
Abstract
The aim of this study was to investigate the effects and the mechanism of diosgenin, a famous plant-derived steroidal sapogenin, on memory deficits in Alzheimer's disease (AD) model mice. Diosgenin-treated 5XFAD mice exhibited significantly improved performance of object recognition memory. Diosgenin treatment significantly reduced amyloid plaques and neurofibrillary tangles in the cerebral cortex and hippocampus. Degenerated axons and presynaptic terminals that were only observed in regions closely associated with amyloid plaques were significantly reduced by diosgenin treatment. The 1,25D3-membrane-associated, rapid response steroid-binding protein (1,25D3-MARRS) was shown to be a target of diosgenin. 1,25D3-MARRS knockdown completely inhibited diosgenin-induced axonal growth in cortical neurons. Treatment with a neutralizing antibody against 1,25D3-MARRS diminished the axonal regeneration effect of diosgenin in Aβ(1–42)-induced axonal atrophy. This is the first study to demonstrate that the exogenous stimulator diosgenin activates the 1,25D3-MARRS pathway, which may be a very critical signaling target for anti-AD therapy.
Similar content being viewed by others


Introduction
We have hypothesized that the enhancement of brain function requires the reinforcement of neuronal networks, including neurite regeneration and synapse formation; therefore, we have been exploring the use of anti-Alzheimer's disease (AD) drugs in reconstructing neuronal networks in the damaged brain. In the last decade, several strategies for lowering Aβ have been studied in basic research and clinical trials as an alternative to enhancing cholinergic function. However, immunotherapy with bapineuzumab, a humanized anti-Aβ monoclonal antibody, did not improve cognitive function in a phase 2 trial1. Although a γ-secretase inhibitor, LY450139, also reduced plasma and cerebrospinal fluid Aβ levels in humans, cognitive amelioration was not detectable2. Neuritic atrophy and loss of synapses underlie the pathogenesis of AD and are located upstream of neuronal death in the Aβ cascade3,4. The dysfunction of neurites and synapses is a direct cause of the memory deficit in AD. Because neurons with atrophic neurites may remain viable and have the potential to be remodeled, the essential event for the achievement of recovery of brain function after injury is the reconstruction of neuronal networks, including neurite regeneration and synaptic reformation5.
5XFAD mice are engineered to co-overexpress and co-inherit mutant human APP (the Swedish mutations: K670N and M671L; the Florida mutation: I716V; and the London mutation: V717I) and PS1 (M146L; L286V) transgenes under the neuron-specific mouse Thy-1 promoter6. Five familial AD mutations act together to additively increase the levels of cerebral Aβ peptides, especially the neurotoxic peptide Aβ42. While the majority of AD transgenic mice require 6–12 months or longer to form amyloid plaques7, 5XFAD mice begin to develop visible amyloid deposits as early as 2 months of age, which is consistent with their dramatically accelerated Aβ42 generation. In addition to the memory deficits in 5XFAD mice, demonstrated by contextual fear conditioning8 and their performance in a Y-maze6, we clarified the impairment of spatial memory9 and object recognition memory10,11 in these mice.
Previously, we found that the herbal drug-derived steroidal sapogenins regenerated neurite atrophy and synaptic loss, leading to memory improvement in AD model mice10,12. Diosgenin is also a steroidal sapogenin and a major constituent in Dioscorea rhizome and other herbal drugs, such as those from Trigonella spp., Polygonatum spp. and Smilax spp. Several biological effects of diosgenin have been reported, such as anti-cancer effects13, anti-food allergy effects14, anti-cognitive deficit effects15 and relief of diabetic neuropathy16. A diosgenin derivative, caprospinol (diosgenin 3-caproate), reduces amyloid deposits and improves memory dysfunction in Aβ1-42-infused AD model rats17. This result led us to hypothesize that diosgenin might also improve memory impairment in 5XFAD mice by decreasing Aβ. A variety of possible signaling pathways for diosgenin have been reported. For example, diosgenin enhanced PI3 kinase activity in melanogenesis18. In contrast, diosgenin attenuated the TNF-α-stimulated phosphorylation of Akt, ERK, JNK and p38 in a vascular smooth muscle cell line19. In hepatocellular carcinoma cell lines, diosgenin inhibited the phosphorylation of STAT3 and downstream c-Src, JAK1 and JAK220. However, the signaling mechanism of diosgenin in neuronal cells and the direct target protein remain unknown. Although several small-molecular-weight compounds derived from medicinal plants exhibit multiple bioactivities, the direct target proteins of those exogenous chemical compounds remain largely unknown. In the present study, we investigated the effects of diosgenin on memory deficits in 5XFAD mice and identified a direct target protein for diosgenin.
Results
To investigate the effects of diosgenin on impaired object recognition memory in 5XFAD mice, diosgenin (10 μmol/kg = 4.14 mg/kg), memantine (200 μmol/kg = 43.15 mg/kg) or vehicle solution was administered i.p. to mice for 20 days. The day after the last administration, an object recognition test was conducted. To assess the appropriate time interval between a training session and a test session, another group of mice had been tested previously and the test session was conducted 10 and 30 min and 24 h after the training session. We had determined the appropriate time interval between a training session and a test session in which control 5XFAD mice cannot memorize an object to be 30 min. Wild-type mice and diosgenin-treated 5XFAD mice showed significantly more frequent exploratory behavior in the presence of a novel object than chance (50%) (Figure 1A). The value of the diosgenin-treated group in the test session was significantly higher than that in the training session. In contrast, the exploratory behavior in the presence of the novel object in the vehicle-treated 5XFAD mice was close to chance (50%). The memantine-treated group showed no improvement in memory. In the open field locomotion test, no significant differences were detected in the moving velocities and distances moved of the four groups in the test (data not shown).

Effects of diosgenin on object recognition memory deficits and AD pathologies in 5XFAD mice.
Diosgenin (10 μmol/kg, i.p.), memantine (200 μmol/kg, i.p.) or vehicle solution was administered for 20 days to mice (females, 6–8 months old). The day after the last administration, an object recognition test was carried out. (A) The preferential indices of the training and test sessions are shown. (*p < 0.05 vs. vehicle-treated 5XFAD mice, one-way ANOVA post hoc Dunnett's test; #p < 0.05 vs. the same group in the training session, paired t-test; n = 4–6 mice). (B–D) The total area of amyloid plaques and PHF-tau in the cerebral cortex and hippocampus were measured in vehicle-treated wild-type, vehicle-treated, diosgenin-treated and memantine-treated 5XFAD mice. (B) The photographs are representative images of Aβ(1−40/42)-positive plaques and PHF-rau in the cerebral cortex. (C) The total area of amyloid plaques per 1 μm2 was quantified in the cerebral cortex and hippocampus. (D) The total area of PHF-tau per 100 μm2 was quantified in the cerebral cortex and hippocampus. The regions associated with amyloid plaques and distal to amyloid plaques were measured separately (*p < 0.05, **p < 0.001, one-way ANOVA post hoc Dunnett's test; the number of measured areas is shown in each column).
