed by miRNA family categories for predictive purposes or otherwise. For example, a recent study (Kamanu, T. K. K., PhD thesis, King Abdullah University of Science and Technology, 2012) has developed an miRNA gene discovery system based on miRNA family categories that enables species-independent prediction of unknown miRNA genes from arbitrary nucleotide sequences. Gerlach et al. (2009)12 and Ding et al. (2011)15 have proposed supervised models for predicting miRNA family membership given unlabeled or unclassified miRNA sequences. Our study aims to determine the genomic distribution of family-annotated miRNA genes in a given species. Knowledge about such species-specific miRNA gene distribution is important when modeling miRNA-regulation, especially for clustered miRNA, co-expressed miRNA genes and mirtrons21,22,23,24,25. miRNA gene distribution may influence the annotation of miRNA promoter regions which are yet to be fully understood26.
The aim of the miRNAVISA system is to provide a user-friendly interactive web interface to enable comparison and exploration of different miRNA families for the purpose of generating hypotheses about their properties. miRNAVISA allows inquiry of and comparison of the genomic distribution of family-annotated genes in a given species, as well as comparison between species. miRNA gene distribution for a given family are comparable in closely related species and can be used to provide clues about the miRNA function and roles in biochemical pathways. For instance, are miRNA and/or miRNA families chromosome-specific? The spatial location of miRNA genes has been implied to preferentially influence miRNA function in different chromosome-linked diseases such as Down's Syndrome27. Recently, human chromosome 19 miRNAs have been suggested to safeguard the integrity of fetal-maternal communication and therefore their mimics offer themselves as candidates for treatment of autoimmune diseases28,29.
miRNAVISA can also be used to query the existence of sub-classes of miRNA genes by generating data-dependent relationships that may define such miRNA sub-classes. In contrast to other tools such as miRNAMap30, which is specific to metazoan genomes and does not interrogate miRNA families, miRNAVISA can be used to interrogate the diversity of miRNA families across species and kingdoms.
Here, we demonstrate the use miRNAVISA for hypotheses generation about miRNA families, their genes and function in different species. Moreover, miRNAVISA can be used to infer new roles of miRNA genes in a given miRNA family.
Results
Hypotheses generation using miRNAVISA
Seven species are used to illustrate hypotheses generation using miRNAVISA. These include dog - Canis familiaris (CFA), Gorilla gorilla (GGO), human - Homo sapiens (HSA), monkey - Macaca mulatta (MML), mouse - Mus musculus (MMU), orangutan - Pongo pygmaeus (PPY) and chimpanzee - Pan troglodytes (PTR). The values within the brackets are the miRBase three-letter species codes. The human and mouse genomes were selected because of the major importance of these species as model organisms and since miRNA gene discovery for them is thought to be almost exhausted15.
The other four primate species were chosen to facilitate comparison against human miRNA genes, whereas the dog is used to illustrate the diversity of different miRNA families across species.
Summary of family-annotated miRNA genes
miRNAVISA can enable a concise database summary given a set of sample families and species of interest. Figure 2 is a visualization of the cross-tabular distribution of family-annotated miRNA genes from miRBase R19 across 193 species and 1,543 miRNA families. The distribution of miRNA genes among 24 miRNA families in the seven sample species is shown against the remaining 186 species. The families were selected based on the family sizes and specific attributes of interest. For example, the let-7 miRNA family contains the founding members of the miRNA class and is among the largest families in the miRBase database; and the mir-515 and mir-548 miRNA families are known to be primate-specific28,29,31,32 (see also Figure 2).

Inter-species comparisons to interrogate the diversity of miRNA families across species, lineage and/or kingdoms.
Summary of the miRBase R19 database comprising of 193 species and 1,543 miRNA families. The row-wise totals in blue show the number of species-specific miRNA genes in the 24 miRNA families analyzed in the figure. Totals under the “FAM” and “ALL” column labels are the number of miRNA genes in other miRNA families that were not selected for analysis and the total (family-annotated and non-family-annotated) number of registered miRNA genes in a given species, respectively. There are 1,600 registered human (Homo sapiens) among which 61.2% (229 plus 750) have been classified into different miRNA families. All the miRNA families mir-515, mir-548 and mir-663 genes have only been reported in primates and similarly the most miRNA family mir-466 genes have been observed in mice (Mus musculus). A majority (>90%) of genes in the mir-467 family are mouse-specific. The disproportionate magnitude of homologous miRNA genes reflects the bias of most computational and experimental methods to favor miRNA annotation in certain species.
