
Abstract
At present, few available drugs can be used to either improve pathological features or prevent the progression of Alzheimer’s disease (AD). DL0410 ((1,1′-([1,1′-biphenyl]-4,4′-diyl) bis (3-(piperidin-1-yl) propan-1-one) dihydrochloride) is a multiple-target small molecule that has been found to reverse cognitive impairment in different animal models of AD. In this study we evaluated the cognition-improving effects of DL0410 in APP/PS1 transgenic mice and explored the underlying mechanisms. APP/PS1 transgenic mice were administered DL0410 (3, 10, 30 mg· kg−1· d−1, ig) for 2 months. We found that DL0410 administration significantly ameliorated cognitive deficits in both the nest-building and Morris water maze tests. In electrophysiological analysis of hippocampal slices, we showed that DL0410 administration significantly enhanced the field EPSP slope and HFS-induced LTP in CA1 area. Furthermore, we revealed that DL0410 administration significantly increased the phosphorylation of AKT and the activity of GSK-3β in the hippocampus and cortex. Moreover, DL0410 administration dose-dependently increased the expression level of phosphorylated ERK1/2 in the hippocampus and cortex. In addition, DL0410 dose-dependently decreased the neuronal loss by decreasing the production of Aβ deposition, inhibited glial overactivation, and the production of inflammatory cytokines such as TNF-α, IL-1β, and IL-6. We conclude that DL0410 ameliorates cognitive deficits in APP/PS1 transgenic mice by promoting synaptic transmission via activating the AKT/GSK-3β and MAPK/ERK signaling pathway and reducing neuronal loss. DL0410 may be an effective agent for AD treatment in the future.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a complex and progressive neurodegenerative disease. It is estimated that over 2.1 billion people will be affected worldwide by the year 2050 [1], and it is becoming the great health care challenge in the 21st century. At present, little is known about the cause of AD, and no curative treatments are available. β-Amyloid protein (Aβ) deposits and neurofibrillary tangles (NFTs) are the typical pathological features of AD, but clinical trials targeting Aβ, including MK-8931 and solanezumab, have all failed [2,3,4,5]. Hence, it is important and necessary to identify treatment strategies besides single-target drugs based on the Aβ cascade hypothesis. A growing number of researchers have determined that multitarget drugs may be potentially helpful for curbing the progression of AD.
Increasing evidence has demonstrated that the progressive loss of synapses is an important characteristic of AD. Synapses are the sites of memory storage in the nervous system and the mechanism by which information is transferred, suggesting a relationship between synaptic plasticity degeneration and cognitive decline in AD [6, 7]. Long-term potentiation (LTP) has been widely used for evaluating synaptic plasticity both in vitro and in vivo. In terms of pathological mechanisms, the PI3K/AKT/GSK-3β and MAPK/ERK signaling pathways play an important role in AD. AKT (also known as protein kinase B) is a serine/threonine kinase that is believed to promote cell viability in many different cell types, including neurons, by phosphorylating and then inactivating downstream molecules such as the serine/threonine kinase glycogen synthase kinase-3 (GSK-3β) and the apoptosis-inhibiting factor Bcl-2. GSK-3 has also been reported to be an important kinase in tau phosphorylation in the central nervous system [8,9,10,11]. In addition, the MAPK/ERK signaling pathway and downstream transcription factors are associated with the activation of genomic programs involved in synaptic plasticity and the encoding of long-term memories [12, 13].
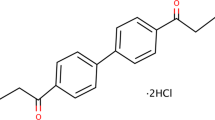
DL0410 is a multitarget small molecule that was selected from more than 100 000 compounds using high-throughput screening assays for AChE inhibitors, BuChE inhibitors and H3R antagonists [14,15,16], and its chemical structure is shown in Fig. 1. DL0410 has been shown to reverse cognitive impairments in AD animal models, including an Aβ1–42-induced amnesia mouse model, an APP/PS1 transgenic mouse model and a scopolamine-induced mouse model [17,18,19,20]. DL0410 can significantly improve learning and memory ability in the above mouse models. An investigation of the pathology and mechanisms of action indicated that DL0410 can reduce amyloid plaque deposition and mitochondrial damage, increase cholinergic neurotransmission and upregulate the neurotrophic CREB/BDNF signaling pathway. However, the mechanism of action of DL0410 in improving learning and memory ability is still unclear.

a The chemical structure of DL0410. b Schematic timeline of DL0410 treatment behavioral tests.
In the present study, we explored the effect of DL0410 on cognitive impairments of APP/PS1 mice and further investigated the potential mechanism by which DL0410 promotes synaptic transmission through the AKT/GSK-3β and ERK signaling pathways and reduces neuronal loss by decreasing Aβ deposition and inhibiting neuroinflammation and neuronal apoptosis. This study provides strong evidence for the anti-AD effect of DL0410.
Materials and methods
Drugs and reagents
DL0410 (purity > 98%) was obtained from the Institute of Materia Medica, Chinese Academy of Medical Sciences (Beijing, China). Donepezil hydrochloride was purchased from the Ji-nan Chenghui Shuangda Chemical Industry Limited Company (Ji-nan, China). Memantine was purchased from Shanghai Jingchun Chemical Industry Limited Company (Shanghai, China). Primary antibodies against β-amyloid precursor protein (APP) and brain-derived neurotrophic factor (BDNF) were purchased from Abcam (Cambridge, UK). Primary antibodies against AKT, p-AKT, GSK-3β, p-GSK-3β (Ser-9), Bax, Bcl-2, and p-ERK were purchased from CST (Danvers, MA, USA). An anti-rabbit IgG HRP-linked antibody and an anti-mouse IgG HRP-linked antibody were purchased from Shanghai Yisheng Biotechnology Limited Company (Shanghai, China). TNF-α, IL-1β, and IL-6 ELISA assay kits were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Animals and treatment
APP/PS1 double transgenic mice (strain name, B6C3-Tg (APPswe/PEN1dE9) 85Dbo/J) and wild-type (WT) C57/BL6J mice were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Mice were housed 4–5 per cage at 23 ± 2 °C under a 12/12-h day/night cycle and free access to food and water. All experimental procedures were conducted in accordance with institutional guidelines and ethics and were approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College.