The day after the novel recognition test, the brains of the mice were removed and slices were prepared for immunohistochemistry. We measured the parietal area containing the parietal cortex and hippocampus at the bregma in 1.4–2-mm sections of the brain. The area of the amyloid deposits was measured in the brain slices. A large number of amyloid deposits were primarily localized in the cerebral cortex and hippocampus of 5XFAD mice. As shown in Figure 1B, the large Aβ(1−40/42)-positive stains (greater than 50 μm in width) were measured as extracellular amyloid deposits, although intracellular inclusions were also observed infrequently in the 5XFAD mice. In the age-matched wild-type mice, no amyloid plaque was observed. The diosgenin-treated 5XFAD mice exhibited a significant reduction of the amyloid plaques in the cerebral cortex and hippocampus (Figure 1C). Memantine also significantly reduced amyloid plaques in the cortex and hippocampus (Figure 1C).
Intense paired helical filament (PHF)-tau expression was observed very close to the amyloid plaques, as well as in the distal area of the amyloid plaques in the 5XFAD mice (Figure 1B). In the wild-type mice, no PHF-tau expression was observed. Diosgenin-treated 5XFAD mice showed a significant reduction of PHF-tau expression in the cortex (associated with and distal to the plaques) and the hippocampus (distal to the plaques) (Figure 1D). Memantine yielded no reduction of PHF-tau expression (Figure 1D).
Bulb-like axons, which were limited to the amyloid plaques, were markedly observed (Figure 2A). Abnormal axon structures, such as bulb shapes and dense staining by the pNF-H antibody, were reported in early- and end-stage AD patients3. In addition, in the PSAPP transgenic mouse AD model, abnormally swollen varicosities of neurites inside fibrillar amyloid deposits were shown in degenerated and dying axons21. With the exception of amyloid plaques in the brain, no abnormal structures were detected in the vehicle-treated 5XFAD and diosgenin-treated 5XFAD mice. The areas of bulb-like swollen axons in the cortex and hippocampus were significantly reduced by diosgenin treatment (Figure 2B). However, memantine treatment did not reduce the swollen axons (Figure 2B).

Effects of diosgenin on axonal and presynaptic abnormalities associated with amyloid plaques.
(A) Double labeling with antibodies to Aβ(1−40/42) (red) and pNF-H (green) or Aβ(1−40/42) (red) and synaptophysin (green) was carried out. Bulb-like axonal structures positive for pNF-H antibody and swollen puncta positive for synaptophysin antibody were localized only in the amyloid plaque areas. Representative images of the cerebral cortex of vehicle-treated wild-type (Wild/Veh) mice, as well as 5XFAD (5XFAD/Veh), diosgenin-treated 5XFAD (5XFAD/Dios) and memantine-treated 5XFAD (5XFAD/Mema) mice, are shown. (B) The quantified areas of abnormal axons per 100 μm2 of amyloid plaque are shown in the cerebral cortex and hippocampus. (C) The quantified areas of abnormal presynapses per 100 μm2 of amyloid plaque are shown in the cerebral cortex and hippocampus (*p < 0.05, **p < 0.001, one-way ANOVA post hoc Dunnett's test; the number of measured areas is shown in each column).
Abnormally swollen presynaptic boutons closely associated with amyloid plaques were also reported as degenerative alterations in 5XFAD mouse cortex22 and aged monkey cortex23. Similar presynaptic alterations were observed in the current study. As shown in Figure 2A, synaptophysin-positive staining in the distal area of amyloid plaques exhibited a minute dot-like pattern in the 5XFAD mice. However, synaptophysin-positive staining associated with amyloid plaques in the vehicle-treated 5XFAD mouse was very dense and exhibited large, swollen dots. In contrast, the abnormal presynaptic structures were significantly reduced by diosgenin treatment in the cerebral cortex but not the hippocampus (Figure 2C). Memantine showed no effects on the reduction of presynaptic degeneration (Figure 2C).
We used the target identification using drug affinity responsive target stability (DARTS) method to identify a direct target protein for diosgenin in neurons24. Cell lysate of mouse cortical neuron primary culture was added to diosgenin or vehicle solution and was then reacted with thermolysin. A 57-kDa protein band in the diosgenin-treated lysate was shown to be thicker than in the vehicle-treated lysate on a silver-stained SDS-PAGE gel (Figure 3A). LC/MS/MS analysis indicated that the band was possibly the protein disulfide-isomerase A3 (Pdia3). The Pdia3 protein has many synonyms, including ERp57, GRP58 and the membrane-associated rapid response steroid-binding receptor (1,25D3-MARRS). This protein is present in multiple subcellular locations, including the endoplasmic reticulum, plasma membrane, cytosol and nucleus25,26. 1,25D3-MARRS was reported to be expressed on the cell surface and to mediate the rapid response of 1α,25-dyhydroxyvitamin D3 (DHVD3)27. The genomic effects of DHVD3 are mediated by a nuclear receptor, nVDR, which operates through binding to DNA and activating gene expression. In contrast, the rapid and non-genomic cell response after the stimulation of DHVD3 is mediated by the cell surface function of 1,25D3-MARRS28 and activates numerous signal transduction cascades, including protein kinase C (PKC), protein kinase A (PKA), extracellular-response-activated kinase (ERK) and phosphoinositide 3-kinase (PI3K)27,29,30. As the studies of 1,25D3-MARRS have primarily used intestinal and osteoblast cells, its function in the brain has never been investigated. The conformation state of DHVD3 is 6-s-trans, which causes it to associate mainly with nVDR31. In contrast, the 6-s-cis conformation of DHVD3 and a 6-s-cis locked derivative of DHVD3, 1α,25-dyhydroxylumisterol, stimulated a rapid response32. Because the core structure of diosgenin is similar to that of the derivative 1α,25-dyhydroxylumisterol, we focused on the possibility that 1,25D3-MARRS might mediate the diosgenin signal as a cell surface receptor.

1,25D3-MARRS as a direct target protein of diosgenin.
