
Abstract
Neuronal activity-dependent synaptic plasticity, a basis for learning and memory, is tightly correlated with the pattern of increase in intracellular Ca2+ concentration ([Ca2+]i). Here, using combined application of electrophysiological experiments and systems biological simulation, we show that such a correlation dynamically changes depending on the context of [Ca2+]i increase. In a cerebellar Purkinje cell, long-term potentiation of inhibitory GABAA receptor responsiveness (called rebound potentiation; RP) was induced by [Ca2+]i increase in a temporally integrative manner through sustained activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). However, the RP establishment was canceled by coupling of two patterns of RP-inducing [Ca2+]i increase depending on the temporal sequence. Negative feedback signaling by phospho-Thr305/306 CaMKII detected the [Ca2+]i context and assisted the feedforward inhibition of CaMKII through PDE1, resulting in the RP impairment. The [Ca2+]i context-dependent dynamic regulation of synaptic plasticity might contribute to the temporal refinement of information flow in neuronal networks.
Similar content being viewed by others


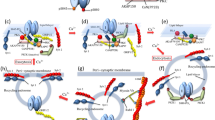
Introduction
Some forms of learning such as episodic and procedural memory require alteration of neuronal computation according to the causality in a series of events1. Although synaptic plasticity has been regarded as a basis for learning and memory2,3,4,5, it remains elusive how temporally sequenced neuronal activities are associated and reflected in the modulation of neuronal information processing. Extensive studies have clarified the close relation between the synaptic plasticity induction and the patterns of increase in intracellular Ca2+ concentration ([Ca2+]i)2,3,4,5,6,7,8,9,10,11,12,13,14,15. For example, at hippocampal excitatory synapses, a large [Ca2+]i increase caused by brief high frequency stimulation leads to long-term potentiation (LTP), while a small [Ca2+]i increase caused by prolonged low frequency stimulation induces long-term depression (LTD). One mechanism to regulate synaptic strength according to the temporal sequence of neuronal activity is spike timing-dependent plasticity (STDP)16,17,18. By producing different intensities of [Ca2+]i increase, the temporal order of pre- and postsynaptic neuronal firing within tens of milliseconds determines whether LTP or LTD is established. However, it remains enigmatic how the temporal pattern of [Ca2+]i increase reflecting a longer period of neuronal activity affects synaptic plasticity. Here, we have addressed this issue by focusing on LTP at inhibitory synapses on a cerebellar Purkinje cell (PC).
At GABAergic synapses on a PC, potent excitatory inputs such as those from a climbing fiber heterosynaptically induce LTP of GABAA receptor (GABAAR)-mediated fast inhibitory synaptic transmission, called rebound potentiation (RP)19. RP is induced by the [Ca2+]i increase and the resultant activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII)19,20,21. The RP induction is controlled by the balance of activity between protein kinases such as CaMKII and protein kinase A (PKA) and protein phosphatases (PPs) such as PP-1 and calcineurin (also called PP-2B)20,21,22,23,24, as hippocampal LTP/LTD (See Fig. 1a). We previously developed a kinetic simulation model of the signaling cascades regulating RP24 and demonstrated that PDE1 (a Ca2+/calmodulin-dependent phosphodiesterase) raises the Ca2+ threshold, so that RP is not induced by tiny [Ca2+]i increases. However, how the temporal context of [Ca2+]i increase affects the RP induction is unclear. Here we attempted to elucidate the relation between the temporal pattern of [Ca2+]i increase and the establishment of RP. Combined application of systems biological model simulation and electrophysiological experiments demonstrated that the temporal order of different patterns of [Ca2+]i increase for several seconds to a few minutes determines whether RP is induced or not. Further, the context-dependent RP regulation was shown to be mediated by the interplay of two negative signaling components, the feedforward inhibition of CaMKII through PDE1 and a negative feedback mechanism through CaMKII Thr305/306 autophosphorylation.

Temporal integration of Ca 2+ signal in RP induction.
(a) Signaling cascades regulating RP induction. There are two feedforward inhibitory pathways to CaMKII through calcineurin and PDE1, two positive feedback pathways consisting of CaMKII autophosphorylation at Thr286 (PF) and CaMKII-mediated inhibition of PDE1 activation and one negative feedback signaling through CaMKII autophosphorylation at Thr305/306 (NF). (b, c) Representative images (b) and time courses (c) of fluorescence ratio (F340/F380) of fura-2 before and after the conditioning depolarization. The strong conditioning consisted of 5 depolarization pulses to 0 mV for 500 msec, while the weak conditioning was composed of 5 [weak (10s)] or 40 [weak (80s)] pulses to 0 mV for 40 msec. n = 9 for each. (d, e) Representative current traces (d) and time courses of the amplitudes (e) of GABA responses before and after the conditioning depolarizations. n = 5 for each. (f, g) Simulated time courses of the amount of active CaMKII (f) and phospho-GABAARs (g) before and after the 3 different patterns of [Ca2+]i increase. Strong (2 μM for 10 sec) or weak (0.35 μM for 10 or 80 sec) [Ca2+]i increase, which imitated that caused by each conditioning depolarization in a PC shown in (c), was applied.
