
Abstract
The abundant pigment-protein membrane complex photosystem-I (PS-I) is at the heart of the Earth’s energy cycle. It is the central molecule in the “Z-scheme” of photosynthesis, converting sunlight into the chemical energy of life. Commandeering this intricately organized photosynthetic nanocircuitry and re-wiring it to produce electricity carries the promise of inexpensive and environmentally friendly solar power. We here report that dry PS-I stabilized by surfactant peptides functioned as both the light-harvester and charge separator in solar cells self-assembled on nanostructured semiconductors. Contrary to previous attempts at biophotovoltaics requiring elaborate surface chemistries, thin film deposition and illumination concentrated into narrow wavelength ranges the devices described here are straightforward and inexpensive to fabricate and perform well under standard sunlight yielding open circuit photovoltage of 0.5 V, fill factor of 71%, electrical power density of 81 µW/cm2 and photocurrent density of 362 µA/cm2, over four orders of magnitude higher than any photosystem-based biophotovoltaic to date.
Similar content being viewed by others


Introduction
PS-I precisely orchestrates 96 chlorophyll molecules with electron donors and acceptors1 ( Fig 1 a ) achieving efficient coherent energy transfer2 and near-unity charge separation quantum yield at ambient temperatures3,4. This is a feat unmatched by any man-made photoelectronic device and has led to PS-I being studied as a candidate for many nanobioelectronic applications5,6,7,8, as well as being the original inspiration behind the dye-sensitized solar cell (DSC)9. So far, research on PS-I biophotovoltaics has focused on proof-of-principle devices, studying immobilized PS-I complexes and isolated reaction centers (RC) in self-assembled monolayers (SAMs) on flat electrodes5,7,8.

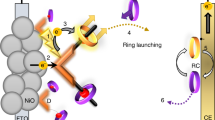
Schematic of PS-I in cellular membrane and in two types of biophotovoltaic cells.
(a) PS-I in ∼30Å thick cellular bilipid membrane (grey). Arrows indicate direction of electron travel with acceptor side facing down. The core subunits are shown in grey and the only prosthetic groups are the core electron transport associated cofactors including the P700 chlorophyll (Chl) dimer in the center, the four associated Chl a molecules (green), the two phyloquinone acceptors (orange) and the three FeS centers Fx, Fa and Fb (yellow) (sulfur) and brown (Fe). The ribbon diagram of stromal subunits PsaD, PsaC and PsaE is shown protruding outside of the membrane and colored blue, red and purple respectively. (b) The natural redox mediators cytochrome c and ferredoxin are absent, replaced by Z813 electrolyte and either a TiO2 nanocrystalline sintered paste (left) or ZnO nanowires (right). Left: stabilized PS-I physisorbed to TiO2 on fluorine-doped tin oxide (FTO) coated glass. Right: (bioengineered) PS-I self-assembled in the presence of an overabundance of PsaE-ZnO subunit on ZnO nanowires grown on ITO glass. In both cases, energy levels are matched to favor electron transfer from electrolyte to photoanode5,6,7,8.
Two main obstacles hinder biophotovoltaics from being a more widely studied technology, constantly improved by many independent researchers. Firstly, while extracting PS-I from a variety of abundant sources is easy, drying this extract on electrodes results in rapid loss of function due to denaturation. Secondly, the electrical power output of biophotovoltaics to date has been so low5,6,7,8, that they were of little practical interest and the characterization necessary to improve their performance required cumbersome, expensive to iterate-optimize methods. For instance, in order to obtain measureable photocurrents it was necessary to make up for the low absorption cross sections of the nearly transparent active SAMs. In prior studies this was addressed by using either laser light with power equivalent to 100 times that of standard air-mass 1.5 (AM1.5) sunlight5, or incoherent monochromatic light6 in both cases precisely tuned to the pigment absorption maxima –an unrealistic emulation of real-world conditions requiring elaborate instrumentation.