(A) Mouse cortical neuron primary cultures (ddY, E14) were maintained for 24 h without drug treatment. Cell lysate was added to diosgenin or vehicle solution and incubated for 30 min at 4°C. The mixture was proteolysed using thermolysin and electrophoresed. The separated proteins were stained. The band in the diosgenin-treated lysate (diosgenin lane) that was thicker than that in the vehicle-treated lysate (Cont lane) was cut out and prepared for mass spectrometry analysis. (B) Double labeling with antibodies to 1,25D3-MARRS (Ab099 clone, red) and MAP2 (green) was carried out in primary cultures of rat cortical neurons. (C) The dose-dependency of cell surface binding of DHVD3 in primary culture cortical neurons. Diosgenin (0–100 μM) was added to cells and incubated for 10 min. The DHVD3 binding to the cell-surface was detected using immunocytochemistry with an anti-DHVD3 antibody. (D) The replacement of the cell-surface binding of DHVD3 with diosgenin. DHVD3 (0.1 μM) and diosgenin (0.1 or 10 μM) were added to the cells and incubated for 10 min. The DHVD3 binding to the cell surface was detected by immunocytochemistry using an anti-DHVD3 antibody.
The expression of 1,25D3-MARRS was investigated in cortical cell primary culture using a specific antibody for the 1,25D3-MARRS, Ab099 clone. An Ab099-positive signal was distributed mainly in neurons but not astrocytes (Figure 3B). A cell binding assay was performed to detect the binding activity of diosgenin to cell-surface 1,25D3-MARRS. Without triton-X, the binding of DHVD3 on cortical neurons was detected using an immunoluminescence assay. DHVD3 binding was detected in a dose-dependent manner (0.0001–100 μM) (Figure 3C). Because 0.1 μM DHVD3 exhibited submaximal binding, the replacement of DHVD3 (0.1 μM) with diosgenin was investigated. The simultaneous treatment with diosgenin (0.1 and 10 μM) reduced the DHVD3 (0.1 μM) binding (Figure 3D). The binding sites of 1,25D3-MARRS and nVDR to DHVD3 and diosgenin were predicted using the docking suite Autodock 4.0 (http://autodock.scripps.edu/). Figure 4 shows the calculated binding structure of 1,25D3-MARRS to diosgenin. The groove area between the α and β domains of 1,25D3-MARRS contacts diosgenin (Figure 4). The docking scores of diosgenin and DHVD3 to 1,25D3-MARRS were −8.4 and −7.9, respectively (Table 1) and the docking scores of diosgenin and DHVD3 to nVDR were −8.2 and −12.6, respectively (Table 1). These results suggest that the binding potency of diosgenin to 1,25D3-MARRS is slightly higher than that of DHVD3 and that the binding potency of DHVD3 to nVDR is much higher than that of diosgenin.

Docking simulation of diosgenin to 1,25D3-MARRS.
The predicted binding complex site of 1,25D3-MARRS and diosgenin is shown (A). The groove area between the α domain and the β domain of 1,25D3-MARRS contacts diosgenin (B).
To investigate the function of 1,25D3-MARRS in diosgenin-induced axonal extension, 1,25D3-MARRS was knocked down using siRNA transfection. Two days after the transfection of siRNA for 1,25D3-MARRS (400 nM), the 1,25D3-MARRS level was markedly reduced in cortical neurons (Figure 5A). In contrast, nVDR expression in cortical neurons was not changed by the transfection of siRNA for 1,25D3-MARRS, suggesting that the knockdown did not affect the genomic signaling of DHVD3 (Figure 5A). Therefore, 2 days after siRNA transfection, denosomin (1 μM), DHVD3 (1 μM) or vehicle solution was applied to the cells. After incubation for 4 additional days, the cells were double-immunostained for pNF-H and MAP2 to measure the axonal length per neuron. In cortical neurons, after the transfection of control siRNA, the density of axons was significantly increased by diosgenin and DHVD3 treatments (Figure 5B). However, both diosgenin-induced and DHVD3-induced axonal growth were almost completely inhibited in neurons after the transfection of 1,25D3-MARRS siRNA (Figure 5B). These data indicate that 1,25D3-MARRS is essential for the diosgenin-induced extensions of axons.

The effect of 1,25D3-MARRS knockdown on diosgenin-induced axonal outgrowth.
(A) siRNA for Pdia3 (400 nM) or control siRNA (400 nM) was transfected into rat cortical neurons. Two days later, double labeling with antibodies to 1,25D3-MARRS (Ab099 clone) and MAP2, or to nVDR and MAP2, was carried out. The expression levels of 1,25D3-MARRS and nVDR in neurons were quantified (*p < 0.05, **p < 0.001, one-way ANOVA post hoc Dunnett's test; the number of measured areas is shown in each column). (B) siRNA for Pdia3 (400 nM) or control siRNA (400 nM) was transfected into cortical cells. Two days later, the cells were incubated with 1 μM diosgenin, 1 μM DHVD3 or the vehicle solution (Veh, 0.1% DMSO). Four days after drug administration, the cells were fixed and double-immunostained for pNF-H and MAP2. The density of pNF-H-positive axons per MAP2-positive neuron was quantified for each treatment (*p < 0.05, one-way ANOVA post hoc Dunnett's test, n = 12–14).
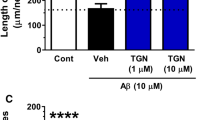
To investigate which protein kinases are involved in diosgenin-induced axonal growth signaling, a specific inhibitors for PI3K, MEK1, PKC or PKA was co-applied with diosgenin. The doses of inhibitors used were determined from the known IC50 values. The PI3K inhibitor LY294002 (10 μM), the MEK1 inhibitor PD98059 (1 and 10 μM), the protein kinase C inhibitor Gő6970 (10 and 100 nM) and the protein kinase A inhibitor PKI (0.1 and 1 μM) completely inhibited diosgenin-induced axonal growth (Figure 6A). DHVD3-induced axonal growth was also completely inhibited by LY294002 (1 and 10 μM), PD98059 (1 and 10 μM), Gő6970 (100 nM) and PKI (0.1 and 1 μM).

Effects of protein kinase inhibitors on diosgenin-induced axonal growth.
Cortical neurons were cultured for one day and then treated with 1 μM diosgenin (A), DHVD3 (B) or vehicle solution. Simultaneously, the PI3K inhibitor LY294002 (10 μM), the MEK1 inhibitor PD98059 (1 and 10 μM), the protein kinase C inhibitor Gő6970 (10 and 100 nM) or the protein kinase A inhibitor PKI (0.1 and 1 μM) was applied to the cells. Four days after the treatment, the cells were fixed and double-immunostained for pNF-H and MAP2. The density of pNF-H-positive axons per MAP2-positive neuron was quantified for each treatment (*p < 0.05, **p < 0.001, one-way ANOVA post hoc Dunnett's test, n = 6–7).
We investigated the effects of diosgenin on axonal regeneration in vitro and the effects of neutralizing 1,25D3-MARRS on diosgenin-induced axon regeneration in rat cortical neurons. Three days after treatment with Aβ(1–42), normal IgG as a control or Ab099, a specific neutralizing antibody for 1,25D3-MARRS28 was applied to the cells. After the 10-min incubation, cells were treated with diosgenin (0.1 and 1 μM) or vehicle solution. Aβ(1–42) treatment markedly reduced the density of axons. In contrast, treatment with Ab099 completely diminished the axonal growth induced by diosgenin (Figures 7A and 7B).