The order preserved in Figure 2 is based on the magnitude of the family sizes (decreasing). Column-wise normalization was done based on family sizes marked in red. The total number of miRNA genes in the 23 selected miRNA families in each species is shown by the non-bracketed numerals on the right-most y-axis, while the total number of species-specific family-annotated miRNA genes is indicated in brackets. The difference between these numeric values represents genes in the remaining 1,519 miRNA families that are not included in our current analysis.
Inter-species comparisons
Inter-species comparison of family-annotated miRNA genes are based on all the 193 species where miRNAs have been reported. For illustration, the following hypotheses can be postulated based on the output from miRNAVISA in Figure 2 given input of the selected species and miRNA families.
-
H1. miRNA family species-specificity : mouse and chicken: The miRNA family mir-466 is specific to mouse and chicken.
-
H2. miRNA family primate-specificity : The miRNA family mir-663 is specific to primate species.
The miRNAVISA web interface allows for customized user-defined selection of miRNA families to evaluate the diversity of miRNA families and their genes across species, lineage or kingdoms (Supplementary Fig. S2 online).
Intra-species comparisons
Intra-species comparisons of family-annotated miRNA genes only accommodate the species where their genome co-ordinates are available. miRNAVISA presents intra-species comparisons in a user-specified genome as miRNA genes/family maps. Each genome miRNA genes/family map shows the chromosomal distribution (rows) and per-family distribution (columns). The rows and columns in a genome miRNA genes/family map are ordered based on the total number of genes indicated on the axes. miRNA genes that are not mapped to any chromosome are reported under a predefined label ‘NoMap/Chunks'. Figure 3 illustrates the genomic distribution of family-annotated miRNA genes in the human genome.

Genomic distribution of family-annotated miRNA genes in the human (Homo sapiens) genome.
The row-wise totals in blue show the number of chromosome-specific miRNA genes (hairpins) in the 50 miRNA families analyzed in the figure. Totals under the “FAM” and “ALL” column labels are the number of miRNA genes in other miRNA families that were not selected for analysis and the total (family-annotated and non-family-annotated) number of registered miRNA genes on a given chromosome, respectively. A total of 135 and 113 miRNA genes are mapped on chromosome 1 and X, respectively. About 56.3% (24 plus 52) and 85.8% (48 plus 49) of all miRNA genes on chromosomes 1 and X have been annotated into different miRNA families. There are 1,600 registered human miRNA genes in miRBase R19 but only 61.2% (367 plus 612) have been annotated into different miRNA families. Genes in miRNA families mir-515 (chromosome 19), mir-154 (chromosome 14), mir-379, mir-743, mir-329, mir-368, mir-500 and say mir-188 are each co-located on a specific chromosome. Some co-located, chromosome 14, miRNA genes in different miRNA families exist as a large cluster (see Supplementary Table S1 online).
The order preserved in the miRNA genes/family map respects both the species-specific miRNA family sizes and total number of family-annotated miRNA genes on each chromosome.
The following hypotheses can be postulated on basis of the genome miRNA genes/family maps (Fig. 3 and Supplementary Figs. S3–S8 online) determined using miRNAVISA in different genomes:
-
H3: miRNA family chromosome-specificity: miRNA genes constituting the miRNA families that correspond to the chromosomes shown in red boxes in Figure 4 are chromosome-specific in different species. Similarly, miRNA genes in the miRNA families that correspond to the green boxes in Figure 4 are predominantly located in the respective chromosomes.
Figure 4 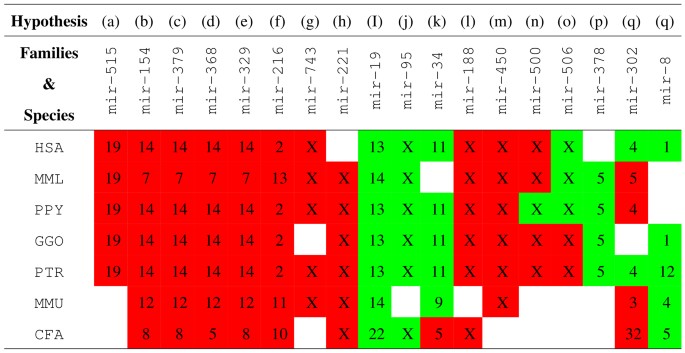
Summary of chromosome-specificity vis-a-vis miRNA families (see Fig. 3 and Supplementary Figs. S3–S8 online).
Genes in miRNA families that correspond to the chromosomes shown in red boxes are chromosome-specific. Genes in miRNA families that correspond to the green boxes are predominantly located in the respective chromosomes.
-
H4: miRNA family chromosome strand-specificity: miRNA genes constituting the miRNA family mir-515 are chromosome- and/or strand-specific.
-
H5:</emph> Clan of miRNA families (miRNA subclass) regulated by genomic imprinting: miRNA families whose genes reside on imprinted chromosomal domains exhibit both chromosome- and strand-specific bias.