Results
We have removed these two obstacles by designing a PS-I biophotovoltaic whose IV characteristics can be easily studied under regular sunlight and its design and fabrication are amenable to low-cost, iterative optimization. To avoid denaturation, we treated PS-I with designer peptide surfactants1. To improve photovoltaic performance we increased the light absorption cross-section without changing the footprint by departing from the traditional flat electrode geometry in favor of mesoscopic, high-surface area semiconducting electrodes (TiO2 nanocrystals and ZnO nanowires). Finally, we showed how high affinity peptide motifs10 bioengineered to promote selective adsorption to specific substrates can enhance photovoltaic performance. These materials, geometries and design resulted in simple, robust biophotovoltaic devices of unprecedented performance.
Photochemically active, trimeric PS-I was isolated and characterized from the thylakoids of the thermophilic cyanobacteria Thermosynechococcus elongatus as described in detail in Fig. 2 of Iwuchukwu et al.11. This PS-I was stabilized for several months in solution12 and in dry form by designer peptide surfactants1 (
Fig. 2
). To build devices, PS-I molecules were air-dried on nanostructured semiconducting substrates and the circuits were completed by liquid electrolyte and platinized glass as is common for conventional DSCs (
Fig. 1 b
). We used nanocrystalline TiO2 and ZnO nanowires to provide a large effective surface area (Aeff) for PS-I adsorption and light harvesting (
Fig. 3
) and without any further optimization, these devices achieved electrical power outputs Pout of up to 81 µW/cm2 and area-normalized short-circuit current densities IscNorm of up to 362 µA/cm2 (
Fig. 4
). These parameters are to be compared to the over 10,000 times lower IscNorm (∼30 nA/cm2) reported previously with monochromatic illumination tuned to the ∼800 nm absorption peak of a monolayer of PS-I on a thin film of gold (actual power efficiency not reported)7and to the up to 120 µA/cm2 IscNorm observed when isolated RCs were chemically bound to a series of evaporated metallic and semiconducting thin films and illuminated with 10 W/cm2 laser light (one-hundred times the power density of AM1.5 sunlight) all concentrated into the 808 nm absorption peak of RCs5. While there have been various estimated and theoretical maximum efficiencies (e.g. an upper possible limit of 20% efficiency for RC-biophotovoltaics5), there have been no reports of conversion efficiency η for PS-I (where  , Pin the total power of the incident light and Poutthe total resulting electrical power). Our photocurrent measurements were carried out under AM1.5 standard simulated sunlight with precisely controlled active surface areas (0.159 cm2) and continuously-calibrated, spectral-mismatch corrected sunlamps, as is the standard in the conventional photovoltaic industry13. Under these conditions, that closely emulate outdoor sunlight, the total external efficiency of conversion of incident sunlight to useable electricity was η ∼ 0.08%. This must not to be confused with the sometimes very high quantum or internal efficiencies routinely reported for organic optoelectronics.
, Pin the total power of the incident light and Poutthe total resulting electrical power). Our photocurrent measurements were carried out under AM1.5 standard simulated sunlight with precisely controlled active surface areas (0.159 cm2) and continuously-calibrated, spectral-mismatch corrected sunlamps, as is the standard in the conventional photovoltaic industry13. Under these conditions, that closely emulate outdoor sunlight, the total external efficiency of conversion of incident sunlight to useable electricity was η ∼ 0.08%. This must not to be confused with the sometimes very high quantum or internal efficiencies routinely reported for organic optoelectronics.

(a) To promote attachment and orientation of the entire PS-I complex to ZnO nanowires, we fused the ZnO-binding peptide tag RSNTRMTARQHRSANHKSTQRARS10 (expressed in E.coli) to the N-terminus of the PsaE subunit. Upon exchanging native PsaE in favor of PsaE-ZnO and self-assembly, the modified PS-I preferentially binds to ZnO nanowires by the electron acceptor side, minimizing distance between electron acceptor and electrode and maximizing electron transfer. (b) The marked increase in the rate of methyl viologen (MV)-mediated oxygen reduction by PS-I in the presence of the designer surfactant peptide A6K, indicates that A6K maintains the ability of PS-I to catalyze photochemical charge separation and MV-mediated O2 uptake relative to the control treatment with either the non-ionic detergent Triton X-100 (middle) or DDM present in the isolation buffer (left). Increased O2 uptake activity cannot be due to free chlorophyll-mediated O2 consumption (via3chl) since treating cyanobacterial PS-I with strong detergents (1% SDS) leads to only minimal loss of chlorophyll from PS-I22. All activity tests are normalized per mol of PS-I. (c) Low-temperature fluorescence of PS-I self-assembled in the presence of excess PsaE-ZnO (red) peaks at the same two wavelengths as unaltered PS-I extract (blue) indicating that bulk chlorophyll organization is preserved12 and the stabilizing interaction with A6K is likely similar in both cases.