Effect of the 1,25D3-MARRS neutralizing antibody on diosgenin-induced axonal regeneration.
Cortical neurons were cultured for three days and then treated with or without aggregated Aβ(1–42) (5 μM). Three days after the administration of Aβ(1–42), the cells were treated with Ab099 antibody (Ab099 ab) or normal rabbit IgG (Control ab). After a ten-minute incubation period, diosgenin (0.1 and 1 μM) or vehicle solution (0.1% DMSO, Veh) was administered to the cells. Five days after treatment, the cells were fixed and double-immunostained for pNF-H and MAP2. The density of pNF-H-positive axons per MAP2-positive neuron was quantified for each treatment (A) (*p < 0.05, one-way ANOVA post hoc Dunnett's test, n = 7–9). (B) Representative images of each treatment are shown. The concentration of diosgenin was 1 μM.
Discussion
The 5XFAD mouse model reveals AD pathologies at an early age. Memory dysfunctions in the 5XFAD mice, including deficits in object recognition memory10,11, spatial memory9 and contextual fear memory8, were detected at 4 months old. The present study showed for the first time that diosgenin administration improved the object recognition memory deficit in 5XFAD mice. Diosgenin administration inhibited several signs of neuronal degeneration, including presynaptic degeneration associated with amyloid plaques in the cortex, axonal degeneration associated with amyloid plaques in the cortex and hippocampus and PHF-tau expression associated with and distal to amyloid plaques in the cortex and hippocampus. In addition, amyloid plaques were decreased by diosgenin treatment. It was reported that the diosgenin derivative, caprospinol (diosgenin 3-caproate), reduced amyloid deposits and improved memory dysfunction in Aβ1-42-infused AD model rats17. Therefore, it is possible that the amyloid plaque decreasing effect of diosgenin results in the protection of axonal degeneration and the hyperphosphorylation of tau. However, diosgenin can enhance axonal growth in normal neurons (Figures 5 and 6) and can induce the regrowth of axons in Aβ-treated neurons following treatment (Figure 7). In addition, diosgenin shows no direct binding activity to Aβ33. These results indicate that diosgenin may contribute to axonal extension through a direct pathway, as well as through the amyloid plaque-lowering pathway. The approved anti-AD drug memantine caused no improvement in object recognition (Figure 1B), the degeneration of axons and presynapses (Figure 1D) or PHF-tau expression (Figures 2B and 2C), but it did reduce amyloid plaques (Figure 1C). The reduction of amyloid plaques by memantine was previously reported in the AD model Tg2576 mice at 10 and 20 mg/kg34 and APP/PS1 mice at 10 mg/kg35, although the mechanism by which it occurred is unknown. Conflicting results have been reported concerning memory improvement due to memantine. Fear-conditioned memory is not improved by treatment with 10 or 20 mg/kg memantine for 6 months in Tg2576 mice, despite the reduction of amyloid plaques34. The administration of 10 mg/kg memantine for 4 months improved object recognition in APP/PS1 mice35, but the administration of 10 mg/kg memantine for 1 week did not affect the spatial memory deficit in APP23 mice36. Surprisingly, memantine treatment significantly increased the number of degenerated axons in the dentate gyrus of Tg2576 mice34. Additionally, in our study, memantine increased the number of degenerated axons in the hippocampus (Figure 2). These results suggest that memantine may have both neuroprotective and neurotoxic effects. In our previous study, a higher dose of diosgenin (100 μmol/kg) was administered to 5XFAD mice (male, 7–9 months old) by i.p. injection for 19 days. In object recognition test, diosgenin-treated 5XFAD mice showed memory improvement. Since the efficacy of diosgenin at a dose of 100 μmol/kg was almost same to that of 10 μmol/kg diosgenin, we consider that 10 μmol/kg diosgenin is enough to improve memory dysfunction.
Figure 2 shows that drastic axonal and presynaptic morphological changes occurred and were associated with amyloid plaques in 5XFAD mice. Other groups have also reported these axonal and synaptic degenerations in AD patients3, 5XFAD mice22 and other AD model mice21. Because a bulb-shaped axon overlaps with a swollen presynapse in the early and late stages of AD37, a terminal of the degenerated axon may also be degenerated and synaptophysin-positive synaptic vesicles may be diffused. The core structure of degenerated axons and presynapses is called a neuritic plaque, which is one of the pathological hallmarks of AD. These morphological degenerations of axons and presynapses may lead to the disruption of neuronal networks. It is possible that Aβ accumulation is closely related to the dystrophy of neurites and synapses. For example, the application of an anti-Aβ antibody to the exposed brain surface of the PDAPP mouse, an AD model, reduced the number and size of swollen dystrophic neurites around the site of amyloid plaques38. In addition, axonal dystrophy triggers the production and release of Aβ, leading to the progression of neuritic plaque formation22. Axonal dystrophy is recognized as a critical degeneration associated with amyloid plaques. Because diosgenin-induced reductions of degenerated axons and presynapses were observed in each amyloid plaque, these reductions may be independent of the decrease in amyloid plaques caused by diosgenin. Memantine reduced amyloid plaques but did not provide amelioration of axonal and presynaptic degeneration, also suggesting that decreasing the number of amyloid plaques is not necessary or sufficient for normalizing axonal structure and function. We then investigated the diosgenin signaling pathway to clarify the mechanism of restoration of axonal degeneration in AD.