The hypotheses H1, H2 and H3(a)–(q) (see Fig. 4) derived in this study are directly supported by the data in miRBase R19. Hypothesis H4 is inferred based on the strand preference of the totality of known and predicted miRNA family mir-515 genes [Supplementary Figs. S9 and S10 online; Supplementary Tables S2–S6 online]. The predictions are based on a novel approach for genome-wide species-independent miRNA discovery (reported separately) that relies on miRNA family categories in contrast to existing method which often over-emphasize and use homology preconditions as a basis for discovery (Kamanu, T. K. K., PhD thesis, King Abdullah University of Science and Technology, 2012). Supplementary Figure S9 online and Supplementary Tables S2–S6 online show the detection of all the known miRNA family mir-515 genes using the species-independent approach in the human genome and similarly the recovered known and predicted novel genes for the same family in the other primate genomes. This data is also used to explore hypotheses H3 which may also be supported by evidence the literature as discussed below. Hypothesis H5 generalizes hypothesis H4 to include other miRNA families whose genes exhibit both chromosome- and strand-specificity given the data in miRBase R19.
Discussion
Co-localization of miRNA genes can imply a common regulatory role and the co-localized miRNA genes can be similarly regulated8. Often genes in the same miRNA family have similar function due to a conserved sequence and structural configuration. Therefore, establishing, linking and jointly interrogating intra- and inter-species distribution of miRNA gene and miRNA family attributes is important to give clues about the function of family-specific mature miRNA, their co-expression and to uncover potential inter-miRNA family relationships (miRNA sub-classes). Support for this assertion in the literature is evaluated below.
miRNA database summaries enabled through miRNAVISA (Fig. 2) are useful to infer the diversity of miRNA families across species and kingdoms, to compare the propensity of miRNA-directed regulation amongst species and to contrast and test potent epigenetic regulation models of miRNA in closely related organisms. Our results (Fig. 2) corroborate other findings in the literature28,29,31,32 that miRNA with a shared target register can exhibit evolutionary innovations in their genes that are restricted to a species (hypothesis H1) or lineage e.g., primates (hypothesis H2). Nonetheless, our study generalizes miRNA and miRNA gene characteristics to existing family categories irrespective of their target register, although such general characteristics should be interpreted and weighed against the knowledge that the miRNA repertoire in some species is still under-explored. In particular, although mice genes from miRNA family mir-467 comprise the largest proportion (Fig. 2) of known genes associated to this family, we argue that this reflects a bias of experimental and computational miRNA gene discovery methods that favors the model organisms. Nonetheless, we postulate here that the totality of miRNA and their genes in some miRNA families such as the mir-515 family (Supplementary Fig. S9 online; Supplementary Tables S2–S6 online) is almost fully explored in primate species.
Somewhat surprisingly, all known and predicted mir-515 genes are exclusively located on the forward strands [denoted as FWD or (+1) strand; Supplementary Figs. S9 and S10 online] in chromosomes 19 of the interrogated primate genomes (hypothesis H3 and H4). The mir-515 miRNA genes are organized as a contiguous (~87 Kb in human) block on the telomeric regions of the chromosomes which have been described before as the chromosome 19 miRNA clusters (C19MC)28,29. This result is puzzling because miRNA genes are known to be strand-independent10,33,34,35. The miRNA genes in C19MC have other interesting attributes. Firstly, they derive from imprinted chromosomal domains which are known to escape conventional Mendelian inheritance dynamics11,28. Secondly, they are exclusively or mono-allelically expressed from the paternally inherited chromosome28,36,37, which could explain the observed chromosome-strand specificity (hypothesis H4). Thirdly, they are normally silenced except in embryonic stem cells and placenta, but their deregulation has been linked to several types of cancer3,38,39 (see miRNAVISA family query). Lastly, the C19MC miRNA genes are transcribed by either RNA pol-II or RNA pol-III5,28.
Genes in miRNA families mir-154, mir-379, mir-368, mir-329, mir-329, mir-216, mir-743, mir-188 and mir-450 were also noted here to be chromosome-specific (hypothesis H3, Fig. 3 and Supplementary Figs. S3–S8 online). Human miRNA genes in miRNA families mir-154, mir-379, mir-329, mir-368 a subset of a large (~44 Kb) known7,33,34,36,37 cluster, abbreviated here as C14MC, which comprises co-located miRNA genes from different families (Supplementary Table S1 online). Human chromosome 14 may be equivalent in synteny to chromosomes 12 and 7 of the mouse and monkey genomes (Fig. 4), respectively. Generally, it is know that intronic miRNA genes can reside in conserved regions of synteny34.