SEM of nanostructured TiO 2 and ZnO photoanodes and schematic of an ideal ultra-low cost biophotovoltaic arrangement.
(a) 3.8 µm-thick, 60 nm-pore TiO2 nanocrystalline photoanode of roughness factor ρTiO2 ∼200 (i.e. surface area increases by roughly fifty times per µm of film thickness) fabricated as described previously13. (b) 3 µm tall, ZnO nanowires grown on Zn-nanoparticle-seeded ITO-glass as described elsewhere15, ρZnO∼30. Round graphic at top left of inserts represents a PS-I trimer drawn to scale. (c), (d), (e) ideal arrangement of PS-I and designer surfactant peptide stabilizers on ZnO nanowires that could be grown at room temperature on a variety of flexible and inexpensive substrates16.

Photocurrent measurements of PS-I biophotovoltaic devices under AM1.5 simulated insolation at 298°K.
Illuminated surface 0.159 cm2 (a) 40 µl of PS-I (0.2 mg/mL) stabilized by 1∶1 0.1%w/v designer surfactant peptide A6K (resulting in a total of 8 µg of protein) dried on a 3.8 µm thick layer of 60 nm-pore TiO2 produces an IV curve typical of a DSC. Fill factor (ff) ranged from 64% to 71% (b) Eliminating ultraviolet (UV) wavelengths below 350 nm resulted in a ∼20% reduction in the normalized short circuit current (JscNorm) and a ∼10% reduction in the open circuit voltage (Voc) indicating that 80% of the total electrical power generated is due to PS-I (the rest due to UV photovoltaic response of TiO2). These photocurrents cannot be attributed to sensitization of TiO2 by leached chlorophyll derivatives1,17. A blank control containing A6K generated no power when exposed to UV-less sunlight of any intensity, neither did controls built with PS-I denatured by boiling for 10 minutes, nor devices built with PS-I not treated with A6K (data not shown). Total incident-light to electrical external power conversion efficiency η was 0.08% with UV, 0.07% without. (c) Linearity test of PS-I photocurrent at intensities from 0.01x to 1.0x AM1.5 shows behavior typical of a DSC. (d) IV of PS-I self-assembled in the presence of an overabundance of PsaE-ZnO electron-accepting subunit yields a total power conversion efficiency, η = 0.03%. (e) Control: IV of PS-I self-assembled with an overabundance of non-ZnO specific histidine-tag containing PsaE subunit yields lower Voc, JscNorm and η = 0.00% as expected, suggesting that the PsaE-ZnO tag either enhanced binding of PS-I to the ZnO nanowires or favored the optimal orientation, or both. Z813Co(II)/Co(III) electrolyte14 and platinized glass were used to complete all devices.
PS-I Biophotovoltaic Solar Cell
In photosynthetic organisms, PS-I catalyses light-driven electron transfer from reduced plastocyanin located in the lumen, to ferredoxin in the stroma providing a path across the membrane consisting of a chain of cofactors ( Fig. 1a ). Light absorption results in excitation of the primary electron donor (P700), transfer to primary and secondary electron acceptors and finally crossing of the membrane. In our biophotovoltaic solar cell, the role of plastocyanin was played by the Co(II)/Co(III) ion-containing electrolyte Z81314 and ferredoxin was replaced by either nanocrystalline TiO2 ( Fig. 1 b left) or ZnO-nanowires ( Fig. 1 b right) to provide large-surface area electron acceptors.
When using TiO2, we chose the pore size of the nanocrystalline film to be double the diameter of our PS-I particles to ensure a high probability of physisorption. When using ZnO nanowires, we substituted (by a self-assembly exchange reaction) the naturally-occurring electron acceptor PsaE subunit with one that contained an amino acid sequence with high affinity for ZnO: RSNTRMTARQHRSANHKSTQRARS10 (PsaE-ZnO) thus promoting adhesion and minimizing the distance that electrons must travel to the anode ( Fig. 2 a ).