Results
Temporal integration of Ca2+ signal in the RP induction
We first examined whether prolonged duration of [Ca2+]i increase lowers the intensity of [Ca2+]i increase required for the RP establishment by whole-cell patch clamp recordings from a cultured PC. Different durations (500 or 40 msec) and/or numbers (5 or 40 times) of depolarization pulses to 0 mV at 0.5 Hz were applied to cause different patterns of [Ca2+]i increase. As shown in Fig. 1b and c, [Ca2+]i imaging using fura-2 (50 μM) confirmed that different intensities and durations of [Ca2+]i increase were caused by different conditionings (fluorescence ratio F340/F380 at the end of 5 pulses for 500 msec, 2.77 ± 0.19; 5 pulses for 40 msec, 0.98 ± 0.07; 40 pulses for 40 msec, 1.24 ± 0.05). Using these patterns of stimulation, RP was examined by recording current responses to GABA iontophoretically applied to the proximal dendrite. In accordance with previous studies19,20,21,22,23,24, strong conditioning depolarization (5 pulses for 500 msec) potentiated the amplitude of the GABA response for longer than 30 minutes (172 ± 16 % at 30 minutes), whereas weak conditioning depolarization (5 pulses for 40 msec) did not (109 ± 4 %) (Fig. 1d and e). Thus, RP was induced only by the strong [Ca2+]i increase for about 10 seconds. On the other hand, when the duration of [Ca2+]i increase was prolonged to about 80 seconds by increasing the number of depolarization pulses, the weak conditioning (40 pulses for 40 msec) effectively induced RP (171 ± 10 %). The extent of potentiation at 30 minutes was comparable to that induced by the strong conditioning, although the initial phase of potentiation soon after the conditioning was small. These results indicate that both the intensity and the duration of [Ca2+]i increase cooperatively induce RP, suggesting a temporal integration mechanism of the Ca2+ signal in RP induction, as LTD at excitatory synapses on a PC8.
This temporal integrative property of RP induction by [Ca2+]i increase was supported by the simulation of systems biological model we previously developed24. Short strong [Ca2+]i increase (2 μM from the basal 0.1 μM for 10 seconds) applied to the model persistently activated CaMKII until the end of simulation (0.019 μM in the basal condition to 4.88 μM at 30 minutes) (Fig. 1f). This sustained activation of CaMKII was shown to rely on two positive feedback loops in the signaling cascades, namely CaMKII autophosphorylation at Thr286 (Thr287 in βCaMKII) and the negative regulation of PDE1 by CaMKII-mediated phosphorylation24,25,26. CaMKII-mediated phosphorylation of GABAARs also persistently increased, reflecting the establishment of RP in the model (0.03 μM in the basal condition to 0.88 μM at 30 minutes) (Fig. 1g). Phosphorylation of GABAARs by CaMKII was reported to potentiate the responsiveness in both neurons and a heterologous expression system27,28. Thus, in our simulation model, the CaMKII activity was used as readout of the RP establishment. When a weak [Ca2+]i increase to 0.35 μM was applied for 10 seconds, CaMKII was not effectively activated (0.019 μM at 30 minutes) (Fig. 1f). On the other hand, a weak [Ca2+]i increase with prolonged duration of 80 seconds brought about sustained CaMKII activation (1.15 μM) (Fig. 1f). Thus, the intensity of [Ca2+]i increase required for CaMKII activation was reduced when the duration of [Ca2+]i increase became longer.
Using the model, we systematically analyzed the relation between the duration and/or intensity of [Ca2+]i increase and the activity of CaMKII. The threshold levels of amplitude or temporal integration of [Ca2+]i increase required for the sustained CaMKII activation exhibited large variations with various durations of [Ca2+]i increase (Fig. 2a, b). On the other hand, the signal obtained by leaky temporal integration of [Ca2+]i (defined by a time constant τ of 1.7 sec) showed the minimum variation of threshold values for CaMKII activation (Fig. 2c). Thus, CaMKII activity level seemed to be determined through a leaky integrative mechanism of [Ca2+]i signaling, as LTD at excitatory synapses on a PC8. Taken together, our results indicated that the CaMKII activation and hence RP are triggered by the cooperative effects of the intensity and duration of [Ca2+]i increase.

CaMKII activation through temporal integration of [Ca2+]i signal with leak.