Nine-month-old APP/PS1 mice and age-matched WT mice were randomly divided into seven groups: the WT group, APP/PS1 group, 3 mg/kg memantine group, 3 mg/kg donepezil group and 3, 10, and 30 mg/kg DL0410 groups. DL0410, memantine, and donepezil were dissolved in distilled water and were administered to the mice by oral gavage once daily for 8 weeks. Meanwhile, the WT mice received distilled water in the same manner. The body weight of each mouse was recorded twice per week (Fig. 1).
Nest-building test
To evaluate the social behavior of the mice, a nest-building test was performed as previously described [17, 21, 22]. APP/PS1 mice and WT mice were singly housed in for 24 h for acclimation. On the first day of testing, 12 pieces of soft paper (4 cm × 4 cm) were placed in the cage for nesting. The next day, nesting building was scored based on a 5-point scale as follows: 1 = paper not noticeably touched, 2 = paper partially torn up, 3 = paper mostly shredded on one side but no perceivable nest shape, 4 = a recognizable but flat nest, 5 = a nearly perfect or perfect nest.
Morris water maze test
To determine the spatial learning and memory abilities of the mice, the Morris water maze test was used [19, 23]. The water maze apparatus consisted of a circular water tank (100 cm in diameter, 35 cm high). The tank was divided into four quadrants and filled with white opaque water at 23 ± 1 °C. A small hidden platform (10 cm × 6.5 cm × 21.5 cm) was placed in the center of the northeast quadrant of the tank and submerged 1 cm below the water surface. In the acquisition trial, each mouse was subjected to two trials per day for five consecutive days. The latency required for the mice to find the hidden platform was recorded. After 5 days of training, the platform was removed, and the mice were allowed to swim freely for 60 s. The time for the mouse to first cross the area in which the platform had been located before and the distance traveled in the platform quadrant was recorded. All data were recorded with an automatic video system.
Electrophysiological recordings
After several behavioral tests, the mice were anaesthetized with 10% chloral hydrate (0.04 mL/10 g), and the brains were removed, cut into 400-μm thick hippocampal slices and then incubated in oxygenated artificial cerebrospinal fluid containing 124 mM NaCl, 3 mM KCl, 2.4 mM CaCl2, 1.3 mM MgSO4, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose (pH 7.4) at room temperature [24]. The slices were allowed to recover for 1–4 h in the artificial cerebrospinal fluid before being placed in the recording chamber for experiments. Stimulation intensity was adjusted to evoke ~50% of the maximal response of the field excitatory postsynaptic potential (fEPSP) [25, 26]. Stimulus responses and input/output (I/O) functions were generated using systematic variation of the stimulus current (100–1000 μA) to evaluate synaptic potency before the induction of LTP. fEPSPs were evoked in the DG region using 0.1 Hz stimulation. After obtaining a steady state baseline response, LTP was induced using high-frequency stimuli protocols of 100 Hz (10 bursts of 20 stimuli, 0.2 ms stimulus duration, 10 s interburst interval) at a stimulus intensity that evoked an fEPSP of ~80% of the maximum response. The fEPSP slope was recorded for the periods of 40, 50, 60, 70, and 80 min after HFS to determine any changes in the synaptic response of CA1 neurons.
Congo red staining
For Congo red staining, paraffin sections (5 μm thick) were used as described [27]. Briefly, coronal sections were deparaffinized in xylene and rehydrated. Then, the slides were treated with a working sodium chloride solution (sodium chloride-saturated 80% alcohol containing 0.01% sodium hydroxide) for 20 min at room temperature, stained with Congo red solution (0.2% in 80% ethanol) for 1 h, dehydrated with 95% and 100% alcohol and covered with a coverslip. In this staining protocol, amyloid deposits were stained red. For the quantification of senile plaques, three fields of the hippocampus or cortex were chosen from each slide. Each individual field was imaged at ×400 magnification and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics Inc., Bethesda, MD, USA).
Immunohistochemical analysis
Three mice per group were selected randomly and anesthetized by i.m. injection of 10% chloral hydrate (0.04 mL/10 g). The mice were perfused with saline solution followed by 4% paraformaldehyde. The brains were removed and fixed in 0.05 M phosphate buffer (pH 7.4) containing 4% paraformaldehyde overnight.
Brain slices (3 μm thick) were blocked with 5% goat serum for 1 h, and the slices were then treated with anti-glial fibrillary acidic protein (GFAP) (1:200, Abcam, Cambridge, MA, USA) and anti-ionized calcium-binding adapter molecule 1 (Iba-1) (1:50, Abcam) antibodies overnight at 4 °C. After rinsing with PBS, the slides were incubated with secondary antibody for 1 h at room temperature. For the quantification of the mean integral optical density, three fields of the hippocampus were chosen from each slide. Each individual field was imaged at ×400 magnification. Imaging analysis was performed using Image-Pro Plus 6.0 software.
Nissl staining
After fixation, the tissues were dehydrated and embedded in paraffin. Sections (4 μm) were prepared and subjected to Nissl staining based on the manufacturer’s instructions. Neurons were distinguished from glial cells by the presence of a nucleolus. Each individual field was imaged at ×200 magnification. The number of neurons was determined by using Image-Pro Plus 6.0 software.
ELISA assay
TNF-α, IL-6, and IL-1β levels in the mouse brains were measured with a sensitive and specific ELISA according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). The absorbance was read at 450 nm with a 96-well plate reader.
Western blot assay
Mouse brain tissues were lysed in RIPA lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 20 mM EDTA, 50 mM sodium fluoride, 1 mM sodium orthovanadate, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate (SDS) with a protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany). After centrifugation at 13 000 × g for 20 min at 4 °C, the supernatants were collected.
Thirty micrograms of the protein samples were separated on 10% SDS-polyacrylamide gels and transferred to PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% BSA and 0.05% Tween-20 in TBS for 1 h at 37 °C The membranes were incubated with the following primary antibodies overnight at 4 °C: anti-β-amyloid precursor protein, anti-BDNF (1:10000, Abcam), anti-AKT, anti-AKT, anti-GSK-3β, anti-phospho-GSK-3β (Ser-9), anti-Bax, anti-Bcl-2, and anti-phospho-ERK1/2 (1:1000, CST). After washing, the immunoblots were incubated with HRP-conjugated secondary antibodies (anti-mouse and anti-rabbit, 1:2000, Cell Signaling Technology) for 2 h at room temperature. The proteins were detected by ECL Western blotting kits (CoWin Bioscience Co., Beijing) using the ChemiDoc-ItTM imaging system (UVP, CA, USA). The results were quantified using Quantity one software.