Stabilization of Native and Bioengineered PS-I
Stabilization of dry PS-I extract on glass and on the transparent conductor Indium-Tin-Oxide (ITO) for at least three weeks has been described elsewhere1. Here, we mixed PS-I at 0.4 mg/mL in a 1∶1 ratio with 0.1% (w/v) of the 2.4 nm long cationic peptide surfactant Ac-AAAAAAK-NH2 (A6K) consisting of six alanines and a lysine at the amidated C-terminus and observed enhanced stability ( Fig. 2 b ). The bioengineered, self-assembled PS-I containing PsaE-ZnO exhibited low-temperature fluorescence peaks identical to unmanipulated PS-I extract undergoing identical treatment ( Fig. 2 c ), indicating that subunit substitution did not adversely affect structure and we expect the photochemical-activity enhancing effect of A6K to be similar in both cases.
Nanocrystalline TiO2 and ZnO Nanowires as Large Surface Area Photoanodes
Isc is directly proportional to electrical power output and is controlled by light absorption. To increase the useful light incidence angle and optical cross-section of our biophotovoltaics, we used two types of rough, large surface area semiconductors as photoanodes. This made our devices able to absorb light from nearly a 2π solid angle and provided an increased effective area for PS-I SAM adsorption: Aeff ∼200 times that of the flat footprint for TiO2 nanocrystals (Fig 3 a) and ∼30 times with ZnO nanowires (Fig 3 b). In addition to providing an inexpensive alternative to TiO2, the charge carrier mobility of ZnO nanowires is one-hundred times faster than TiO215 and large-scale, ambient temperature solution-growth of ZnO nanowires is simple, requiring fewer steps, less energy and is easily adaptable to flexible conducting substrates16. However, ZnO DSC photoanodes have so far always underperformed15 when compared with identically sensitized TiO2. This is due to their lower roughness factor ρ, poor dye loading and the shunting of the photocurrent by the corrosion of ZnO by common DSC dyes and electrolytes. The IV behavior of our biophotovoltaics indicated that tagging PS-I with an amino acid sequence that binds to ZnO promoted orientation and/or binding to ZnO nanowires with η = 0.03%, for PsaE-ZnO, while η = 0.00% for the histidine-tagged control (Fig. 4 d and e respectively). The Isc achieved with ZnO (Fig. 4 d) is roughly a factor of ten lower than that with TiO2, consistent with the ratio of the two Aeff (ρZnO∼30, ρTiO2∼200) suggesting that Aeff is the primary control of Isc.
Photocurrent Measurements Under Realistically Simulated Solar Illumination
The shapes of the IV curves ( Fig. 4 ) obtained upon exposing our devices to AM1.5 sunlight for both PS-I complexes on TiO2 and bioengineered PS-I on ZnO are similar to conventional DSCs. After accounting for the UV-excited photocurrent ( Fig. 4b ) the following four types of control devices, containing all buffer components and electrolyte, did not yield any additional photovoltaic action: i) devices made with denatured PS-I (boiled for 10 min), ii) blanks without PS-I or A6K , iii) blanks without PS-I but with A6K, iv) devices with PS-I but no A6K. This leaves stabilized, non-denatured PS-I as the only possible agent behind 80% of the maximum measured photocurrent (the remaining 20% being due to UV excitation of TiO2 and ZnO). We emphasize that these photocurrents cannot be attributed to sensitization by leached chlorophyll, because unless precisely organized by the PS-I scaffold, chlorophyll alone does not act as a photovoltaic sensitizer1, the interaction between the chlorophyll ester units with the TiO2 surface is weak and chlorophyll does not adsorb17 on TiO2 or ZnO .