Interestingly, both C14MC and C19MC have similar gene organization, are derived from intronic regions and are associated with imprinted chromosomal domains that have similar epigenetic regulation mechanisms28,29. In contrast to C19MC, C14MC is expressed from maternally inherited chromosomal domains28,36,37,40. The control elements and imprinting mechanism say for the C19MC and C14MC clusters are not fully understood, including whether or not the two loci are micro-imprints or subsets of larger imprinted chromosomal domains.
We noted preferential chromosome and/or strand-specificity (to either the forward or reverse complementary strand) of genes in miRNA families that are speculated to reside on imprinted chromosomal domains. For example, with the exception of one known Rattus norvegicus gene belonging to the miRNA family mir-368, all the genes in miRNA families mir-329, mir-379 and mir-154 have been annotated only on the FWD (+1) strand in different species (hypothesis H3 and H4). Genes belonging to miRNA families mir-1193, mir-134, mir-412, mir-485, mir-541, mir-654, mir-668 and mir-889 have been annotated only on the +1 strand in different species (see Supplementary Table 1 online for miRNA families belonging to human C14MC and the miRNAVISA family keyword query at http://www.cbrc.kaust.edu.sa/mirnavisa/family.php). Taken together, this observation implies that miRNA families with genes residing on imprinted genomic loci may define a subclass of the miRNA class that exhibits preferential strand-biased expression and therefore genes in such subclasses may have similar but currently unknown transcriptional and epigenetic regulation mechanisms. These miRNA families show strict chromosome-specificity in different species as indicated in Figure 4 (hypothesis H5).
Existing tools for predicting miRNA function are based on the interaction of mature miRNA and mRNA and not on miRNA family groups. The function of mature miRNA in model species, such as human and mouse, has been well studied. PhenomiR, miRdSNB, HMDD, miRNAMap and miRBase (see “Methods”) provide useful links of the functional mature miRNA to their genes and associated diseases, but do not associate miRNA and miRNA gene characteristics to existing miRNA family categories irrespective of their mRNA target register. The miRNA families by definition and curation have no species association; they can therefore be used to exploit the gains made in annotating the miRNA function in model species to infer the general function of family-specific mature miRNA across species. Our tool miRNAVISA addresses this issue and complements these tools as a first step towards understanding the general functions of mature miRNA derived from genes in the same family, under a “guilt by association” presumption. We also considered the inference of the function of mature miRNA derived from co-located/clustered miRNA genes belonging to different miRNA families.
Intra-species comparisons (see Fig. 3 and Supplementary Figs. S3–S8 online) enabled by miRNAVISA allow for evaluation of genome organization and inference of the function of co-located and clustered miRNA genes. Genes in a miRNA family can have about 65% or more sequence identity, portray partial or full conservation of the seed sequence and at times 100% conservation of the functional mature miRNA8,41. Using the assertion of “guilt by association”, we hypothesize that miRNA genes in the same family have similar roles in different biological processes and pathways across diverse species. This presumption is supported by the evidence in the literature as well as results of miRNA family keyword queries enabled by miRNAVISA. For example, Zehavi et al. (2012)37 experimentally verified that three (miR-376a-1, miR-376a-2 and miR-376c) mature miRNA derived from the four-member miRNA family 368 on human chromosome 14 (Fig. 3) act as melanoma tumor suppressors/biomarkers. The mature miRNA derived from the other miRNA family mir-368 gene [i.e., miR-376b derived from hsa-mir-376b; Fig. 3 and hypothesis H3(d)] can therefore be inferred as an equivalent melanoma tumor to miR-376a-1, miR-376a-2 or miR-376c in different species (Supplementary Figure S11) on the basis of miRNA family association. miRNAVISA can thus be used as a first step in deriving hypotheses about the biological functions of genes in miRNA families.
Some of the C14MC genes in Supplementary Table S1 online exist as a structural miRNA cluster that are derived from complex, intermediary polycistronic transcripts of lengths 1–2 kb that exhibit co-expression and shared functional roles6,8,33. Structural miRNA clusters can be thought of as independent miRNA genes that belong to different miRNA families which may have similar biological function, could be involved in similar biochemical pathway, or could be co-regulated. Intra-species miRNA genes/family maps determined using miRNAVISA can be used to examine miRNA families whose genes may be transcribed as structural miRNA clusters. For example, Figure 3 indicates the implicit relationships of co-located/clustered miRNA genes in miRNA families mir-17 and mir-19 (chromosome 13), mir-17, mir-19 and mir-25 (chromosome X) and mir-17 and mir-25 (chromosome 7) that have been confirmed to be co-expressed and involved in different cancer pathways8. Moreover, some genes in miRNA families mir-145, mir-368 and mir-493 (co-located with the mir-368 family genes on human C14MC) have been shown to target 3'-UTR of the IGF1R gene that is implicated in tumorigenesis37. Given that human C19MC (miRNA family mir-515) genes are closely co-located (downstream and upstream intergenic distance of 1.5 Kb and 25 Kb, respectively) with genes belonging to mir-506, mir-1323, mir-498 and mir-290 in the region spanning about 121 Kb, we postulate here that these miRNA families may exhibit coordinated expression and may similarly be deregulated in different experimental conditions in a tissue-, development-stage- or species-specific manner.
