
Abstract
Despite significant improvements in lentivirus (LV) vector-based gene therapy there are still several safety risks using LV vectors including the potential formation of replication-competent LV particles. To address this shortcoming, we constructed a novel and safer gene transfer system using modified SIN-based LV gene transfer vectors. Central to our approach is a conditional deletion of the Ψ packaging signal after integration in the target genome. Here we demonstrate that after transduction of target cells, conventional SIN-based LV transfer vectors can still be mobilized. However mobilization is rendered undetectable if transductions are followed by a Cre/loxP-mediated excision of Ψ. Thus conditional elimination of the packaging signal may represent another advance in increasing the safety of LV vectors for gene therapeutic treatment of chronic diseases.
Similar content being viewed by others

Introduction
Gammaretro (RV) - and lentivirus (LV) vectors are among the most advanced tools for the delivery of therapeutic DNA into patient cells1,2. However, with the arrival of virus-based gene transfer techniques in clinical trials, numerous safety concerns have been raised after adverse effects have been observed in laboratory animals and in patients treated with RV- and LV-modified cells3. While reported adverse effects are mainly due to insertional mutagenesis with resulting oncogene activation and clonal expansion, production of replication-competent retroviruses (RCRs4, remains a significant concern even after the development of self-inactivating (SIN) transfer vector systems with LTR-based enhancer deletions5. Specifically, Logan et al6. noted residual LTR-based promoter activity in a 293 derived cell line and cryptic low-level vector mobilization while investigating a SIN-based LV gene transfer system. So far, no practical example for the production of replication-competent LV vectors (RCLs) have been described in clinical settings7 yet this may be due to the relatively small numbers of patients having received LV-based gene therapy. Possible scenarios of e.g. HIV-mediated LV mobilization in a patient as a clinical pitfall of LV therapy have been simulated in vitro in gene-modified T cells8. Furthermore, RCRs have been found with components originating from endogenous retroviruses9. The concern of endogenous retroviruses being involved in the vector mobilization process has also been raised for LV vector systems10. Thus it is important to maximize safety of the LV gene transfer system with respect to controlling vector molecule assembly in target cells.
To prevent inadvertent mobilization when using a LV gene transfer system, we designed LV vector genomes with conditional presence of the required viral RNA packaging signal Ψ while still maintaining the established SIN-based alterations that minimize activation of host genes in the periphery of the viral integration site.
Results
Functional LV vector particles after Ψ translocation into a cleavable loxP-flanked cassette
In order to evaluate whether the change in the location of the HIV packaging signal would still allow for LV vector production we designed a construct where Ψ was translocated into a cleavable loxP flanked cassette. The resulting transfer vector was then compared to a construct of similar design with the native packaging signal (Fig. 1). Viral production was carried out by transfection of 293T cells with these constructs 512 (LeGO-IG- mCherry- loxP-eGFP) or 521 (ΔΨ-LeGO-IG- mCherry- loxP-Ψ-eGFP), plus co-transfected HIV helper constructs expressing Gag, Pol and the VSV-G envelope. FACS analyses of transduced reporter cells (HT1080) revealed successful transduction of recipient cells with transfer vector 521, albeit at a 100 fold reduced titer when compared to the titer obtained in vector production using the parental transfer vector construct 512 (data not shown).

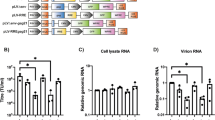
Structure of the double fluorescent lentivirus vectors analysed in this study: The LV vectors 512 and 521 contain SIN long terminal repeats (LTR), the packaging signal Ψ, a LV cis-acting region (CAR), an EF1α(short) promoter (EF1a) driving expression of an mCherry reporter gene and an SFFV promoter (SFFV) together with an internal ribosome entry site (IRES) driving expression of a GFP reporter gene.
The woodchuck hepatitis virus post transcriptional regulatory element (w) is also indicated.