Discussion
Using inexpensive raw materials and simple processes, we have achieved record biophotovoltaic performances. We isolated PS-I from thermophilic cyanobacteria, but the structural similarity between this and higher plant PS-I suggests that many other abundant sources of highly pigmented thylakoids can also be used including the normally discarded leafy parts of common agricultural crops or timber. PS-I is indeed a promising raw material for ultra-low-cost biophotovoltaics as postulated by LaVan et al.4 but significant optimization challenges must be met. The devices characterized here indicate merely the lowest limit of PS-I biophotovoltaic performance possibilities. Since we did not perform any optimization, significant efficiency gains can be expected from increased loading, better oriented and more tightly coupled PS-I to photoanode, customization of stabilizing agents and better matching of bio-friendly electrolytes with photoanode/photocathode substrates. The short-term stability and photoactivity of various treatments of PS-I is summarized in Fig. 2 b and discussed in detail elsewhere1,12 and is encouraging. Clearly, tracking biophotovoltaic performance over the long-term is an important future step but was beyond the scope of the present study18. While we used centrifugation, equally pure PS-I can be isolated from plant or bacterial extracts by porous membrane bioseparation, an inexpensive method that can be scaled up to industrial levels by simple yet highly efficient affinity binding with protein-specific epitope tags19. The design and methodology principles described here are suitable to biophotovoltaics that can be characterized under ordinary sunlight. We hope these results encourage optimization efforts to deliver biosolar power that is truly “green”.
Methods
Cloning and expression of ZnO-binding subunits
While we here show data on PsaE only ( Fig. 2c ), plasmids coding for PsaE (UniProt accession number Q62007) and also PsaD (UniProt accession number P34982) from Cyanobacterium Mastigocladus laminosus were expressed and studied. Both self-assemble near the final electron acceptor and ejection site of the PS-I complex and are ideally placed to appropriately attach and orient the molecule to a photoanode. The peptide tag RSNTRMTARQHRSANHKSTQRARS10, was fused to the N-terminal of the respective coding sequence via a five-residue glycine linker and a six residue histidine tag was fused to the C-terminal by PCR ( Fig. 2a ) and cloned into expression plasmid pET-DEST42, Invitrogen (Carlsbad, CA, USA), according to the manufacturer’s instructions. The plasmids were transformed into the Escherichia coli (E. coli) expression strain BL21 (DE3) pLysS. Protein expression was carried at 37°C in LB media and the soluble protein fraction isolated by centrifugation after lysing cells by sonication. The proteins were purified using His-Trap column (GE Healthcare, Uppsala, Sweden) using an ÄKTA purifier chromatography system (GE Healthcare), according to the manufacturer’s instructions.
Subunit exchange
To exchange the native PsaE (or PsaD) in PS-I to the recombinant PsaE-ZnO (or PsaD-ZnO), the recombinant proteins were incubated with PS-I in a 50∶1 molar ratio for 2 hours at 4°C. Free PsaE (or PsaD) was removed by centrifuging the sample over an YM-100 filter (Millipore) leaving unbound PsaE (or PsaD) in the flow-through ( Fig. 2a ). These exchange reaction protocols are expected to yield exchange efficiency of >65%. Since we used a dual isolation system using both IMAC and sucrose density exchange, we know with certainty that our efficiency is higher than 65%.
Stability test of immobilized PS-I via low temperature fluorescence spectroscopy
Low-temperature fluorescence spectroscopy1 was used to ascertain stability of the PS-I complex when immobilized on ZnO nanowires. Each 1 cm × 1 cm ZnO nanowire chip covered in PS-I (drops standardized to contain a total of 2.9 µg of protein) was placed on a custom made Teflon holder and cooled under liquid nitrogen (−196.15°C) in a cryostat with glass windows at right angles. Fluorescence was excited optically using a 408 nm laser incident at a −45° angle to the normal to the chip surface. Steady-state emission spectra were recorded using a CCD spectrometer (slit width 20 µm) with an optical fiber input oriented +45° to the normal thus detecting at a right angle from the excitation beam. The fluorescence intensity of each sample was qualitatively normalized and the peaks were found to be identical to those of native PS-I ( Fig. 2c ), as expected if no structural changes resulted from the exchange reaction or immobilization on the ZnO nanowires.