Statistical analysis
All data are presented as the mean ± SEM. Statistical analysis was performed by using SPSS software (version 16.0, SPSS Inc., Chicago, IL, USA). Differences in the escape latency in the Morris water maze test were analyzed using two-way ANOVA with repeated measures. The other data were analyzed by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. P < 0.05 was considered significant for all tests.
Results
DL0410 improved the learning and memory of APP/PS1 transgenic mice in the behavioral tests
DL0410 improved the nesting score of the mice in the nest-building test
The nest-building test reflects the ability of mice to perform daily activities, which is reported to be reduced significantly in AD mice due to Aβ-related neurotoxicity. In this study, the nesting ability of APP/PS1 mice was significantly reduced compared with that of WT mice (P < 0.05), while DL0410 (3 mg/kg) increased the nesting ability (P < 0.05, Fig. 2).

Data are expressed as mean ± SEM. a Images of nest-building results of DL0410-treated mice and wild-type groups. b Nest score of DL0410-treated mice and wild-type groups. *P < 0.05 vs WT group, #P < 0.05, ##P < 0.01 vs APP/PS1 group.
DL0410 rescued the cognitive ability of mice in the Morris water maze test
The Morris water maze test is widely used to evaluate spatial learning and memory ability in rodents. In the acquisition training, the escape latency was dependent on both time and group. The escape latency in all groups decreased from d 1 to d 5, and there was a significant difference between the APP/PS1 group and the WT group. DL0410 reduced the escape latency significantly on the fourth and fifth days (P < 0.05 for 10 mg/kg DL0410 and P < 0.05 for 30 mg/kg DL0410 on the fourth and fifth days, Fig. 3a). The swimming speed was not significantly different among the groups in the acquisition training (Fig. 3b).

a DL0410 reduced the escape latency significantly. *P < 0.05, **P < 0.01, ***P < 0.001 vs WT group, #P < 0.05, memantine vs APP/PS1 group, ^P < 0.05, donepezil vs APP/PS1 group, &P < 0.05, DL0410 (10 mg/kg) vs APP/PS1 group, $P < 0.05, DL0410 (30 mg/kg) vs APP/PS1 group. b Swimming speed was not significantly different among the groups in the acquisition training. c The moving trajectory of mice in each group on d 6. d DL0410 decreased the escape latency (first time crossing the area which the platform had been located before) significantly in the probe trial. ***P < 0.001 vs WT group, #P < 0.05, ##P < 0.01 vs APP/PS1 group. e DL0410 showed rising tendency in the crossing numbers in the target quadrant. *P < 0.05 vs WT group, ##P < 0.01 vs APP/PS1 group. f DL0410 increased the searching time significantly in the target quadrant in the probe trial. ***P < 0.001 vs WT group, #P < 0.05, ##P < 0.01, ###P < 0.001 vs APP/PS1 group. g DL0410 increased the searching distance significantly in the target quadrant in the probe trial. ***P < 0.001 vs WT group, #P < 0.05, ##P < 0.01, ###P < 0.001 vs APP/PS1 group.
In the probe trial, APP/PS1 mice presented significantly higher escape latency (the time for the mouse to first cross the area in which the platform had been located before) than that of WT mice (P < 0.001). DL0410 (10 mg/kg and 30 mg/kg) decreased the escape latency (P < 0.01 for 10 mg/kg DL0410 and P < 0.05 for 30 mg/kg DL0410, Fig. 3d). As shown in Fig. 3e–g, APP/PS1 mice exhibited significantly fewer target crosses and shorter search time and distance traveled in the target quadrant (P < 0.001). DL0410 showed a trend of increasing the number of crosses, search time and distance traveled in the target quadrant. Moreover, 3 mg/kg DL0410 significantly increased the search time (P < 0.05), and 3 mg/kg and 10 mg/kg DL0410 significantly increased the distance traveled in the target quadrant (P < 0.01 for 3 mg/kg DL0410 and P < 0.05 for 10 mg/kg DL0410).
DL0410 promoted synaptic transmission in APP/PS1 transgenic mice
DL0410 enhanced LTP in the hippocampal CA1 region
Synaptic transmission, which is reflected by LTP, a hallmark of synaptic plasticity, is the most relevant factor for learning and memory. To assess the effects of DL0410 on LTP, we measured excitatory postsynaptic potentials (EPSP) in hippocampal slices (six slices per mouse). The average traces were recorded before and after the induction of LTP in the hippocampal CA1 region in the WT, APP/PS1, and DL0410 groups (Fig. 4a). As shown in Fig. 4b, c, the EPSP slope in the WT group increased after HFS, and the EPSP slope in the APP/PS1 group was lower than that in the WT group (P < 0.01). DL0410 (30 mg/kg) significantly enhanced the EPSP slope at 10, 20, 30, 40, and 50 min after HFS.

Data are expressed as mean ± SEM (n = 6). a The average traces were recorded before and after induction of LTP in hippocampal CA1 of hippocampal slice. b Time curves of the EPSP slope after HFS. c Cumulative histograms of averaged EPSP slopes measured at 10, 20, 30, 40, 50 min post-HFS. The averaged EPSP slope in WT group increased after HFS, and the averaged EPSP slope in APP/PS1 group was lower with respect to that in WT group (**P < 0.01 vs WT group). DL0410 could significantly enhance the averaged EPSP slope (#P < 0.05, ##P < 0.01, ###P < 0.001 vs APP/PS1 group).