To the best of our knowledge no definition of the term ‘miRNA family' exists in the literature at present, although the same has been used in miRNA gene annotation. A clarification of the term is necessary since its use has been inconsistent: for example it has been used to denote the entire miRNA class42 or even describe a set of mature miRNA with similar seed sequences43. A miRNA family can be defined as an organization of pre-miRNAs according to their homologous relationships such that the pre-miRNA in a given miRNA family have: a/ similar ancestry or exhibit evolutionary conservation in sequence15,16,19; b/ similar motif usage that reflects secondary structures conservation6,15,16; c/ similar mature miRNA/miRNA* and thus probable shared functional characteristics or biological function15,41; and/or d/ conserved mature miRNA-seed-target relationships15,44,45. The union of all individual miRNA families, sub-classes of related miRNA families (clans) and other non-categorized miRNA genes, can therefore be thought of as the “miRNA class”. Class members in general have no distinctive homology at sequence level, but still share common structural and functional properties16,33.
This study presented miRNAVISA, a web tool that we developed for exploration of properties of miRNA families. miRNAVISA links the spatial genomic distribution of known miRNA genes to the intrinsic properties encoded by set(s) of genes in a miRNA family in order to facilitate interrogation of their properties and generation of hypotheses about function/role of the constitutive genes. The study advanced the notion that the genomic organization and in particular co-localization of genes in different miRNA families favors their coordinated expression say in a tissue-, developmental-stage- and/or species-specific manner; and in some cases there is clear evidence of miRNA conservation in synteny across species.
An interesting insight derived when using miRNAVISA is that miRNA families can be both chromosome-specific and their constituent miRNA genes can as well exhibit strand-specific expression. This result has consequences to the benchmarks for the annotation of miRNAs in miRBase. This finding indicates that it is important to consider similar potential miRNA gene candidates residing on different strands as independent miRNA genes. To the best of our knowledge, there are only about 20 human strand-independent, repeat-related and palindromic (Kamanu, T. K. K., PhD thesis, King Abdullah University of Science and Technology, 2012) miRNA genes registered in miRBase R19 (e.g., the hsa-mir-3130 gene with Accession Numbers MI0014147 and MI0014148, on chromosome 2).
This study demonstrated that miRNAVISA intra-species miRNA genes/family maps can guide the inference and discovery of the regulatory roles of clustered or co-localized miRNA genes. It is possible to use these maps to improve experimental designs that seek to assess and infer relationship and possible roles of clustered miRNA genes. Experimental designs may be restricted in some miRNA families to a specific lineage. The limited access to the relevant experimental tissue, say in primates, for functional analysis can benefit from conservation in synteny (Fig. 4) which may suggest other suitable organisms.
Our results assert that miRNA families may encode broader intrinsic attributes that: can be used to define miRNA family sub-classes, may imply shared roles in certain biochemical pathways, and/or may imply similarity in miRNA regulation mechanisms, but such attributes are not yet fully understood. Potential sub-classes may include genome-imprinting-associated, lineage/primate-specific and repeat-related miRNA families. The miRNA families mir-548 and mir-467 are known to constitute a miRNA sub-class that is defined by repeat-derived miRNA genes32,46,47. Equally important is the sub-class of miRNA families whose genes are predominantly located near fragile sites because of their importance to the understanding of cancer and other diseases48,49,50,51.
On the whole, although the miRNA genes/family maps provide some useful insights discussed above, they also raise other interesting questions that may be useful to ponder but are not addressed here. For instance, is there a correlation between function and synteny of miRNA and their genes? Does the relative genome-wide distribution of miRNA genes between families correlate with their functions and importance in different species? What is the biological significance of the conservation of seed and mature miRNA relative to that of their genes to the extent and distribution of the associated family-annotated miRNA genes? Does the latter have any significance to phenotypic expression in vivo?