Development of a transient packaging cell line containing HIV based vector components
Our next objective was to establish a transient 293T packaging cell line containing at least one LV transfer vector genome with either the deleted Ψ (via vector 521) or the native counterpart (via vector 512). To accomplish this task, transduced GFP+ target cells were enriched by sorting: the cell populations transduced with authentic LV transfer vector genome indicated by their double GFP and mCherry positive phenotype reached similar levels of 68.0% (LV 512, Fig. 2 A, upper middle panel) and 69.7 % (LV 521, Fig. 2 A, upper right panel). The sorted GFP+ cell populations were subsequently transfected with a Cre expression plasmid (pCMV-Shuttle-Cre). Cre mediated deletions of the loxP cassettes in cells transduced with vectors 512 or 52111,12 resulted in the concomitant deletion of GFP but not mCherry. Thus we monitored fluorescent reporter gene expression on day two and five post transfection. After the fifth day, approximately 20 % 293T LV 512 and 12% 293T LV 521 of the mCherry/GFP double positive cells had converted to a GFP-deficient phenotype (Fig. 2, compare percentage of GFP-negative populations in the left part of the upper histograms (“− Cre”) with the percentage of GFP-negative populations in the left part of the lower histograms (“+ Cre”). These GFP-negative but still mCherry-positive cells were again enriched by cell sorting (Fig. 2 B). Consequently producer cells containing the 521 transfer vector no longer contain either Ψ or GFP while producer cells with the transfer vector 512 only lost the GFP gene.

(a) Excision of Ψ via loxP sites monitored by disappearance of GFP reporter gene expression. 293T cells transduced with LV 512 (upper middle panel) and LV 521 (upper right panel), respectively, were transfected with pCMV-Shuttle-Cre and analyzed for GFP and mCherry reporter gene expression six days post transfection (lower panels, left: 293T LV 512, right 293T LV 521). (b) Cre-loxP-treated enriched cell populations after transfection with pCMV-Shuttle-Cre. Left: 293T-LV 512, right: 293T-LV 521.
LV vector production with cells containing HIV-based vector components
In order to determine whether producer cells already containing an integrated SIN - modified LV transfer vector could inadvertently mobilize this LV genome during packaging and if a concomitant deletion of Ψ could abrogate this potential mobilization, we performed transient transfections utilizing 293T LV-521 and 293T LV-512 modified producer cells. Specifically, the GFP negative, mCherry positive 293T LV-512 and 293T LV-521 cells were transfected with HIV Gag, Pol and VSV-G envelope helper plasmids and a titration series of a transfer vector construct 528 containing a BFP fluorescent tag. Two days post transfection supernatants were harvested by centrifugation and HT1080 reporter cells were transduced with equal volumes of the producer cell supernatants. Three days post transduction the recipient HT1080 cells were then analysed for BFP and mCherry reporter gene expression (Fig. 3). As expected HT1080 target cells transduced with suboptimal amounts of the BFP encoded transfer vector (Fig. 3 B–E) yielded a progressively lower number of BFP-positive cells when compared to cells transduced with supernatants obtained from vector productions carried out under optimal conditions (Fig. 3A). However, under suboptimal concentrations of the transfer vector construct 528, we were also able to detect a low percentage of mCherry positive BFP negative cells within the HT1080 pool containing viral vector preparations from the supernatant of 293T LV-512. These single positive mCherry cells however were not detectable in the HT1080 pool transduced with the supernatant from 293T LV-521 cells (deleted Ψ) under the same conditions, thus providing evidence of abrogated mobilization.

Cryptic transduction of HT1080 cells with SIN mCherry vectors can be abolished by deletion of the HIV packaging signal.
512 LV 293T and 521 LV 293T cell pools were transfected with mixtures of HIV Gag-Pol and VSV-G envelope expression plasmids (pCMV D R8.74 und pMD2.G) and a10 fold dilution series of BFP vector plasmid pRSCPBw. Two days post transfection supernatants were harvested by centrifugation (1200 g, 10 min) and applied onto HT1080 target cells (upper row, “+Ψ”, : LV512 supernatants, lower row, “-Ψ”,: LV521 supernatants). Three days post transduction HT1080 cells were FACS analysed for BFP (ordinate) and mCherry (abscissa) expression.