Fabrication of sealed solar cells, current-voltage (IV) and control measurements
Devices ( Fig 1 ) were made by allowing a 40 µL drop of PS-I solution to air dry at room temperature on two different nanostructured semiconducting electrodes: a TiO2 of 60 nm average pore size, 3.8 µm film thickness, roughness factor: ρTiO2 ∼200 (i.e. surface area increases by x50 per µm of film thickness) fabricated as described previously13 ( Fig 3a ) and a mat of 3 µm tall, ZnO nanowires (grown on Zn-nanoparticle-seeded ITO-glass as described elsewhere1) with ρZnO ∼30 ( Fig 3b ). Cells were sealed with 40 µm-thick heat-treated Syrlyn gaskets and Z813 Co(II)/Co(III) electrolyte14 composed of: 0.5 M Co(Ophen)3(bis(trifluoromethanesulfonyl)imide)2 0.05M Co(Ophen)3(bis(trifluoromethanesulfonyl)imide)3 0.2 M LiClO4 dissolved in a 60%Ethylene Carbonate and 40% acetonitrile (v/v) solvent, which was introduced by capillary action. Platinum-coated, fluorine-doped tin-oxide (FTO) glass was used as the counter electrode. In all cases, photomasks of 0.159 cm2 where used and IV curves, JscNorm and total power were normalized to this area. All measurements were performed using a continuously thermopile-calibrated solar simulator with neutral density filters as described in detail by Ito et al.13. 60 devices total were tested, the results reported in Fig 4 belong to individual devices exhibiting typical behavior, not averages over many devices. The error bars in all IV curves are included in the plots but are smaller than the size of the data point markers. Control devices made with TiO2 and electrolyte but containing no PS-I and exposed to full sunlight (including UV) gave Pout 28 µW/cm2, with 277 µA/cm2 Isc and 257 mV Voc with a fill factor of 0.39. It was impossible to obtain IPCE curves of control unsensitized devices due to very low currents, as expected (see supplemental materials).
PS-I Purification
Our PS-I was identical to that used in Iwuchukwu et al.11. Briefly, PS-I was extracted from the thylakoid membranes of the thermophilic cyanobacteria T. elongatus. Bacteria were grown in 2 L airlift fermenters (Bethesda Research Labs, Bethesda MD) to late log phase at 56°C. Bacterial growth was followed by incubation with 0.25% (w/v) lysozyme for 2 hours at 37°C under gentle agitation. Cells were lysed with the French press; unlysed cells were removed at 3,000 x g for 5 min and membranes were collected at 20,000 RPM. The membranes were washed and solubilized as in Fromme and Witt20 with the exception that in the final wash 3 M NaBr was used. Then the supernatant was loaded on a 10-30% linear sucrose gradient (20 mM MES pH 7.0, 10 mM MgCl2, 10 mM CaCl2 and 0.3% w/v n-dodecyl-b-D-maltopyranoside (DDM), for 16 hours at 24,000 RPM. The lower green band was collected (see Fig. 2 of Iwuchukwu et al.11 ), pooled and stored at –20°C. Purity was confirmed by Tris-tricine SDS-PAGE gel electrophoresis. The chlorophyll content of PS-I was measured as described previously21.
References
Kiley, P. et al. Self-assembling peptide detergents stabilize isolated photosystem-I on a dry surface for an extended time. PLoS Biol. 3, 1181–1186 (2005).
Collini, E. et al. Coherently wired light-harvesting photosynthetic marine algae at ambient temperature. Nature 463, 644–647 (2010).
Editorial Live Wires. Nature Nanotechnology 1, 79 (2006).
LaVan, D. A. & Cha, J. N. Approaches for biological and biomimetic energy conversion. PNAS 103, 5251–5255 (2006).
Das, R. et al. Integration of photosynthetic protein molecular complexes in solid-state electronic devices. Nano Letters 4, 1079–1083 (2004).
Trammell, S. A., Wang, L., Zullo, J. M., Shashidhar, R. & Lebedev, N. Orientated binding of photosynthetic reaction centers on gold using Ni-NTA self-assembled monolayers. Biosensors & Bioelectronics 19, 1649–1655 (2004).
Trammell, S. A., Spano, A., Price, R. & Lebedev, N. Effect of protein orientation on electron transfer between photosynthetic reaction centers and carbon electrodes. Biosensors and Bioelectronics 21, 1023–1028 (2006).
Carmeli, I., Frolov, L., Carmeli, C. & Richter, S. Photovoltaic activity of photosystem I-based self-assembled monolayer. J. Am. Chem. Soc. 129, 12352–12353 (2007).
Grätzel, M. Photoelectrochemical Cells. Nature 414, 338–344 (2001).