The exploration tool developed here can generally be extended for analysis of other well-known non-coding RNA classes with existing family groupings and sub-classes such as snRNA (U1, U2, etc.), snoRNA, rRNA, RNase P and tRNAs. For example, snoRNA consists of two distinct sub-classes - H/ACA box and C/D box snoRNA - that arise from grouping together individual snoRNA families16,19. Currently, Rfam has over 100 snoRNA families. The RNase P RNA class also has the RNase MRP sub-class, whereas the rRNA RNA class has the 5 s rRNA sub-class19. It is worth noting that both snoRNA and miRNA may be only classes of non-coding RNA that are regulated by genome imprinting and their genes are often co-located at a genomic locus36,52,53.
The functionality of miRNAVISA is not affected by potential refinement of miRNA families that influence family membership. Such may be the case if a family is split into two or more independent miRNA families, merging two or more existing miRNA families, or the addition/deprecation of some family members when less/more stringent benchmarks are imposed during curation. It is likely that tools such as miRNAVISA will find more prominent use with improved refinement of the miRNA gene set at miRBase or availability of complete genome assemblies.
Methods
Data
The data used in this study was sourced from the miRBase database. The miRNA families in miRBase are manually curated. In contrast, Rfam17,19 is a database compiled by an automated pipeline, for general RNA families and includes 523 miRNA families (version 11, dated August 2012). Our study uses miRBase miRNA families because they are more comprehensive and curated.
miRBase provides per-species miRNA genome coordinates in Generic Feature Format (GFF) files. In some cases, experimentally validated miRNA genes in miRBase may lack genomic coordinates because of non-existent or incomplete genome assemblies, while there are also miRNA genes that have been mapped to unassembled reads, ESTs and/or cDNA. In total, only 85 species in miRBase R19 have accompanying GFF files out of which 61 (~72%) belong to the animal kingdom (Supplementary Fig. S1 online). The genomic distribution of a majority of plant, viral and protist miRNA genes are yet to be determined and this reflects a continuing annotation process as is the case for some miRNA sequence that may not map to available genome assemblies.
Most miRNA are increasingly being implicated in cancer as tumor suppressors or oncogenes i.e., up-regulated or down-regulated, respectively. In order to facilitate inquiry of expression trends, the current version of miRNAVISA integrates functional mature miRNA expression and miRNA disease associations data from manually curated public databases i.e., miR2Disease3, PhenomiR39, human miRNA disease database (HMDD)20 and the miREnvironment38,54 database. These databases separately give references to the supporting experimental evidence of miRNA deregulation in different diseases and biochemical pathways. Future versions of miRNAVISA will integrate disease-associated single nucleotide polymorphisms (SNPs) and miRNA data in the miRdSNP55 database.
Implementation of miRNAVISA
miRNAVISA enables cross-tabulation of miRNA genes/family data based on user inputs or variables which include species and miRNA family names. The resulting statistics that are organized as matrices are conveniently represented in graphical form as circles in an array of unit squares (cells) to allow exploration of relationships between variables. miRNAVISA normalizes matrix objects by columns and represents the scaled matrix in proportion to observed miRNA gene statistics. The relative radii of circles in a normalized column are proportional to the magnitude of the corresponding matrix cells and hence can be compared. The scaled matrix data is ranked and represented in different colors according to the relative magnitude determined using vector algebra. miRNAVISA graphical back-end is written in the R programming language and uses the base, graphics and plotrix packages56,57. A legend comprising a ramp of interpolated colors between red, blue, green is provided for interpretation in percentage terms. Red (100%), blue (50%) and white (0%) imply proportionately high, intermediate and low score, respectively, along a given column. The row and column totals are provided in the graphical output to enable approximate recovery of the absolute magnitude (number of miRNA genes) in each matrix cell. The total number of family-annotated (denoted as “FAM”) and the total number (denoted as “ALL”) of known miRNA genes across a row category are also recorded on each plot window.
Usage of miRNAVISA
miRNAVISA web tool (Supplementary Fig. S2 online) is freely available at www.cbrc.kaust.edu.sa/mirnavisa. It is implemented in PHP and uses modern HTML5 and CSS with JavaScript libraries like jQuery for handling asynchronous HTTP (Ajax) calls to Perl and R scripts. Three forms are provided to enable either inter-species comparison, intra-species comparison, or a keyword query on the basis of standard miRBase nomenclature5,13 for miRNA genes or families. The user interactively selects genomes or species, miRNA family categories, or a keyword. Results are returned in as Scalable Vector Graphics (SVG) and supporting tables also packaged for simple one-click download and these tables are easy to import to spreadsheet programs. A user manual is also available with worked-out examples. Future versions will integrate chromosome specific density charts of miRNA genes based on the specifications of miRNA family categories.
References
Bartel, D. P., Lee, R. & Feinbaum, R. MicroRNAs: genomics, biogenesis, mechanism and function. Cell 116, 281–297 (2004).