DL0410 activated the AKT-GSK-3β signaling pathway in the hippocampus and cortex
AKT, also known as PKB, plays an important role in synaptic plasticity and memory formation [28]. GSK-3β, a subtype of GSK-3, is a substrate of AKT. GSK-3β is a major regulation of NMDA receptor-dependent LTP, which is involved in learning and memory [29]. It has been reported that the inhibition of GSK-3β prolongs long-term memory and enhances LTP [30]. In this study (as shown in Fig. 5), the expression of phosphorylated AKT in the cortex and hippocampus was significantly reduced in the APP/PS1 group compared with the WT group (P < 0.05 in the cortex and hippocampus), and DL0410 (10 mg/kg and 30 mg/kg) increased the expression of phosphorylated AKT in the cortex and hippocampus (P < 0.05 for 10 mg/kg in the cortex, P < 0.01 for 10 and 30 mg/kg in the hippocampus). In addition, compared with that in the brains of WT mice, the activity of GSK-3β was enhanced in the cortex and hippocampus of the brains of APP/PS1 mice (P < 0.05 in the cortex, P < 0.01 in the hippocampus). DL0410 (10 mg/kg and 30 mg/kg) decreased the activity of GSK-3β (P < 0.05 for 10 mg/kg in the cortex, P < 0.05 for 10 and 30 mg/kg in the hippocampus).

Data are expressed as mean ± SEM (n = 3). a The Western blotting results of the effects of DL0410 on AKT-GSK-3β signal pathway in the cerebral cortex and hippocampus. b The Western blotting results of the effects of DL0410 on AKT-GSK-3β signal pathway in the hippocampus. c–d Quantification analysis of Western blotting results both in cerebral cortex and hippocampus. The expression of phosphorylated AKT in the cortex and hippocampus was significantly reduced in APP/PS1 group, and DL0410 increased the expression of phosphorylated AKT in the cortex and hippocampus. The activity of GSK-3β was enhanced in the cortex and hippocampus, while DL0410 decreased the activity of GSK-3β. *P < 0.05 **P < 0.01 vs WT group; #P < 0.05 vs APP/PS1 group.

Data are expressed as mean ± SEM (n = 3). a The Western blotting results of the effects of DL0410 on phosphorylated ERK1/2 in the cerebral cortex and hippocampus. b The Western blotting results of the effects of DL0410 on phosphorylated ERK1/2 in the hippocampus. c–d Quantification analysis of Western blotting results both in cerebral cortex and hippocampus. DL0410 increased the level of phosphorylated ERK1/2 expression in the hippocampus significantly, while only the rising tendency of phosphorylated ERK1/2 expression was observed in cerebral cortex. *P < 0.05 vs WT group; #P < 0.05 vs APP/PS1 group.
DL0410 increased the phosphorylation of ERK in the hippocampus
Extracellular signal-regulated kinase (ERK) is a member of the MAPK family, which plays a key role in neuronal survival and synaptic development. In our study, the western blotting results showed that the expression of phosphorylated ERK1/2 was reduced in the cortex and hippocampus of APP/PS1 mice compared with WT mice (P < 0.05 for the cortex and hippocampus, Fig. 6a–d). DL0410 showed a trend of increasing the level of phosphorylated ERK1/2 expression in the cortex and hippocampus, and 30 mg/kg DL0410 significantly increased this level in the hippocampus (P < 0.05).
DL0410 reduced neuronal loss in APP/PS1 transgenic mice
DL0410 decreased amyloid plaque deposits via inhibiting the expression of APP in the hippocampus and cortex
The accumulation of extracellular Aβ is a classical pathological marker of AD patients, and Congo red dye is a basic dye with high affinity for Aβ plaques. The results showed that the number of senile plaques in the hippocampus of the APP/PS1 mice was significantly increased compared with that in the WT mice (P < 0.01, Fig. 7a). DL0410 (3, 10, and 30 mg/kg) significantly reduced senile plaque deposition (P < 0.05 for 3 and 10 mg/kg, P < 0.01 for 30 mg/kg).

Data are expressed as mean ± SEM (n = 3). a Images of senile plaques stained with Congo red in the hippocampus. b Quantification analysis of amyloid plaques numbers. **P < 0.01 vs WT group; #P < 0.05, ##P < 0.01 vs APP/PS1 group. c–f The expression of APP protein was significantly increased in the cortex and hippocampus. However, no significant differences on the expression of BACE1 protein between WT group and APP/PS1 group were observed. DL0410 decreased APP protein level in cerebral cortex and hippocampus. **P < 0.01 vs WT group; #P < 0.05 vs APP/PS1 group.
The APP protein is through to maintain the development of nerves, promote the formation of synapses and participate in the formation of Aβ, and it is degraded via the nonamyloidogenesis pathway under physiological conditions. However, it is degraded by beta-secretase (BACE1) in the amyloidogenic pathway under pathological conditions, and Aβ, which further aggregates and precipitates in the brain, is produced. We further investigated the expression of APP and BACE1 using Western blotting. Compared with that in WT mice, the expression of the APP protein was significantly increased in APP/PS1 mice (P < 0.01). DL0410 significantly reduced the expression of the APP protein in the cortex and hippocampus of APP/PS1 mice (P < 0.05). The expression of BACE1 showed a tendency to be increased in the cortex and hippocampus of APP/PS1 mice and to be decreased by DL0410, but the differences were not significant.
DL0410 inhibited glial activation and reduced inflammatory factor levels in the hippocampus and cortex
In the brains of AD patients and transgenic AD mice, activation of astrocyte and microglial was observed around Aβ plaque, indicating that neuroinflammation may be involved in brain damage [31, 32]. In this study, the results showed that the expression of GFAP and Iba-1 in the CA1 region of the hippocampus in APP/PS1 mice was significantly upregulated compared with that in WT mice (P < 0.01 for GFAP, P < 0.05 for Iba-1). Moreover, the astrocytes and microglia in APP/PS1 mice were activated and exhibited swelling soma and more processes. DL0410 (30 mg/kg) downregulated the expression of GFAP (P < 0.05) and inhibited the activation of astrocytes and microglia in the brain. However, the inhibitory effect of DL0410 on the expression of Iba-1 was not statistically significant.
TNF-α is produced by monocytes/macrophages and appears to be the earliest and most important inflammatory factor in the inflammatory response. IL-6 can induce B cell differentiation and induce T cell activation, which is an initiator of the inflammatory reaction. IL-1β is derived from microglia and macrophages and is involved in the occurrence of inflammation. In our study, the results showed that the levels of TNF-α and IL-6 in the APP/PS1 group were significantly increased compared with those in the WT group (P < 0.05, Fig. 8b1, b2, b3). DL0410 (10 mg/kg and 30 mg/kg) significantly decreased the levels of TNF-α and IL-6 (P < 0.05). No significant difference was observed in the level of IL-1β.