In order to exclude the possibility that these presumed HT1080 reporter cells may actually represent mCherry positive 293T producer cells, single mCherry-positive cells transduced with the 293T LV-512 supernatant (containing Ψ) were FACS-selected and pooled (Supplemental Fig. S1). After limiting cell dilution we expanded four positive for mCherry expression. We observed clonal LV insertions by LAM-PCR13 with at least three distinct integration profiles (Supplemental Fig. S1 and S2a). These data provide direct evidence that that these cells must have been transduced by cryptically activated LV SIN mCherry transfer vectors within the 293T LV-512 cells. In order to assure that the analyzed clones represent indeed HT1080 reporter cells and not 293T LV-512 cells carried over during the transduction process, we also evaluated these cell clones for their lack of adenoviral DNA detectable in 293T14, but not in HT1080 genomic DNA. For reference, we selected a GAPDH DNA fragment which was detectable by PCR in all genomic DNA samples. Adenoviral DNA was not amplifiable in any of our analysed clones. Thus, these data also support our assertion that the isolated clones are of HT1080 and not 293T origin (Supplemental Fig. S2b).
Since FACS analyses with HT1080 cells transduced with the supernatant of 293T LV-521 cells provided no evidence of mCherry-positive viral particles (Fig. 2), data directly support the premise that deleting the packaging signal after successful transfer into target cells can substantially increase the safety of LV vector-based gene delivery.
Discussion
In order to guard against potential LV vector mobilization as a result of RCL formation we improved the safety of the SIN-based LV gene delivery system by allowing for conditional deletion of the packaging signal Ψ. Here we moved Ψ further downstream between the loxP sites and immediately adjacent to a GFP reporter gene. This manipulation allows for a conditional removal of Ψ after delivery of the vector into recipient cells that can be monitored by the concomitant loss of GFP. To our knowledge these data are the first demonstration that upon removal of Ψ within the recipient genome cryptic vector mobilization can be abrogated.
Our initial goal was to investigate if inadvertent mobilization of lentiviral vectors would be detectable in 293T cells, commonly used to produce viral vector particles. Our data support earlier findings by Logan and coworkers6 that SIN-based transfer vectors can undergo cryptic mobilization in the presence of LV helper constructs. These findings are of great concern as recent retroviral gene therapy endeavors favor the use of LV vectors. The use of third generation helper plasmids15 during LV vector production addresses a safety concern for separation of viral helper genes in order to reduce the risk of the development of replication-competent LVs. The use of SIN-based transfer vectors for LV transductions reduces the risk of undesired gene activation in the vicinity of the viral integration site. However, if an integrated viral vector can still be mobilized, additional insertional and putatively mutagenic events may occur following vector activation and packaging into capsids provided by either possible endogenous retroviruses or unrelated pathogenic viral infections. Phenotypic mixing16 is well known and utilized when pseudotyping RV and LV vectors with VSV-G for example. Thus our aim was to eliminate the possibility of vector mobilization and viral dissemination by deleting the LV packaging signal Ψ following a Cre/loxP mediated excision in SIN-ΔΨ-LVs after successful gene transfer into recipient cells. In addition to rendering LV vector integrations more stable in a clinical setting, deletions encompassing Ψ would also allow for safer manipulation from a bioengineering perspective. For example, LV helper constructs could be introduced into the cellular genome of producer cells and optimized for expression without concomitant concern of RCL formation.
Here we have shown that Ψ can be deleted post-transduction in target cells. This manipulation also opens the possibility of therapeutic cell expansion by coupling Ψ with homeobox transcription factors. HoxB4 for instance, a potential candidate for therapeutic stem cell expansion, could be used with hematopoietic stem and progenitor cells in a clinical setting17 and with this vector system. Since constitutive expression of HoxB4 is associated with leukemia18,19, an in vitro strategy to simultaneously delete HoxB4 and Ψ after gene transfer and successful cell expansion could be of great therapeutic potential.