Chiang, H. & Schoenfeld, L. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24, 992–1009 (2010).
Jiang, Q. et al. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37, D98–D104 (2009).
Jones-Rhoades, M. W., Bartel, D. P. & Bartel, B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 57, 19–53 (2006).
Erson, A. E. & Petty, E. M. MicroRNAs in development and disease. Clin Genet 74, 296–306 (2008).
Kaczkowski, B. et al. Structural profiles of human miRNA families from pairwise clustering. Bioinformatics 25, 291–294 (2009).
Hertel, J. & Stadler, P. F. Hairpins in a Haystack: recognizing microRNA precursors in comparative genomics data. Bioinformatics 22, e197–e202 (2006).
Mathelier, A. & Carbone, A. Large scale chromosomal mapping of human microRNA structural clusters. Nucleic Acids Res 41, 4392–4408 (2013).
Mendes, N. D., Freitas, A. T. & Sagot, M.-F. Current tools for the identification of miRNA genes and their targets. Nucleic Acids Res 37, 2419–2433 (2009).
Golan, D., Levy, C., Friedman, B. & Shomron, N. Biased hosting of intronic microRNA genes. Bioinformatics 26, 992–995 (2010).
Morison, I. M., Ramsay, J. P. & Spencer, H. G. A census of mammalian imprinting. Trends Genet 21, 457–465 (2005).
Gerlach, D., Kriventseva, E. V., Rahman, N., Vejnar, C. E. & Zdobnov, E. M. miROrtho: computational survey of microRNA genes. Nucleic Acids Res 37, D111–D117 (2009).
Ambros, V. A uniform system for microRNA annotation. RNA 9, 277–279 (2003).
Kozomara, A. & Griffiths-Jones, S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39, D152–D157 (2011).
Ding, J., Zhou, S. & Guan, J. miRFam: an effective automatic miRNA classification method based on n-grams and a multiclass SVM. BMC Bioinformatics 12, 1–11 (2011).
Will, S., Reiche, K., Hofacker, I. L., Stadler, P. F. & Backofen, R. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol 3, 1–12 (2007).
Daub, J. et al. The RNA WikiProject: community annotation of RNA families. RNA 14, 2462–2464 (2008).
Ohler, U., Yekta, S., Lim, L. P., Bartel, D. P. & Burge, C. B. Patterns of flanking sequence conservation and a characteristic upstream motif for microRNA gene identification. RNA 10, 1309–1322 (2004).
Gardner, P. P. et al. Rfam: updates to the RNA families database. Nucleic Acids Res 37, D136–D140 (2009).
Lu, M. et al. An analysis of human microRNA and disease associations. PLoS One 3, 1–5 (2008).
Berezikov, E., Chung, W., Willis, J., Cuppen, E. & Lai, E. C. Mammalian mirtron genes. Mol Cell 28, 328–336 (2009).
Chan, S.-P. & Slack, F. J. And now introducing mammalian mirtrons. Dev Cell 13, 605–607 (2007).
Ha, M., Pang, M., Agarwal, V. & Chen, Z. J. Interspecies regulation of microRNAs and their targets. Biochim Biophys Acta 1779, 735–742 (2008).
Kai, Z. S. & Pasquinelli, A. E. MicroRNA assassins: factors that regulate the disappearance of miRNAs. Nat Struct Mol Biol 17, 5–10 (2010).
Yu, X., Lin, J., Zack, D. J., Mendell, J. T. & Qian, J. Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res 36, 6494–6503 (2008).
Schmeier, S., Schaefer, U., MacPherson, C. R. & Bajic, V. B. dPORE-miRNA: polymorphic regulation of microRNA genes. PLoS One 6, 1–6 (2011).
Kuhn, D. E. et al. Human chromosome 21-derived miRNAs are over-expressed in Down Syndrome brains and hearts. Biochem Bioph Res Co 370, 473–477 (2008).
Noguer-Dance, M. et al. The primate-specific microRNA gene cluster (C19MC) is imprinted in the placenta. Hum Mol Genet 19, 3566–3582 (2010).
Bullerdiek, J. & Flor, I. Exosome-delivered microRNAs of “chromosome 19 microRNA cluster” as immunomodulators in pregnancy and tumorigenesis. Mol Cytogenet 5, 1–4 (2012).
Hsu, S.-D. et al. miRNAMap 2.0: genomic maps of microRNAs in metazoan genomes. Nucleic Acids Res 36, D165–D169 (2008).
Bentwich, I. et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37, 766–770 (2005).
Piriyapongsa, J. & Jordan, I. K. A family of human microRNA genes from miniature inverted-repeat transposable elements. PLoS One 2, 1–11 (2007).