(a, b, c) Simulated CaMKII activities (at 30 minutes) caused by various intensities and durations of [Ca2+]i increase are plotted as a function of the intensity of [Ca2+]i increase (a) the temporally integrated [Ca2+]i increase (b) or the temporally integrated [Ca2+]i increase with leak defined by the time constant (τ) of 1.7 sec (c). (d) Stationary CaMKII activity plotted as a function of [Ca2+]i in the model obtained by nullcline analysis.
Temporal coupling of two RP-inducing [Ca2+]i increases cancels the RP establishment
Systematic analysis of the model unveiled an unique relation between the CaMKII activity and the [Ca2+]i increase (see Fig. 2a–c). The [Ca2+]i increase for 80 seconds with an amplitude of 0.4∼0.8 μM resulted in the impairment of CaMKII activation, in spite that either that with shorter duration (10∼40 seconds) or that with smaller amplitude (∼0.35 μM) effectively activated CaMKII. Thus, there might be a Ca2+-dependent negative regulation of CaMKII which becomes evident if the duration of [Ca2+]i increase is long and the intensity is medium. This idea was supported by the nullcline analysis showing the relation between stationary CaMKII activities and [Ca2+]i in the model (Fig. 2d). Curiously, the RP-regulating signaling cascades exhibited two uncontinuous sets of CaMKII activity. Bistability, in which two (high and low) stable CaMKII activity levels exist against a certain [Ca2+]i, was observed in limited ranges of [Ca2+]i (0.09∼0.2 μM and around 0.3 μM), probably due to Ca2+-dependent negative regulators of CaMKII, such as calcineurin and PDE1.
Based on the above theoretical analysis, we hypothesized that the RP induction triggered by the strong [Ca2+]i increase might be affected by the following [Ca2+]i level. To test this idea, two RP-inducing [Ca2+]i increases were coupled in the simulation model (Fig. 3a). Surprisingly, when a strong [Ca2+]i increase (2 μM for 10 seconds) was followed by a weak [Ca2+]i increase (0.35 μM for 80 seconds), the CaMKII activation was not augmented, but rather suppressed (0.019 μM) (Fig. 3b). This result suggests that temporal coupling of two CaMKII-activating [Ca2+]i increases converts their positive effect to negative, impairing the sustained CaMKII activation. Interestingly, reversing the temporal order of the strong and the weak [Ca2+]i increase resulted in effective CaMKII activation (1.35 μM) (Fig. 3a, b). Thus, model simulation demonstrated that the temporal sequence of [Ca2+]i increase is critical for CaMKII activation, predicting the possibility of RP regulation by the context of [Ca2+]i increase.

Temporal context-dependent suppression of RP induction.
(a) The conditioning [Ca2+]i increases used in the simulation. Strong (2 μM for 10 sec)-weak (0.35 μM for 80 sec) or weak-strong [Ca2+]i increase was applied. (b) Simulated time courses of active CaMKII amount before and after the two patterns of coupled [Ca2+]i increase. (c, d) Representative images (c) and time courses (d) of fura-2 fluorescence ratio (F340/F380) before and after the coupled conditioning depolarizations. n = 9 for each. (e, f) Representative current traces (e) and time courses of amplitudes (f) of GABA responses before and after the coupled conditioning depolarizations. n = 5 for each.
Next, we tested this model prediction by whole-cell recording experiments on cultured PCs. When the weak conditioning depolarization (40 pulses for 40 msec) was applied immediately after the strong conditioning depolarization (5 pulses for 500 msec), the RP induction was impaired (98 ± 4 %, p = 0.009 compared with the strong alone, Fig 3c–f). Thus, as suggested by the simulation, RP was cancelled by the strong-weak sequence of [Ca2+]i increase. In contrast, the weak conditioning depolarization preceding to the strong one did not suppress RP (171 ± 7 %, p = 0.97, Fig. 3c–f). Thus, the temporal sequence of [Ca2+]i increase determines the RP establishment.