Several concerns will still have to be addressed before the above ‘proof of principle’ experiments can be implemented in gene therapy. One concern is the observed reduced titer in our study after translocation of the packaging signal Ψ. While this could be a consequence of spontaneous sequence deletions flanked by repetitive loxP elements encompassing Ψ during virus production, it seems unlikely since the region of homology is only 34 bp and our cell population with dual fluorescent tags remained stable in the absence of Cre. Another possibility for the reduced titer is a positional effect due to the translocation of Ψ20,21,22. Thus we are currently investigating viral titer yields after moving the loxP cleavage cassette containing Ψ closer to its native locus. A second concern to be addressed is the reported potential low frequency carcinogenesis due to chromosomal rearrangements after implementation of the cre/loxP system in cells with two or more integrants23,24. Thus, for clinical trials, careful screening for single integration events and/or the absence of chromosomal rearrangements would be required prior to reintroducing the therapeutic construct into a patient. Alternatively, implementation of a conditional ‘suicide gene’ may also serve as a safeguard with implementation of the above cre/lox system.
Methods
Cloning procedures
All PCR-based cloning and analytic procedures were carried out using Platinum Taq polymerase (Invitrogen, Carlsbad, CA). Primers were purchased from Integrated DNA Technologies Inc. (IDT, Coralville, IA). Restriction enzymes and DNA modifying enzymes were purchased either from Invitrogen or New England Biolabs (NEB, Ipswich, MA), respectively.
Construction of double fluorescent LV vectors with Ψ translocation into a removable cassette
As a basis for our experiments we selected pLeGO-iG-SFFV-IRES-eGFP (a generous gift of Dr. Kristoffer Weber, University Medical Center Hamburg, Germany), in which an SFFV promoter – MCS – IRES - GFP expression cassette is flanked by loxP25. We modified this LV gene transfer system by inserting a second fluorescent reporter gene, mCherry driven by the Elongation Factor 1α (EF1α) promoter as follows: A 8144 bp HpaI XhoI fragment from pLeGO-iG-SFFV-IRES-eGFP was recombined with an 1140 bp HpaI-SalI fragment from pWPT-mCherry. Plasmid pWPT-mCherry was derived from a BamHI, SalI substitution of GFP in pWPT-GFP containing the EF 1α promoter with mCherry from pRSC-PCh-W resulting in LeGO-IG- mCherry- loxP-eGFP or construct 512 (Fig. 1A). A DNA fragment encoding the HIV packaging signal Ψ was amplified with construct 512 as a template using primers HIV Ψ sense (5′- CCCGCTAGCTCGACGCAGGACTCGGCTTGC-3′) and HIV Ψ anti (5′- CCCGCTAGCCTCCCCCGCTTAATACTGACG-3′) and Platinum Taq polymerase under the following cycling conditions: 40 cycles with 95°C 30″, 51.3°C 30″, 72°C 60″. The 156 bp amplicon was digested with NheI and transferred into the NheI site of construct 512 located between the 5′ loxP site and the SFFV promoter leading to construct 512-Ψ-shift. Deletion of the native HIV Ψ signal in p512-Ψ-shift was performed as follows: A PCR fragment encoding for the CMV promoter and the RU5 region but lacking the packaging signal Ψ was generated with construct 512 as template using primers Ψ-del-sense (5′-TGCTTAGGGTTAGGCGTTTTGCGCTGCTTC-3′) and Ψ-del-anti (5′-GCGCGCTCTTCCTCTATCCCTTTCGCTTTCAAGTCC -3′) with Platinum Taq polymerase (Invitrogen). at 35 cycles with 95°C for 30″, 50°C for 30″ and 2′ at 72°C. After digestion with NruI and SfoI the PCR fragment was substituted for a 1003 bp NruI flanked fragment in construct 512 leading to ΔΨ-LeGO-IG- mCherry- loxP-Ψ-eGFP or construct 521 (Fig. 1B). Sequence integrity for all sequence manipulations was verified by standard sequence analysis (Sequencing facility FHCRC, Seattle).