Altuvia, Y. et al. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 33, 2697–2706 (2005).
Weber, M. J. New human and mouse microRNA genes found by homology search. FEBS J 272, 59–73 (2005).
Lai, E. C., Tomancak, P., Williams, R. W. & Rubin, G. M. Computational identification of Drosophila microRNA genes. Genome Biol 4, 1–20 (2003).
Seitz, H., Royo, H. & Bortolin, M. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res 14, 1741–1748 (2004).
Zehavi, L. et al. Silencing of a large microRNA cluster on human chromosome 14q32 in melanoma: biological effects of mir-376a and mir-376c on insulin growth factor 1 receptor. Mol Cancer 11, 1–15 (2012).
Yang, Q., Qiu, C., Yang, J., Wu, Q. & Cui, Q. miREnvironment database: providing a bridge for microRNAs, environmental factors and phenotypes. Bioinformatics 27, 3329–3330 (2011).
Ruepp, A. et al. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol 11, 1–11 (2010).
Suzuki, M. & Hayashizaki, Y. Mouse-centric comparative transcriptomics of protein coding and non-coding RNAs. Bioessays 26, 833–843 (2004).
Lewis, B. P., Shih, I., Jones-Rhoades, M. W., Bartel, D. P. & Burge, C. B. Prediction of mammalian microRNA targets. Cell 115, 787–798 (2003).
He, L. & Hannon, G. J. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5, 522–531 (2004).
Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Chen, K. & Rajewsky, N. The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8, 93–103 (2007).
Friedman, R. & Farh, K. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 (2009).
Yuan, Z., Sun, X., Liu, H. & Xie, J. MicroRNA genes derived from repetitive elements and expanded by segmental duplication events in mammalian genomes. PLoS One 6, 1–13 (2011).
Borchert, G. M. et al. Comprehensive analysis of microRNA genomic loci identifies pervasive repetitive-element origins. Mob Genet Elements 1, 8–17 (2011).
Calin, G. A. et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101, 2999–3004 (2004).
Mazière, P. & Enright, A. J. Prediction of microRNA targets. Drug Discov Today 12, 452–458 (2007).
Gregory, R. I. & Shiekhattar, R. MicroRNA biogenesis and cancer. Cancer Res 65, 3509–3512 (2005).
Bueno, M. J. & Castro, I. P. De & Malumbres, M. Control of cell proliferation pathways by microRNAs. Cell Cycle 7, 3143–3148 (2008).
Ender, C. et al. A human snoRNA with microRNA-like functions. Mol Cell 32, 519–528 (2008).
Scott, M. S., Avolio, F., Ono, M., Lamond, A. I. & Barton, G. J. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput Biol 5, 1–13 (2009).
Qiu, C., Chen, G. & Cui, Q. Towards the understanding of microRNA and environmental factor interactions and their relationships to human diseases. Sci Rep 2, 1–7 (2012).
Bruno, A. E. et al. miRdSNP: a database of disease-associated SNPs and microRNA target sites on 3'UTRs of human genes. BMC Genomics 13, 1–7 (2012).
R Development Core Team. R: A Language and Environment for Statistical Computing. (2013). at http://www.r-project.org/
Lemon, J. Plotrix: a package in the red light district of R. R-News 6, 8–12 (2006).
Author information
Authors and Affiliations
Contributions
T.K.K.K. developed the rationale of miRNAVISA, preprocessed the data, wrote the miRNAVISA back-end software and prepared manuscript. A.R. designed and programmed the miRNAVISA web server front-end. J.A.C.A. guided the biological enquiry and interpretation. V.B.B. headed the research group and guided the preparation of the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Figure S1-S11 and Supplementary Table S1:
Supplementary Information
Supplementary Table S2
Supplementary Information
Supplementary Table S3
Supplementary Information
Supplementary Table S4
Supplementary Information
Supplementary Table S5
Supplementary Information
Supplementary Table S6
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kamanu, T., Radovanovic, A., Archer, J. et al. Exploration of miRNA families for hypotheses generation. Sci Rep 3, 2940 (2013). https://doi.org/10.1038/srep02940
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02940
This article is cited by
-
Effect of topical motesanib in experimental corneal neovascularization model
International Ophthalmology (2023)
-
Hsa-mir-548 family expression in human reproductive tissues
BMC Genomic Data (2021)
-
Single-molecule amplification-free multiplexed detection of circulating microRNA cancer biomarkers from serum
Nature Communications (2021)
-
Secreted breast tumor interstitial fluid microRNAs and their target genes are associated with triple-negative breast cancer, tumor grade, and immune infiltration
Breast Cancer Research (2020)
-
Quantification of purified endogenous miRNAs with high sensitivity and specificity
Nature Communications (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