Data are expressed as mean ± SEM (n = 3). a The immunohistochemistry results were analysed by an astrocytic marker GFAP and microglia marker Iba-1. a1 Represensive images of GFAP and Iba-I positive immunoreactive staining in the CA1 region of hippocampus. Magnification: ×400. a2–a3 Quantification analysis of immunoreactivity. The expression of GFAP and Iba-1 in CA1 region of hippocampal in APP/PS1 mice was upregulated significantly. DL0410 could downregulate the expression of GFAP. *P < 0.05, **P < 0.01 vs WT group; #P < 0.05 vs APP/PS1 group. b1–b3 The levels of TNF-α and IL-6 in the APP/PS1 group were significantly increased, while only rising tendency was observed in the levels of IL-1β. DL0410 could significantly decrease the levels of TNF-α and IL-6. *P < 0.05 vs WT group; #P < 0.05 vs APP/PS1 group.
DL0410 ameliorated apoptosis-related protein expression and neuronal loss in the hippocampus and cortex
Bcl-2 and Bax are important molecules for apoptosis; Bcl-2 inhibits apoptosis, and Bax promotes apoptosis. In this study, the expression of Bcl-2 and Bax was investigated, as shown in Fig. 9a. Compared with that in WT control mice, the ratio of Bcl-2/Bax in the cortex and hippocampus was significantly reduced in APP/PS1 mice (P < 0.01 for the cortex and hippocampus), and apoptosis was induced. DL0410 (30 mg/kg) increased the ratio of Bcl-2/Bax in the cortex and hippocampus (P < 0.05 for the cortex and hippocampus) and inhibited apoptosis.

Data are expressed as mean ± SEM (n = 3). a1 The Western blotting results of the effects of DL0410 on the expression of Bcl-2/Bax in the cerebral cortex. a2 The Western blotting results of the effects of DL0410 on the expression of Bcl-2/Bax in the hippocampus. a3–a4 Quantification analysis of Western blotting results both in cerebral cortex and hippocampus. The expression of Bcl-2/Bax was significantly reduced in the cortex and hippocampus in APP/PS1 group, and DL0410 significantly increased the expression of Bcl-2/Bax. b1 The Nissl staining results of the effects of DL0410 on the neuronal cells in the cerebral cortex and hippocampus. b2–b3 Quantification analysis of the number of neuronal cells both in cerebral cortex and hippocampus. **P < 0.01, ***P < 0.001 vs WT group; #P < 0.05 vs APP/PS1 group.
Nissl staining was employed to detect histomorphological changes in the cerebral cortex and neurons in the CA3 region of the hippocampus. Photomicrographs of the cerebral cortex and the CA3 region of the hippocampus were taken under a ×20 objective lens, as shown in Fig. 9b. Compared with that in WT mice, the number of neuronal cells was decreased in the cerebral cortex and the CA3 region of the hippocampus in APP/PS1 mice (P < 0.01 for the cortex and hippocampus). In addition, neuronal cells in the cerebral cortex and the CA3 region of the hippocampus were loosely arranged. The number of neuronal cells in the 30 mg/kg DL0410-treated group was increased both in the cerebral cortex and the CA3 region of the hippocampus (P < 0.05 for the cortex and hippocampus).
Discussion
AD is the most prevalent neurodegenerative disorder, and the pathogenesis is still incompletely unclear. Until now, only four cholinesterase inhibitors and one NMDAR antagonist have been approved by the FDA for AD treatment. Synaptic dysfunction and neuron loss, which are the basis of cognitive impairments, occur throughout the pathogenesis of AD. It has been reported that DL0410 can improve cognitive defects, reduce Aβ deposition, inhibit oxidative reactions, and ameliorate mitochondrial function in a number of AD models [17, 19, 20]. Based on a previous study, we further clarified the beneficial effects of DL0410 in AD via promoting synaptic function and reducing neuronal loss in APP/PS1 mice.
Nine-month-old APP/PS1 mice were subjected to 8 weeks of treatment with DL0410 (ig), and two commonly used behavioral methods, the nest building and MWM tests, were conducted to evaluate cognition and spatial memory. The nest-building test reflects the ability of mice to perform daily activities. The instinctive behavior is lost in mice with cognitive deficits [33, 34]. In this test, APP/PS1 mice lost the ability to build nests, and the nesting scores were lower than those of wild-type mice. DL0410 (3 mg/kg) restored the ability to perform daily activities, and DL0410-treated mice built fine nests. The Morris water maze is a typical test measuring spatial learning and memory [23]. In the acquisition trial, APP/PS1 mice spent more time finding the platform. In the probe trial, APP/PS1 mice presented higher escape latencies (the time for the mice to first cross the area in which the platform had been located before), fewer crosses, shorter search times and shorter distances traveled in the target quadrant. DL0410 (10 mg/kg and 30 mg/kg) effectively decreased the escape latency, 30 mg/kg DL0410 increased the search time, and 3 mg/kg and 10 mg/kg DL0410 increased the distance traveled in the target quadrant, indicating that DL0410 improved spatial learning and memory ability. Moreover, our results showed that the therapeutic effect of 3 mg/kg DL0410 on ameliorating learning and memory impairments matched that of the positive control drugs (3 mg/kg donepezil and 3 mg/kg memantine), and the therapeutic effect of 10 mg/kg and 30 mg/kg DL0410 was even better than that of the positive control drugs.
Synaptic damage is the pathological basis of cognitive dysfunction. Several studies have shown a strong relationship between synaptic deficits and the degree of memory impairment in AD. Moreover, synapse changes have been detected in the neocortex and hippocampus of AD patients, even in very early stages, and in mild cognitive impairment (MCI) patients [35]. Histological studies have demonstrated that the hallmarks of synaptic alterations include decreased synaptic density, impaired synaptic transmission and defected synaptic plasticity [36, 37]. Hence, the concept of synaptic repair has been proposed to prevent the pathophysiology of AD [38]. In previous studies, we found that the number of synapses and the thickness of the PSD in the hippocampal CA1 region are reduced in APP/PS1 mice and that DL0410 reverses these changes by ameliorating synaptic structure [20]. Synaptic plasticity is the basis of learning and memory, and LTP is considered the gold standard of synaptic plasticity in the hippocampus [39, 40]. Based on previous studies, we further detected LTP to confirm its effect. The results demonstrated that LTP was significantly inhibited in the hippocampal CA1 area in APP/PS1 mice and that DL0410 remarkably enhanced LTP and thus enhanced synaptic plasticity.