Kjærgaard, K., Sorensen, J. K., Schembri, M. A. & Klemm, P. Sequestration of zinc oxide by fimbrial designer chelators. Appl. Env. Microbiol. 66, 10–14 (2000).
Iwuchukwu, I., J. et al. Self-organized photosynthetic nanoparticle for cell-free hydrogen production. Nature Nanotechnology 5 (2009).
Matsumoto, K., Vaughn, M., Bruce, B. D., Koutsopoulos, S. & Zhang, S. Designer Peptide Surfactants Stabilize Functional Photosystem-I Membrane Complex in Aqueous Solution for Extended Time. J. Phys. Chem. B 113, 75–83 (2008).
Ito, S. et al. Fabrication of screen-printing pastes from TiO2 powders for dye-sensitised solar cells. Prog. Photovoltaics 15, 603–612 (2007).
Herve-Nusbaumer,. Zakeeruddin, S. M., Moser, J.-E. & Graetzel, M. An alternative efficient redox couple for the dye-sensitized solar cell system. Chem. Eur. J. 9, 3756–3763 (2003).
Law, M., Greene, L. E., Johnson, J. C., Saykally, R. & Yang, P. Nanowire dye-sensitized solar cells. Nature Materials 4, 455–458 (2005).
Xu, F. et al. Simple approach to highly oriented ZnO nanowire arrays: large scale growth, photoluminescence and photocatalytic properties. Nanotechnology 17, 588–594 (2006).
Kay, A. & Graetzel, M. Artificial Photosynthesis. 1. Photosensitization of TiO2 solar Cells with chlorophyll derivatives and related natural porphyrins. J. Phys. Chem. B 97, 6272–6277 (1993).
Butler, D. Thin Films: ready for their close-up? Nature 454, 558–559 (2008).
Liu, J., Lu, J., Zhao, X., Lu, J. & Cui, Z. Separation of glucose oxidase and catalase using ultrafiltration with 300-kDa polyethersulfone membranes. Journal of Membrane Science 299, 222–228 (2007).
Fromme, P. & Witt, H. T. Improved isolation and crystallization of Photosystem I for structural analysis. Biochimica Et Biophysica Acta-Bioenergetics 1365, 175–184 (1998).
Demarsac, N. T. & Houmard, J. Complementary Chromatic Adaptation - Physiological Conditions and Action Spectra. Methods in Enzymology 167, 318–328 (1988).
Lundell, D. J., Glazer, A. N., Melis, A. & Malkin, R. Characterization of a cyanobacterial photosystem-I complex. The Journal of Biological Chemistry 260, 646–654 (1985).
Acknowledgements
We thank the Intel Corporation for their unrestricted gift partially seed-funding this work. LK gratefully acknowledges her fellowship by the Knut and Alice Wallenberg foundation. SZ gratefully acknowledges the John Simon Guggenheim Foundation for his Guggenheim Fellowship to pursue this research. We are grateful to Sloan Kulper for creating panels c, d & e of Figure 3. We are indebted to S. M. Zakeerrudin, Jun-Ho Yum, Peter Chen and Jiang Liu for their assistance. BB and SZ were partially supported from an NSF NIRT award. BB was partially supported from a gift from the Gibson Family Foundation.
Author information
Authors and Affiliations
Contributions
AM, BB and SZ wrote the main manuscript text. AM, KM, LK, DY, MV, MN, BB performed the experiments, AM, KM, LK, MV, BB prepared figures 1–4. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Mershin, A., Matsumoto, K., Kaiser, L. et al. Self-assembled photosystem-I biophotovoltaics on nanostructured TiO2 and ZnO. Sci Rep 2, 234 (2012). https://doi.org/10.1038/srep00234
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00234
This article is cited by
-
Plant-mediated Z-scheme ZnO/TiO2-NCs for antibacterial potential and dye degradation: experimental and DFT study
Scientific Reports (2024)
-
Arraying of microphotosynthetic power cells for enhanced power output
Microsystems & Nanoengineering (2022)
-
Printable logic circuits comprising self-assembled protein complexes
Nature Communications (2022)
-
Biohybrid photoelectrodes for solar photovoltaic applications
Bulletin of Materials Science (2022)
-
Competition between intra-protein charge recombination and electron transfer outside photosystem I complexes used for photovoltaic applications
Photochemical & Photobiological Sciences (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