Interestingly, our results indicated that the receptor mediating DHVD3 signaling also works as a diosgenin receptor. Although the function of 1,25D3-MARRS in neurons and neurodegenerative diseases was not unknown, the receptor, as a rapid and non-genomic response mediator, was considered to interact with multiple signaling pathways in intestinal and osteoblast cells, including PKA27,28, PKC27,29,39, ERK29, PI3K30 and STAT326. Our results also show that diosgenin stimulates PI3K, ERK, PKC and PKA (Figure 6). However, no reports have yet elucidated why the multiple signaling pathways can be activated by 1,25D3-MARRS. It also remains unknown how 1,25D3-MARRS is associated with the plasma membrane, although 1,25D3-MARRS has one myristoylated site at amino acid residues 315–32040. Although a neurite outgrowth effect of DHVD3 was reported in embryonic rat hippocampal neurons, it is unknown whether the effect is mediated by a genomic or non-genomic response41. The PI3K-Akt pathway is well known to regulate local protein translation via the mTOR pathway, thus playing an important role in axon regeneration42. PI3K also regulates Cdc42, which is a key regulator of cytoskeletal reorganization in axonal tips43. ERK signaling is also required for local axon assembly44 and local protein translation at the growth cone45. The phosphorylation of GAP-43 by PKC in growth cones is required for axonal outgrowth46. The PKA pathway is also known to be associated with axonal extension in cortical neurons47. The non-genomic action of DHVD3 is known to enhance the cerebral clearance of the Aβ(1–40) peptide in mice48 and ERp57 is one of the carrier proteins that prevents the aggregation of Aβ in cerebrospinal fluid49, suggesting that the diosgenin-stimulated 1,25D3-MARRS pathway may contribute to Aβ elimination from the brain. The PI3K-Akt and MEK-ERK pathways are reported to promote nonamyloidogenic cleavage of APP, resulting in a decrease in Aβ50. Although we need to investigate other possible causes of Aβ lowering by diosgenin, such as β-secretase, γ-secretase, Aβ accumulation inhibitors and Aβ-degrading enzymes, the multiple signaling pathways stimulated by diosgenin may contribute to the enhancement of axonal growth and Aβ reduction. Our unpublished study for identifying phosphorelated proteins by diosgenin using 5XFAD mice showed that downstream molecules of PI3K activation (PDK1, Akt1 and XIAP), a PKA substrate (CREB), a PKC substrate (MARCKS) and upstream molecules of MEK1 (Raf1 and MEKK1) and a downstream molecule (RSK2) were phosphorylated 30 min after diosgenin treatment in the hippocampus (data not shown). Those results suggest that diosgenin may stimulate multiple signal pathways also in vivo.
The nVDR-mediated genomic action of DHVD3 against axonal damage has not been elucidated, although nVDR expression is rich in most areas of the brain51. It has been reported that the in vitro exposure of cortical neurons to Aβ1-42 reduced nVDR expression and induced cytotoxicity and the addition of DHVD3 up-regulated nVDR expression and inhibited cell death52. However, there are no reports demonstrating any relation between axonal degeneration and nVDR. In our study, nVDR was not involved in diosgenin- or DHVD3-induced axonal growth in cortical neurons (Figure 5), suggesting that the 1,25D3-MARRS cascade is related to axonal growth and regrowth events.
A docking simulation suggested that the binding potency of diosgenin to 1,25D3-MARRS is greater than that of DHVD3. In contrast, the binding potency of diosgenin to nVDR is less than that of DHVD3. Although the conformation of DHVD3 is flexible, approximately 95% of it exists in the non-steroidal and extended 6-s-trans form31,53. In addition, the biological concentration of DHVD3 is strictly controlled and metabolized to 1α,24,25-dihydroxyvitamin D354 because hyper DHVD3 causes adverse effects, such as hypercalcemia. Therefore, diosgenin is an exogenous stimulator that allows the 1,25D3-MARRS cascade to work more effectively than the endogenous agonist.
This is the first report of diosgenin successfully recovering the memory deficit in 5XFAD mice and restoring axonal and presynaptic degeneration in the cerebral cortex and hippocampus. We also found that diosgenin may function as an exogenous stimulator of 1,25D3-MARRS and induce axonal growth and regrowth, even under Aβ-induced damaging conditions. These results suggest that the 1,25D3-MARRS pathway is activated by the exogenous stimulator diosgenin and may be a very critical signaling pathway for anti-AD therapy.
Methods
All experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Sugitani Campus of the University of Toyama. All protocols were approved by the Committee for Animal Care and Use of the Sugitani Campus of the University of Toyama. The approval number for the animal experiments is S2008-INM-6 and the confirmation number for the gene recombination experiments is 20-1. All efforts were made to minimize the number of animals used.
Materials
Diosgenin (Wako, Osaka, Japan), memantine hydrochloride (Tokyo Chemical Industry, Tokyo, Japan) and 1α,25-dihydroxy vitamin D3 (DHVD3), Cayman Chemicals, Ann Arbor, MI, USA) were dissolved in dimethyl sulfoxide (DMSO). Aβ(1–42) (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in sterile distilled water and incubated for 4 days at 37°C. The phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (Cayman Chemicals), MEK1 inhibitor PD98059 (LC laboratories) and protein kinase C inhibitor Gő6970 (LC laboratories, Woburn, MA, USA) were dissolved in DMSO. The protein kinase A inhibitor PKI (14–22) amide (myristoylated) (Enzo, Farmingdale, NY, USA) was dissolved in distilled water.
Animals
Transgenic mice (5XFAD) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The 5XFAD mice have the following five mutations: Swedish (K670N and M671L), Florida (I716V) and London (V717I) in human APP695 cDNA and human PS1 cDNA (M146L and L286V) under the transcriptional control of the neuron-specific mouse Thy-1 promoter6. They were maintained by crossing hemizygous transgenic mice with B6/SJL F1 breeders. To investigate the effect of diosgenin on 5XFAD, the study used hemizygous 5XFAD mice (female, 6–8 months old) and non-transgenic littermate wild-type mice (female, 6–8 months old), which were obtained by crossing a hemizygous 5XFAD mouse and a B6/SJL F1 mouse. All mice were housed with free access to food and water and were kept in a controlled environment (22 ± 2°C, 50 ± 5% humidity, 12-h light/dark cycle starting at 7:00 am).
Novel object recognition test
Diosgenin or memantine hydrochloride was dissolved in DMSO at 10 times the final concentration and the stock solution was diluted in physiological saline to the final concentration immediately before treatment. The drug or vehicle solution (10% DMSO in physiological saline) was intraperitoneally administered once a day for 20 days. On the last day of drug administration, mice were individually habituated to an open-field box composed of polyvinyl chloride (33 cm×28 cm; height, 26.5 cm) for 10 min. Their paths were tracked using a digital camera system. The distance moved for 10 min was analyzed as the locomotion activity with EthoVision 3.0 (Noldus, Wageningen, The Netherlands). The next day, a novel object recognition test was performed as described previously10,11. Testing was carried out in a dimly illuminated room.