AKT-GSK-3β and Ras-MEK-ERK1/2 signaling cascades may be involved in synaptic strength and promote long-term neuroprotection against a variety of insults. AKT is a serine/threonine kinase that is an important downstream protein in the phosphatidylinositol-3-kinase (PI3K) signaling pathway. Biochemical analyses have demonstrated that LTP is associated with the activation of the domain of the downstream substrate AKT by PI3K [28]. GSK-3β is the downstream protein of AKT, which can be activated by phosphorylation at Tyr-216 and inhibited by phosphorylation at Ser-9 [41,42,43]. It has been reported that the GSK-3β inhibitors lithium chloride and kenpaullone can enhance synaptic strength by activating NMDA receptors [30, 44]. Electrophysiological data has shown that the upregulation of ERK1/2 phosphorylation can induce LTP in the CA1 region [45]. Previously, we discovered that DL0410 decreases the phosphorylation level of NMDAR2B and increases the phosphorylation level of CAMKII. In the current study, we further found that p-AKT and p-GSK-3β were significantly decreased in the hippocampus and cortex of APP/PS1 mice. DL0410 increased the phosphorylation level of AKT and inhibited GSK-3β activity by increasing the phosphorylation level of GSK-3β (Ser-9). The ERK1/2 pathway is involved in interacting with synaptic structural proteins and participates in synaptic plasticity, ultimately affecting cognitive function [46, 47]. In the present study, the expression of phosphorylated ERK1/2 was significantly decreased in the cortex and hippocampus of APP/PS1 mice, and DL0410 increased the level of phosphorylated ERK1/2 expression. In summary, our results suggested that DL0410 might reverse synaptic dysfunction via the AKT-GSK-3β and ERK1/2 signaling pathways.
Strong evidence has indicated that AD is caused by the toxicity of the Aβ peptide, either in the form of microaggregates or amyloid deposits [36]. All AD-causing mutations in APP, PS1, and PS2 increase Aβ deposition, and excessive levels of Aβ peptide in the brain are responsible for the development and pathology of AD, including amyloid plaques, NFTs, synapse failure, and eventual neuronal death [48, 49]. Animal tests have confirmed that the intracerebroventricular injection of Aβ can induce cognitive impairments, electrophysiological changes, and histopathological injuries [19]. In the current study, we found that DL0410 decreased the expression of the APP protein in the cortex and hippocampus of APP/PS1 mice but did not alter the expression of BACE1, the key enzyme responsible for Aβ production. In addition, Congo red staining results also confirmed that DL0410 effectively removed senile plaque deposits in the hippocampus of APP/PS1 mice. These results are consistent with those of a previous study [20], and we further confirmed that DL0410 did not reduce Aβ deposition by inhibiting the expression of BACE1. Hence, other molecular mechanisms by which DL0410 reduces Aβ deposition and aggregation in APP/PS1 transgenic mice require further research.
In our study, we found abnormal Aβ accumulation, which might be sufficient to induce neuroinflammatory reactions that subsequently contribute to cognitive decline, in APP/PS1 mice [50,51,52]. Reactive gliosis, including astrocyte and microglial activation, occurs in response to neuroinflammation and neurodegeneration [53]. Studies have confirmed that the activation of astrocytes is involved in the formation and development of AD. The elevated expression of GFAP is considered a biomarker of astrogliosis [54]. Microglia are more sensitive than astrocytes to injury, and Iba-1 is the only marker of microglial cells [12]. Furthermore, experimental data have demonstrated that overactivated astrocytes and microglia are the major sources of inflammatory cytokines (such as TNF-α, IL-1β, and IL-6) in AD. Thus, neuroinflammation is considered an important aspect in the development of dementia [55]. In our study, we found that GFAP and Iba-1 were overexpressed in the cortex, while DL0410 attenuated reactive gliosis by reducing the expression of GFAP and Iba-1. In addition, the levels of inflammatory cytokines, including TNF-α and IL-6, were significantly increased in APP/PS1 mice. DL0410 decreased the levels of TNF-α and IL-6. Our results indicated that DL0410 suppressed neuroinflammation via inhibiting astrocyte and microglial overactivation and reducing the expression of inflammatory cytokines.
Neuronal loss is the basis of AD and is considered the most important structural foundation of AD anatomy. A marked reduction in the number of neurons can be found in the entorhinal cortex and hippocampus of AD patients [56, 57]. The loss of neuronal cells is correlated with prominent extracellular amyloid pathology and/or the induction of the neuroinflammatory response [58]. In addition, more than half of hippocampal neurons are lost in APP/PS1 transgenic mice at the age of 10 months, and this neuronal loss is associated with the accumulation of intraneuronal Aβ [59]. Nissl bodies can reflect surviving neurons, and under pathological conditions, these neurons they may dissolve and disappear. As shown by the Nissl staining results, the number of neurons was decreased in the cerebral cortex and the CA3 region of the hippocampus, and these neurons were arranged loosely. DL0410 increased the number of neurons in both the cortex and hippocampus. Cell apoptosis is an important feature of degenerative diseases such as AD. It has been proven that apoptosis is regulated by Bcl-2 protein family members, which can be divided into two main categories: those that support cell survival (e.g., Bcl-2, Bcl-xl, Bcl-w) and those that promote cell apoptosis (e.g., Bax, Bad, Bak, Bid) [60, 61]. In AD, oxidative stress, Aβ deposition, the lack of neurotrophic factors and other insults are responsible for apoptosis. Taken together, these results suggest that DL0410 can increase neuronal cell survival via reducing neuroinflammation and inhibiting apoptosis.
In summary, our results clearly showed that DL0410 ameliorated cognitive deficits in APP/PS1 transgenic mice. DL0410 was effective in improving synaptic dysfunction by activating the AKT/GSK-3β and MAPK/ERK signaling pathways. On the other hand, DL0410 reduced neuronal loss by decreasing senile plaque deposition and inhibiting glial overactivation and neuroinflammation (Fig. 10). In summary, DL0410 may represent a promising candidate agent for AD therapy.