Construction of the Cre expression plasmid pCMV-Shuttle-Cre
Adenovirus 5 encoding Cre Recombinase (Ad-Cre, Eton Bioscience Inc., San Diego, CA) was utilized as DNA template to amplify the Cre recombinase DNA fragment with standard PCR amplification conditions using primers pShuttle-Cre-5′ (5′-AGATCTGCCACCATGTCCAATTTACTGACCGTACAC-3′) and pShuttle-Cre-3′ (5′-GTCGACTAATCGCCATCTTCCAGCA-3′). The resulting PCR product was subcloned into pCR2.1 (Clontech, Mountain View, CA) for sequence analysis (Sequencing facility FHCRC, Seattle) and transferred into BglII, SalI digested pShuttle-CMV26 (Addgene, Cambridge, MA).
Construction of the BFP LV vector plasmid
Plasmid pRSCPBw was obtained by substituting the 722 bp blue fluoresecent protein gene (BFP) cassette flanked by AgeI and SalI sites from pCVL.Sce.IRES.mTagBFP (a kind gift from Dr. Andrew Scharenberg, Seattle Children's Research Institute, Seattle, WA, USA) for the GFP cassette in pRRLsin.cPPT.hPGK.GFP.Wpre29.
Tissue culture, vector production, transduction and fluorescent reporter gene analysis
Human embryonic kidney 293T (HEK 293T27 and HT1080 cells28 were cultivated in 10% FCS (Invitrogen) and DMEM (Invitrogen) with penicillin/streptomycin solution (Invitrogen) at 37°C and 5% CO2 in a humidified atmosphere. LV vector production and was carried out according to previously published procedures29. For viral vector transduction, unwanted HEK 293T producer cells were removed by centrifugation at 1200 x g. Supernatants were applied onto HT1080 reporter cells. FACS analyses of cells expressing fluorescent reporter genes were performed on an LSR-II flow cytometer (Becton Dickinson, BD, San Jose, CA) and resulting data were analyzed with FLOJO flow cytometry analysis software (FLOJO, Ashland, OR). Sorting of fluorescent cells was performed with an ARIA II cell sorter (Becton Dickinson).
Analytical PCR to discriminate between 293T producer cells and HT1080 target cells
For PCR analysis of transduced HT1080 cells primers Adeno-sense (5′-GGTTTACTGGCCCCAATTTT-3′) and Adeno-anti(5′-CACAGCCACGCTTTTCACTA-3′) were used to detect a 304 bp fragment specific for adenoviral DNA found in 293T cells but not HT1080 cells. For reference primers GAPDH sense (5′-CATCACTGCCACCCAGAA-3′) and GAPDH anti (5′-GGGCCATGAGGTCCACCA-3′) were utilized to detect a 552 bp Glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) PCR fragment. Both primer sets were added to 300 ng of genomic DNA and amplified with AmpliTaq Gold under standard PCR conditions (32 cycles with 95°C for 15″, 62°C for 20″ and 7′ at 72°C).
References
Naldini, L. Medicine. A comeback for gene therapy. Science 326, 805–806 (2009).
Naldini, L. Ex vivo gene transfer and correction for cell-based therapies (Review). Nat Rev Genet 12, 301–315 (2011).
Yi, Y., Noh, M. J. & Lee, K. H. Current advances in retroviral gene therapy (Review). Current Gene Therapy 11, 218–228 (2011).
Kappes, J. C. & Wu, X. Safety considerations in vector development (Review). Somat.Cell.Mol.Genet. 26, 147–158 (2001).
Cockrell, A. S. & Kafri, T. Gene delivery by lentivirus vectors (Review). Molecular Biotechnology 36, 184–204 (2007).
Logan, A. C., Haas, D. L., Kafri, T. & Kohn, D. B. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J.Virol. 78, 8421–8436 (2004).
Cornetta, K. et al. Replication-competent lentivirus analysis of clinical grade vector products. Mol Ther 19, 557–566 (2011).
Evans, J. T. & Garcia, J. V. Lentivirus vector mobilization and spread by human immunodeficiency virus. Hum.Gene Ther. 11, 2331–2339 (2000).
Chong, H., Starkey, W. & Vile, R. G. A replication-competent retrovirus arising from a split-function packaging cell line was generated by recombination events between the vector, one of the packaging constructs and endogenous retroviral sequences. J.Virol. 72, 2663–2670 (1998).