Immunohistochemistry
One day after the novel object recognition test, mice were anesthetized and transcardially perfused with cold physiological saline. The brains were carefully removed from the skull, immediately immersed in 10 to 30% sucrose-PBS and stored at −80°C. The brains were cut in 20-μm successive coronal slices every 100 μm in the parietal area (bregma 1.4–2 mm) sections using a cryostat (CM3050S, Leica, Heidelberg, Germany). The slices were fixed with 4% paraformaldehyde and stained with a polyclonal antibody against Aβ(1−40/42) (1∶300) (Chemicon, Temecula, CA, USA), a monoclonal antibody against pNF-H (1∶500) (Covance, Emeryville, CA, USA), a monoclonal antibody against synaptophysin (1∶500) (Sigma-Aldrich) and a monoclonal antibody against PHF-tau (1∶100) (Thermo Scientific, Rockford, IL, USA) at 4°C for 20 h. Alexa Fluor 488-conjugated goat anti-mouse IgG (1∶300) and Alexa Fluor 568-conjugated goat anti-rabbit antibody (1∶300) were used as secondary antibodies (Molecular Probes, Eugene, OR, USA). The fluorescent images for axons, presynapses, PHF-tau and Aβ(1−40/42) were captured using a fluorescent microscope (AX-80) at 324 μm × 430 μm (for pNF-H, synaptophysin and PHF-tau) or 1620 μm×2150 μm (for Aβ(1−40/42)). Three successive brain slices of the frontal cortex and five successive slices of the hippocampus were captured from a mouse for quantification. Extracellular amyloid plaques were determined by the size (greater than 50 μm in width). The area of amyloid plaques was measured using the image analyzing software ImageJ (http://rsbweb.nih.gov/ij). The area of the pNF-H-positive abnormal bulb-like axons was measured using ImageJ for every amyloid plaque in the fluorescent images (324 μm×430 μm). The areas of the synaptophysin-positive presynaptic boutons in every amyloid plaque were measured in fluorescent images (324 μm×430 μm) using Image J. The areas of PHF-tau in every amyloid plaque and outer amyloid plaques were measured in fluorescent images (324 μm×430 μm) using ImageJ.
Primary culture
Embryos were removed from pregnant Sprague-Dawley rats (Japan SLC, Shizuoka, Japan) at 17 days of gestation. The cortices were dissected and the dura mater was removed. The tissues were minced, dissociated and grown in cultures with neurobasal medium (Invitrogen, Grand Island, NY, USA) that included 12% B-27 supplement (Invitrogen), 0.6% D-glucose and 2 mM L-glutamine on 8-well chamber slides (Falcon, Franklin Lakes, NJ, USA) coated with 5 μg/ml poly-D-lysine at 37°C in a humidified incubator with 10% CO2. The seeding cell density was 4.35×104 cells/cm2.
Measurement of axonal density
To measure the density of axons, the cells were treated with 5 μΜ Aβ(1−42) for 3 days (Figure 7) or without Aβ(1−42) for 4 days (Figures 5 and 6) and they were treated with diosgenin or vehicle (0.1% DMSO) for 3 days. The Aβ(1−42) was previously incubated for 4 days at 37°C for aggregation. When treated with the neutralizing antibody, the control antibody (normal rabbit IgG, 1∶5000) or the polyclonal anti-rabbit 1,25D3-MARRS (Ab099 clone, 1∶500) was applied to the cells 3 days after the Aβ(1−42) treatment. Ten minutes after incubation with the antibody, the cells were treated with vehicle solution (0.1% DMSO) or diosgenin (0.1 and 1 μM). The cells were fixed with 4% paraformaldehyde and immunostained at 4°C for 20 h with a monoclonal antibody against pNF-H (1∶500) as an axonal marker and a polyclonal antibody against MAP2 (1∶500) as a neuronal marker. Alexa Fluor 488-conjugated goat anti-mouse IgG (1∶300) and Alexa Fluor 568-conjugated goat anti-rabbit IgG (1∶300) were used as secondary antibodies. The fluorescent images were captured using a fluorescence microscope system (BX61/DP70, Olympus) at 324 μm×430 μm. Twelve to fourteen images (Figure 5B), 6 to 7 images (Figure 6) or 7 to 9 images (Figure 7A) were captured per treatment. The lengths of the pNF-H-positive axons were measured using an image analyzer Neurocyte (Kurabo), which automatically traces and measures neurite length without measuring the cell bodies. The sum of the axon lengths was divided by the number of MAP2-positive neurons. The resulting axon densities were averaged over all the images and are shown in Figures 5B, 6 and 7A.
Identification of target proteins for diosgenin
A mouse cortical neuron primary culture (ddY, E14) was maintained for 24 h without drug treatment. The cells were lysed with M-PER (Thermo Scientific) supplemented with protease and phosphatase inhibitors. After centrifugation (20,000 x g) for 15 min, the protein concentration of the lysates was determined using Quick Start (Bio-Rad Laboratories, Hercules, CA, USA). The cell lysate (5.6 μg each) was added to 2 μl of 10 mM diosgenin of vehicle solution and incubated for 30 min at 4°C. The mixture was proteolyzed using 7.5 μg thermolysin in reaction buffer [50 mM Tris·HCl (pH 8.0), 50 mM NaCl, 10 mM CaCl2] for 10 min at room temperature. To stop the proteolysis, 0.5 M EDTA (pH 8.0) was added to each sample in a 1∶10 ratio. The samples were then mixed well and placed on ice.
The reaction samples were electrophoresed using a 5–20% real gel plate (Bio Craft, Tokyo, Japan). The separated proteins were stained using a negative gel stain MS kit (Wako). The bands in the diosgenin-treated lysate that were thicker than those in the vehicle-treated lysate were cut out and prepared for mass spectrometry analysis. After alkylation using iodpacetamide and in-gel digestion by trypsin, the supernatants were analyzed using nano-LC/MS/MS on a ThermoFisher LTQ Orbitrap XL. The MS/MS data were searched using Mascot (www.matrixscience.com).
Luminescence detection of 1,25D3-MARRS binding
Primary cultured cortical neurons (SD, E17) were harvested in a 96-well white plate at a density of 0.5×105 cells/well. Three days later, the cells were treated with DHVD3 alone or in combination with DHVD3 and diosgenin for 10 min at 37°C. The cells were fixed with 4% paraformaldehyde and incubated at 4°C for 20 h with a monoclonal antibody against DHVD3 (1∶2000, Acris Antibodies, Herford, Germany) in Triton-X-free PBS to detect only the cell-surface binding of DHVD3. Horse radish peroxidase-conjugated goat anti-mouse IgG (1∶2000) was used as a secondary antibody. The chemiluminescence of the antigen-antibody complex was detected using a GENios multi-plate reader (Tecan, Männedorf, Zurich, Switzerland).
siRNA transfection
siRNAs were transfected into rat cortical neurons (SD E18) according to the manufacturer's protocol for nucleofection (Lonza, Bazel, Switzerland). In brief, rat cortical neurons (5.25×106 cells) were mixed with 400 nM siPdia3 (a mixture of three sequences of Stealth siRNA for Pdia3, Invitrogen) or 400 nM control siRNA (Stealth RNAi Negative Control Low GC Duplex #3, Invitrogen) and electroporated with Amaxa Nucleofector (Lonza). Two days after the transfection, cells were fixed and double-immunostained with a monoclonal antibody against MAP2a and 2b (1∶500, NeoMarker, Fremont, CA, USA), as a neuron marker and Ab099 clone, a specific antibody for 1,25D3-MARRS (1∶500, provided as a gift by Dr. Nemere). Rat polyclonal anti-nVDR antibody (1∶200, Millipore, Billerica, MA, USA) was used for the detection of the nuclear receptor for vitamin D3 (nVDR). The appropriate concentration of siRNA and the appropriate duration for knockdown (2 days after transfection) were determined previously. Two days after the transfection of siRNA, the cells were treated with diosgenin (1 μM), DHVD3 (1 μM) or vehicle solution (0.1% DMSO). Four days after treatment, the axon density was measured.