Molecular mechanism of DL0410 in the improvement of cognitive dysfunction in APP/PS1 mice.
References
Gaugler J, James B, Johnson T, Scholz K, Weuve J. Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12:459–509.
Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement Transl Res Clin Interv. 2017;3:367–84.
Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Hong LS, et al. Phase 3 solanezumab trials: Secondary outcomes in mild alzheimer’s disease patients. Alzheimers Dement J Alzheimers Assoc. 2016;12:110–20.
Godyń J, Jończyk J, Panek D, Malawska B. Therapeutic strategies for Alzheimer's disease in clinical trials. Pharmacol Rep. 2016;68:127–38.
Abushakra S, Bairu M, Kesslak P, Kurth M, Miller D, Lyketsos C. Agitation and aggression in Alzheimer's disease (AD): study design and outcome measures in a trial of scyllo-inositol (ELND005 Harmony AD Study). Neurobiol Aging. 2014;35:S1–S.
Li Y, Sun Z, Cao Q, Chen M, Luo H, Lin X, et al. Role of amyloid beta protein receptors in mediating synaptic plasticity. Biomed Rep. 2017;6:379–86.
Hahm ET, Nagaraja RY, Waro G, Tsunoda S. Cholinergic homeostatic synaptic plasticity drives the progression of Aβ-induced changes in neural activity. Cell Rep. 2018;24:342–54.
Orellana AMM, Vasconcelos AR, Leite JA, de Sá Lima L, Andreotti DZ, Munhoz CD, et al. Age-related neuroinflammation and changes in Akt-GSK-3β and Wnt/β-catenin signaling in rat hippocampus. Aging. 2015;7:1094–111.
Tiwari SK, Seth B, Agarwal S, Yadav A, Karmakar M, Gupta SK, et al. Ethosuximide induces hippocampal neurogenesis and reverses cognitive deficits in an amyloid-beta toxin-induced alzheimer rat model via the phosphatidylinositol 3-kinase (PI3K)/Akt/Wnt/beta-Catenin pathway. J Biol Chem. 2015;290:28540–58.
Neill CO. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer's disease. Exp Gerontol. 2013;48:647–53.
Wang PJ, Song RR, He DD. Significance of PI3K/Akt signal in apoptotic cells mimic Alzheimer's disease and the influence of edaravone. J Apoplexy Nerv Dis. 2010;1:12–5.
Kim EK, Choi EJ. Compromised mapk signaling in human diseases: an update. Arch Toxicol. 2015;89:867–82.
Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, et al. Differential activation of neuronal ERK, JNK/SAPPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev. 2001;123:39–46.
Pang X, Fu H, Yang S, Wang L, Liu AL, Wu S, et al. Evaluation of novel dual acetyl- and butyrylcholinesterase inhibitors as potential anti-Alzheimer's disease agents using pharmacophore, 3D-QSAR, and molecular docking approaches. Molecules. 2017;22:E1254.
Fang J, Li Y, Liu R, Pang X, Li C, Yang R, et al. Discovery of multitarget-directed ligands against Alzheimer's disease through systematic prediction of chemical-protein interactions. J Chem Inf Model. 2015;55:149–64.
Fang J, Yang R, Gao L, Zhou D, Yang S, Liu AL, et al. Predictions of BuChE inhibitors using support vector machine and naive bayesian classification techniques in drug discovery. J Chem Inf Model. 2013;11:3009–20.
Lian W, Jia H, Xu L, Zhou W, Kang, Liu A, et al. Multi-protection of DL0410 in ameliorating cognitive defects in D-galactose induced aging mice. Front Aging Neurosci 2017;9:409.
Lian W, Fang J, Xu L, Zhou W, Kang, Xiong W, et al. DL0410 ameliorates memory and cognitive impairments induced by scopolamine via increasing cholinergic neurotransmission in mice. Molecules. 2017;22:E410.
Zhou D, Zhou W, Song JK, Feng ZY, Yang RY, Wu S, et al. DL0410, a novel dual cholinesterase inhibitor, protects mouse brains against Aβ-induced neuronal damage via the Akt/JNK signaling pathway. Acta Pharmacol Sin. 2016;37:1401–12.
Yang RY, Zhao G, Wang DM, Pang XC, Wang SB, Fang JS, et al. DL0410 can reverse cognitive impairment, synaptic loss and reduce plaque load in APP/PS1 transgenic mice. Pharmacol Biochem Behav. 2015;139:15–26.
Deacon RM, Cholerton LL, Talbot K, Nair-Roberts RG, Sanderson DJ, Romberg C, et al. Age-dependent and -independent behavioral deficits in tg2576 mice. Behav Brain Res. 2008;189:126–38.
Otabi H, Goto T, Okayama T, Kohari D, Toyoda A. Subchronic and mild social defeat stress alter mouse nest building behavior. Behav Process. 2016;122:21–5.
Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60.
Wang CY, Wang ZY, Xie JW, Wang T, Wang X, Xu Y, et al. Dl-3-n-butylphthalide-induced upregulation of antioxidant defense is involved in the enhancement of cross talk between CREB and Nrf2 in an Alzheimer's disease mouse model. Neurobiol Aging. 2016;38:32–46.
Reisi P, Babri S, Alaei H, Sharifi MR, Mohaddes G, Noorbakhsh SM, et al. Treadmill running improves long-term potentiation (LTP) defects in streptozotocin-induced diabetes at dentate gyrus in rats. Pathophysiology. 2010;17:33–8.
Babri S, Amani M, Mohaddes G, Alihemmati A, Ebrahimi H. Effect of aggregated beta-amyloid (1-42) on synaptic plasticity of hippocampal dentate gyrus granule cells in vivo. Bioimpacts. 2012;2:189–94.
Zhang D, Fa HB, Zhou JT, Li S, Diao XW, Yin W. The detection of beta-amyloid plaques in an Alzheimer's disease rat model with DDNP-SPIO. Clin Radio. 2015;70:74–80.
Horwood JM, Dufour F, Laroche S, Davis S. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006;23:3375–84.
Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, et al. A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci. 2012;5:13.