Connolly, J. B. Lentiviruses in gene therapy clinical research. Gene Ther. 9, 1730–1734 (2002).
Gossen, M. & Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc.Natl.Acad.Sci.USA 89, 5547–5551 (1992).
Gossen, M. et al. Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769 (1995).
Schmidt, M. et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR). Nature Methods 4, 1051–1057 (2007).
Louis, N., Evelegh, C. & Graham, F. L. Cloning and sequencing of the cellular-viral junctions from the human adenovirus type 5 transformed 293 cell line. Virology 233, 423–429 (1997).
Dull, T. et al. A third-generation lentivirus vector with a conditional packaging system. J.Virol. 72, 8463–8471 (1998).
Zavada, J. Viral pseudotypes and phenotypic mixing (Review). Archives of Virology 50, 1–15 (1976).
Sorrentino, B. P. Clinical strategies for expansion of haematopoietic stem cells (Review). Nat Rev Immunol 4, 878–888 (2004).
Zhang, X.-B. et al. High incidence of leukemia in large animals after stem cell gene therapy with a HOXB4-expressing retroviral vector. J.Clin.Invest. 118, 1502–1510 (2008).
Larochelle, A. & Dunbar, C. E. HOXB4 and retroviral vectors: adding fuel to the fire. J.Clin.Invest. 118, 1350–1353 (2008).
Richardson, J. H., Child, L. A. & Lever, A. M. Packaging of human immunodeficiency virus type 1 RNA requires cis-acting sequences outside the 5′ leader region. J.Virol. 67, 3997–4005 (1993).
Berkowitz, R. D., Hammarskjold, M. L., Helga-Maria, C., Rekosh, D. & Goff, S. P. 5′ regions of HIV-1 RNAs are not sufficient for encapsidation: implications for the HIV-1 packaging signal. Virology 212, 718–723 (1995).
Lu, K., Heng, X. & Summers, M. F. Structural determinants and mechanism of HIV-1 genome packaging (Review). J.Mol.Biol. 410, 609–633 (2011).
Van Deursen, J., Fornerod, M., Van Rees, B. & Grosveld, G. Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc.Natl.Acad.Sci.USA 92, 7376–7380 (1995).
Smith, A. J. et al. A site-directed chromosomal translocation induced in embryonic stem cells by Cre-loxP recombination [Erratum appears in Nat Genet 1996 Jan;12(1):110]. Nat.Genet. 9, 376–385 (1995).
Weber, K., Bartsch, U., Stocking, C. & Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol Ther 16, 698–706 (2008).
He, T. C. et al. A simplified system for generating recombinant adenoviruses. PNAS 95, 2509–2514 (1998).
DuBridge, R. B. et al. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol.Cell.Biol. 7, 379–387 (1987).
Rasheed, S., Nelson-Rees, W. A., Toth, E. M., Arnstein, P. & Gardner, M. B. Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer 33, 1027–1033 (1974).
Beagles, K. E., Peterson, L., Zhang, X., Morris, J. & Kiem, H.-P. Cyclosporine inhibits the development of green fluorescent protein (GFP)-specific immune responses after transplantation of GFP-expressing hematopoietic repopulating cells in dogs. Hum.Gene Ther. 16, 725–733 (2005).
Acknowledgements
We would like to thank Helen Crawford , Bonnie Larson and Laura A. Farren for assisting with the preparation of this manuscript. This work has been supported by National Institutes of Health (Bethesda, MD) grant P30DK056465. Dr. Hans-Peter Kiem is a Markey Molecular Medicine Investigator and the recipient of the Jose Carreras/E.D. Thomas Endowed Chair for Cancer Research.
Author information
Authors and Affiliations
Contributions
RML, JE and HPK jointly prepared the manuscript. JE repositioned the lentiviral packaging signal and RML added the mCherry fluorescent marker for the constructs presented and conducted the experiments described.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Luche, R., Enssle, J. & Kiem, HP. Abrogated cryptic activation of lentiviral transfer vectors. Sci Rep 2, 438 (2012). https://doi.org/10.1038/srep00438
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00438
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