Docking simulation
Autodock 4.0 (AutoDock4 and AutoDockTools4; Molecular Graphics Laboratory. Department of Molecular Biology The Scripps Research Institute; http://autodock.scripps.edu/) was used to predict the conformation of the protein-ligand complexes55. Ligand structures (diosgenin and DHVD3) were constructed and minimized using MOPAC2009 for 2D and 3D conformation56. For the docking studies, the crystal structures of the proteins (PDB ID: NP_036630.1 for RLAR; NP_001619.1 for human AR) were determined from the protein sequence alignment [Brookhaven Protein Data Bank (PDB ID: 3f8u chain A for 1,25D3-MARRS; 1ie9 chain A for nVDR)]. The predicted protein ligand complexes were optimized and ranked according to the empirical scoring function, which estimates the binding free energy of the ligand receptor complex.
Statistical analysis
Statistical comparisons were performed using a one-way analysis of variance (ANOVA) post hoc Dunnett's test and a paired t-test using SigmaStat 3.5 (SYSTAT, Chicago, IL, USA) and Graphpad Prism 5 (Graphpad Software, La Jolla, CA, USA). Values of p < 0.05 were considered significant. The means of the data are presented together with the SE.
References
Salloway, S. et al. Bapineuzumab 201 Clinical Trial Investigators. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070 (2009).
Henley, D. B., May, P. C., Dean, R. A. & Siemers, E. R. Development of semagacestat (LY450139), a functional gamma-secretase inhibitor, for the treatment of Alzheimer's disease. Expert Opin Pharmacother 10, 1657–1664 (2009).
Dickson, T. C. & Vickers, J. C. The morphological phenotype of β-amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience 105, 99–107 (2001).
Terry, R. D. et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annal Neurol 30, 572–580 (1991).
Tohda, C., Kuboyama, T. & Komatsu, K. Search for natural products related to regeneration of the neuronal network. Neurosignals 14, 34–45 (2005).
Oakley, H. et al. Intraneuronal β- amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140 (2006).
Eriksen, J. L. & Janus, C. G. Plaques, tangles and memory loss in mouse models of neurodegeneration. Behav Genet 37, 79–100 (2007).
Ohno, M. Failures to reconsolidate memory in a mouse model of Alzheimer's disease. Neurobiol Learn Mem 92, 455–459 (2009).
Urano, T. & Tohda, C. Icariin improves memory impairment in Alzheimer's disease model mice (5xFAD) and attenuates amyloid β-induced neurite atrophy. Phytother Res 24, 1658–1663 (2010).
Joyashiki, E., Matsuya, Y. & Tohda, C. Sominone Improves Memory Impairments and Increases Axonal Density in Alzheimer's Disease Model Mice, 5XFAD. Int J Neurosci 2011; 121, 181–190.
Tohda, C., Nakada, R., Urano, T., Okonogi, A. & Kuboyama, T. Kamikihi-to (KKT) rescues axonal and synaptic degeneration associated with memory impairment in a mouse model of Alzheimer's disease, 5XFAD. Int J Neurosci 121, 641–648 (2011).
Tohda, C., Hashimoto, I., Kuboyama, T. & Komatsu, K. Metabolite 1 of protopanaxadiol-type saponins, an axonal regenerative factor, stimulates teneurin-2 linked by PI3-kinase cascade. Neuropsychopharmacol 31, 1158–1164 (2006).
Yan, L. L., Zhang, Y. J., Gao, W. Y., Man, S. L. & Wang, Y. In vitro and in vivo anticancer activity of steroid saponins of Paris polyphylla var. yunnanensis. Exp Oncol 31, 27–32 (2009).
Huang, C. H., Ku, A. Y. & Jan, T. R. Diosgenin attenuates allergen-induced intestinal inflammation and IgE production in a murine model of food allergy. Planta Med 75, 1300–1305 (2009).
Chiu, C. S. et al. Diosgenin ameliorates cognition deficit and attenuates oxidative damage in senescent mice induced by D-galactose. Am J Chin Med 39, 551–563 (2011).
Kang, T. H. et al. Diosgenin from Dioscorea nipponica ameliorates diabetic neuropathy by inducing nerve growth factor. Biol Pharm Bull 34, 1493–1498 (2011).
Lecanu, L. et al. Caprospinol reduces amyloid deposits and improves cognitive function in a rat model of Alzheimer's disease. Neuroscience 165, 427–435 (2010).
Lee, J., Jung, K., Kim, Y. S. & Park, D. Diosgenin inhibits melanogenesis through the activation of phosphatidylinositol-3-kinase pathway (PISK) signaling. Life Sci 81, 249–254 (2007).
Choi, K. W. et al. Inhibition of TNF-α-induced adhesion molecule expression by diosgenin in mouse vascular smooth muscle cells via downregulation of the MAPK, Akt and NF-κB signaling pathways. Vascul Pharmacol 53, 273–280 (2010).
Li, F., Fernandez, P. P., Rajendran, P., Hui, K. M. & Sethi, G. Diosgenin, a steroidal saponin, inhibits STAT3 signaling pathway leading to suppression of proliferation and chemosensitization of human hepatocellular carcinoma cells. Cancer Lett 292, 197–207 (2010).
Tsai, J., Grutzendler, J., Duff, K. & Gan, W. B. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nature Neurosci 7, 1181–1183 (2004).
Zhang, X. M. et al. β-secretase-1 elevation in transgenic mouse models of Alzheimer's disease is associated with synaptic/axonal pathology and amyloidogenesis: implications for neuritic plaque development. Eur J Neurosci 30, 2271–2283 (2009).
Cai, Y. et al. β-Secretase-1 elevation in aged monkey and Alzheimer's disease human cerebral cortex occurs around the vasculature in partnership with multisystem axon terminal pathogenesis and β-amyloid accumulation. Eur J Neurosci 32, 1223–1238 (2010).
Lomenick, B. et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci USA 106, 21984–21989 (2009).
Wu, W. et al. Nuclear translocation of the 1,25D3-MARRS (membrane associated rapid response to steroids) receptor protein and NFkappaB in differentiating NB4 leukemia cells. Exp Cell Res 316, 1101–1108 (2010).
Guo, G. G. et al. Association of the chaperone glucose-regulated protein 58 (GRP58/ER-60/ERp57) with Stat3 in cytosol and plasma membrane complexes. J Interferon Cytokine Res 22, 555–563 (2002).