Yi JH, Zhang J, Ko SY, Kwon H, Jeon SJ, Park SJ, et al. Fluoxetine inhibits natural decay of long-term memory via Akt/GSK-3beta signaling. Mol Neurobiol. 2018;55:7453–62.
Mosher KI, Wysscoray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014;88:594–604.
López-González I, Schlüter A, Aso E, Garcia-Esparcia P, Ansoleaga B, Llorens F, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol. 2015;74:319–44.
Greenberg GD, Huang LC, Spence SE, Schlumbohm JP, Metten P, Ozburn AR, et al. Nest building is a novel method for indexing severity of alcohol withdrawal in mice. Behav Brain Res. 2016;302:182–90.
Go J, Park TS, Han GH, Park HY, Ryu YK, Kim YH, et al. Piperlongumine decreases cognitive impairment and improves hippocampal function in aged mice. Int J Mol Med. 2018;42:1875–84.
Masliah E, Mallory M, Alford M, Deteresa R, Hansen LA, Mckeel DW, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127–9.
Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol 2012;4:a005777.
Yu W, Lu B. Synapses and dendritic spines as pathogenic targets in Alzheimer's disease. Neural Plast. 2012;2:247150.
Bai L, Guhan N, Xiaoming G, Nathan PJ, Paul W. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401.
HÖlscher C. Synaptic plasticity and learning and memory: LTP and beyond. J Neurosci Res. 1999;58:62.
Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–56.
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661.
Griffin RJ, Fennelly C, O’Connor R, O’Neill C, Johnston J, Williams CH, et al. Akt signal transduction is impaired in the Alzheimer's disease brain. Biochim Biophys Acta. 2002;192:81.
Rickle A, Bogdanovic N, Volkman I, Winblad B, Ravid R, Cowburn RF. Akt activity in Alzheimer's disease and other neurodegenerative disorders. Neuroreport. 2004;15:955–9.
Lei G, Xia Y, Johnson KM. The role of Akt-GSK-3beta signaling and synaptic strength in phencyclidine-induced neurodegeneration. Neuropsychopharmacology. 2008;33:1343–53.
Krania P, Dimou E, Bantouna M, Kouvaros S, Tsiamaki E, Papatheodoropoulos C, et al. Adenosine A2A receptors are required for glutamate mglur5- and dopamine d1 receptor-evoked ERK1/2 phosphorylation in rat hippocampus: involvement of NMDA receptor. J Neurochem. 2018;145:217–31.
Targowska-Duda KM, Wnorowski A, Budzynska B, Jozwiak K, Biala G, Arias HR. The positive allosteric modulator of alpha7 nicotinic acetylcholine receptors, 3-furan-2-yl-n-p-tolyl-acrylamide, enhances memory processes and stimulates ERK1/2 phosphorylation in mice. Behav Brain Res. 2016;302:142–51.
Ma G, Chen SX, Ba M, Yang H, Lu G. Short-term interleukin-1(beta) increases the release of secreted app(alpha) via MEK1/2-dependent and JNK-dependent alpha-secretase cleavage in neuroglioma U251 cells. J Neurosci Res. 2005;80:683–92.
Hardy J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Jangra A, Kasbe P, Pandey SN, Dwivedi S, Gurjar SS, Kwatra M, et al. Hesperidin and silibinin ameliorate aluminum-induced neurotoxicity: modulation of antioxidants and inflammatory cytokines level in mice hippocampus. Biol Trace Elem Res. 2015;168:462–71.
Prema A, Justin Thenmozhi A, Manivasagam T, Mohamed Essa M, Guillemin GJ. Fenugreek seed powder attenuated aluminum chloride-induced tau pathology, oxidative stress, and inflammation in a rat model of Alzheimer's disease. J Alzheimers Dis. 2017;60:S209–S220.
Zhang F, Jiang L. Neuroinflammation in Alzheimer's disease. Neuropsychiatr Dis Treat. 2015;11:243-56.
Kaushik DK, Mukhopadhyay R, Kumawat KL, Gupta M, Basu A. Therapeutic targeting of krüppel-like factor 4 abrogates microglial activation. J Neuroinflammation. 2012;9:57.
Wu Z, Zhao L, Chen X, Cheng X, Zhang Y. Galantamine attenuates amyloid-beta deposition and astrocyte activation in APP/PS1 transgenic mice. Exp Gerontol. 2015;72:244–50.
Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, et al. Increased white matter inflammation in aging- and Alzheimer's disease brain. Front Mol Neurosci. 2017;10:206.
West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer's disease. Neurobiol Aging. 2004;25:1205–12.
Kril JJ, Patel S, Harding AJ, Halliday GM. Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 2002;103:370–6.
Jimenez S, Torres M, Vizuete M, Sanchez-Varo R, Sanchez-Mejias E, Trujillo-Estrada L, et al. Age-dependent accumulation of soluble amyloid beta (abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J Biol Chem. 2011;286:18414–25.
Casas C, Sergeant N, Itier JM, Blanchard V. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289.
Jang JH, Surh YJ. Beta-amyloid-induced apoptosis is associated with cyclooxygenase-2 up-regulation via the mitogen-activated protein kinase-NF-kappab signaling pathway. Free Radic Biol Med. 2005;38:1604–13.
Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–9.
Acknowledgements
The study was supported by the National Great Science Technology Projects (2014ZX09507003-002), the Drug Innovation Major Project (No. 2018ZX09711001-003-002), CAMS Initiative for Innovative Medicine (CAMS-IZM) (2016-IZM-3-007), the Beijing National Science Foundation (7152103) and the National Natural Science Foundation of China (81673480).
Author information
Authors and Affiliations
Contributions
ALL and GHD designed the experiment and revised the paper; WZ, RY, and WWL performed the experiments; and WZ, LJX, HJ, and LW analyzed the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Zhou, W., Lian, Ww., Yan, R. et al. DL0410 ameliorates cognitive deficits in APP/PS1 transgenic mice by promoting synaptic transmission and reducing neuronal loss. Acta Pharmacol Sin 41, 599–611 (2020). https://doi.org/10.1038/s41401-019-0312-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0312-y
Keywords
This article is cited by
-
DL0410 ameliorates cognitive disorder in SAMP8 mice by promoting mitochondrial dynamics and the NMDAR-CREB-BDNF pathway
Acta Pharmacologica Sinica (2021)