Larsson, B. & Nemere, I. Effect of growth and maturation on membrane-initiated actions of 1,25-dihydroxyvitamin D3-II: calcium transport, receptor kinetics and signal transduction in intestine of female chickens. J Cell Biochem 90, 901–913 (2003).
Nemere, I. et al. Ribozyme knockdown functionally links a 1,25(OH)2D3 membrane binding protein (1,25D3-MARRS) and phosphate uptake in intestinal cells. Proc Natl Acad Sci USA 101, 7392–7397 (2004).
Rosso, A. et al. 1α,25(OH)2-Vitamin D3 stimulates rapid plasma membrane calcium influx via MAPK activation in immature rat Sertoli cells. Biochimie 94, 146–154 (2012).
Buitrago, C., Arango, N. & Boland, R. 1α,25(OH)(2) D(3)-dependent modulation of Akt in proliferating and differentiating C2C12 skeletal muscle cells. J Cell Biochem 113, 1170–1181 (2012).
Okamura, W. H. et al. Chemistry and conformation of vitamin D molecules. J Steroid Biochem Mol Biol 53, 603–613 (1995).
Dormanen, M. C. et al. Nonnuclear effects of the steroid hormone 1 alpha,25(OH)2-vitamin D3: analogs are able to functionally differentiate between nuclear and membrane receptors. Biochem Biophys Res Commun 201, 394–401 (1994).
Lecanu, L. et al. Identification of naturally occurring spirostenols preventing β-amyloid-induced neurotoxicity. Steroids 69, 1–16 (2004).
Dong, H., Yuede, C. M., Coughlan, C., Lewis, B. & Csernansky, J. G. Effects of memantine on neuronal structure and conditioned fear in the Tg2576 mouse model of Alzheimer's disease. Neuropsychopharmacol 33, 3226–3236 (2008).
Scholtzova, H. et al. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res 86, 2784–2791 (2008).
Van Dam, D., Abramowski, D., Staufenbiel, M. & De Deyn, P. P. Symptomatic effect of donepezil, rivastigmine, galantamine and memantine on cognitive deficits in the APP23 model. Psychopharmacol 180, 177–190 (2005).
Dickson, T. C., King, C. E., McCormack, G. H. & Vickers, J. C. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer's disease. Exp Neurol 156, 100–110 (1999).
Brendza, R. P. et al. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest 115, 428–433 (2005).
Nemere, I., Garbi, N., Hämmerling, G. J. & Khanal, R. C. Intestinal cell calcium uptake and the targeted knockout of the 1,25D3-MARRS (membrane-associated, rapid response steroid-binding) receptor/PDIA3/Erp57. J Biol Chem 285, 31859–31866 (2010).
Nemere, I., Safford, S. E., Rohe, B., DeSouza, M. M. & Farach-Carson, M. C. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25D3-MARRS) binding protein. J Steroid Biochem Mol Biol 89–90, 281–285 (2004).
Brown, J., Bianco, J. I., McGrath, J. J. & Eyles, D. W. 1,25-dihydroxyvitamin D3 induces nerve growth factor, promotes neurite outgrowth and inhibits mitosis in embryonic rat hippocampal neurons. Neurosci Lett 343, 139–143 (2003).
Verma, P. et al. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci 25, 331–342 (2005).
Shi, S. H., Jan, L. Y. & Jan, Y. N. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 112, 63–75 (2003).
Atwal, J. K., Massie, B., Miller, F. D. & Kaplan, D. R. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 27, 265–277 (2000).
Campbell, D. S. & Holt, C. E. Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron 37, 939–952 (2003).
Aigner, L. et al. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 83, 269–278 (1995).
Bouchard, J. F., Horn, K. E., Stroh, T. & Kennedy, T. E. Depolarization recruits DCC to the plasma membrane of embryonic cortical neurons and enhances axon extension in response to netrin-1. J Neurochem 107, 398–417 (2008).
Ito, S. et al. 1α,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-β peptide(1–40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS 8, 20 (2011).
Erickson, R. R. et al. In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind beta-amyloid. Biochem Biophys Res Commun 332, 50–57 (2005).
Shi, C. et al. Ginsenoside Rg1 promotes nonamyloidgenic cleavage of APP via estrogen receptor signaling to MAPK/ERK and PI3K/Akt. Biochim Biophys Acta 1820, 453–460 (2012).
Eyles, D. W., Smith, S., Kinobe, R., Hewison, M. & McGrath, J. J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat 29, 21–30 (2005).
Dursun, E., Gezen-Ak, D. & Yilmazer, S. A novel perspective for Alzheimer's disease: vitamin D receptor suppression by amyloid-β and preventing the amyloid-β induced alterations by vitamin D in cortical neurons. J Alzheimers Dis 23, 207–219 (2011).
Mizwicki, M. T. & Norman, A. W. The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling. Sci Signal 2, re4 (2009).
Deeb, K. K., Trump, D. L. & Johnson, C. S. Vitamin D signalling pathways in cancer: potential for anticancer therapeutics. Nat Rev Cancer 7, 684–700 (2007).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31, 455–461 (2010).
Stewart, J. J. Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements. J Mol Model 13, 1173–1213 (2007).
Acknowledgements
This work was partially supported by the Regional Innovation Cluster Program, a Global Type (II) grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Contributions
C.T. designed the study, wrote the protocol, carried out experiments and statistical analyses and prepared the manuscript. T.U. and M.U. carried out experiments and analyses. I.N. prepared and gifted an antibody. T.K. managed the literature searches and analyses. All authors contributed to and have approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tohda, C., Urano, T., Umezaki, M. et al. Diosgenin is an exogenous activator of 1,25D3-MARRS/Pdia3/ERp57 and improves Alzheimer's disease pathologies in 5XFAD mice. Sci Rep 2, 535 (2012). https://doi.org/10.1038/srep00535
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00535
This article is cited by
-
Diosgenin restores memory function via SPARC-driven axonal growth from the hippocampus to the PFC in Alzheimer’s disease model mice
Molecular Psychiatry (2023)
-
Anticancer Activity of Diosgenin and Its Molecular Mechanism
Chinese Journal of Integrative Medicine (2023)
-
Axonal Regeneration Mediated by a Novel Axonal Guidance Pair, Galectin-1 and Secernin-1
Molecular Neurobiology (2023)
-
MEK1/2 inhibition rescues neurodegeneration by TFEB-mediated activation of autophagic lysosomal function in a model of Alzheimer’s Disease
Molecular Psychiatry (2022)
-
Neuroprotective role of Diosgenin, a NGF stimulator, against Aβ (1–42) induced neurotoxicity in animal model of Alzheimer’s disease
Metabolic Brain Disease (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