Context-dependent RP suppression is caused by [Ca2+]i increase with medium intensity and long duration
We next confirmed that [Ca2+]i is responsible for the context-dependent RP cancellation by increasing a Ca2+ chelator (EGTA) from 1.4 to 5 mM in the whole-cell pipette. The RP induction by the strong conditioning depolarization was not affected by 5 mM EGTA (156 ± 14 %) (Fig. 4a), probably due to the slow kinetics of EGTA-mediated Ca2+ chelation. In contrast, the RP suppression by the strong-weak sequence of depolarization was impaired by 5 mM EGTA (161 ± 18 %, p = 0.82 compared with strong alone), suggesting that the RP impairment by the strong-weak conditioning highly depended on [Ca2+]i. To further confirm this idea, the level of weak [Ca2+]i increase was altered by changing the duration of each depolarization pulse. Compared with the [Ca2+]i increase brought about by the weak conditioning (40 pulses for 40 msec, F340/F380: 1.26 ± 0.06 at the end of 40 pulses), depolarization pulses for 100 or 20 msec caused a larger (1.54 ± 0.09) or smaller (1.00 ± 0.08) [Ca2+]i increase, respectively (Fig. 4b, c). Neither larger nor smaller conditioning depolarization canceled the RP induction by the preceding strong depolarization (100 msec, 174 ± 24 %; 20 msec, 163 ± 11 %) (Fig. 4d). Thus, a medium level of [Ca2+]i increase after a strong one cancelled the RP induction. We also tested whether our model supports the experimental results. In contrast to the marked suppression of CaMKII activation by a [Ca2+]i increase of 0.35 μM for 80 sec, neither a [Ca2+]i increase of 0.9 μM nor that of 0.25 μM impaired the sustained CaMKII activation triggered by the strong [Ca2+]i increase (0.9 μM [Ca2+]i, 1.45 μM of active CaMKII; 0.25 μM [Ca2+]i, 1.05 μM) (Fig. 4e). The level of [Ca2+]i required to effectively down-regulate CaMKII (to < 1 μM at 30 minutes) was 0.26 – 0.86 μM (Fig. 4f), supporting the idea that a medium level of [Ca2+]i increase cancels the RP establishment triggered by the preceding strong depolarization.

Medium intensity of [Ca2+]i underlies the context-dependent RP suppression.
(a) Time courses of amplitudes of GABA responses before and after the strong-weak sequence or the strong alone conditioning depolarization in the presence of 5 mM intracellular EGTA. (b, c) Representative images (b) and time courses (c, n = 9 for each) of fura-2 fluorescence ratio (F340/F380). (d) Time courses of GABA response amplitudes (n = 5 for each) before and after the strong-weak sequence of conditioning depolarization consisting of 3 different durations (20, 40, or 100 msec) of weak depolarization pulses. (e) Simulated time courses of active CaMKII amount in response to the strong-weak sequence of [Ca2+]i increases with 3 different amplitudes (0.25, 0.35, or 0.9 μM) of weak [Ca2+]i increase, as shown on the left. Note that the sustained CaMKII activity was suppressed only by medium [Ca2+]i (0.35 μM). (f) Relation between the amount of active CaMKII at 30 minutes and the intensity of weak [Ca2+]i increase.
We next examined the duration of weak [Ca2+]i increase effective for the RP cancellation by altering the number of weak depolarization pulses. As shown in Fig. 5a and b, the RP cancellation by the weak conditioning depolarization (for 40 msec) required a relatively large number (> 20) of depolarization pulses (5 times, 170 ± 14 %; 10 times, 154 ± 11 %; 20 times, 123 ± 10 %; 40 times, 98 ± 4 %; 80 times, 97 ± 6 %). In accordance with these results, simulation showed that long duration was needed for the weak [Ca2+]i increase (0.35 μM) to cancel the CaMKII activation (Fig. 5c, d). Comparison of CaMKII activities caused by the strong-weak and weak-strong stimuli suggested that the context-dependent suppressive effect of the weak stimulation was limited to [Ca2+]i increase with long duration and low amplitude (Fig. 5e and Supplementary Fig. S1). Taking all these results together, we conclude that RP establishment requires the [Ca2+]i to return to the basal level relatively rapidly after the strong increase; otherwise, the once-triggered RP will be cancelled.

Long duration of [Ca2+]i underlies the context-dependent RP suppression.
(a) Time courses of GABA response amplitudes before and after the strong-weak sequence of conditioning depolarization consisting of different numbers (5, 10, 20, 40, or 80) of weak depolarization pulses. n = 5 for each. (b) Relation between the extent of RP at 30 minutes and the duration of the conditioning weak depolarizations caused by different numbers of 40 msec depolarization pulse. (c) Simulated time courses of active CaMKII amount in response to the strong-weak sequence of [Ca2+]i increases consisting of different durations (10, 20, 40, 80 or 160 sec) of weak [Ca2+]i increase. (d) Relation between the amount of active CaMKII at 30 minutes and the duration of the conditioning weak [Ca2+]i increases following the strong one. (e) A heatmap showing the weak [Ca2+]i effective for the context-dependent regulation of CaMKII activation. Ratio of CaMKII activity (at 30 minutes) caused by the strong-weak and weak-strong [Ca2+]i increase ([weak-strong]/[strong-weak]) is plotted as a function of the amplitude and duration of weak [Ca2+]i increase. Strong [Ca2+]i increase (2 μM for 2 sec) was applied before or after the weak one.
PDE1 mediates the Ca2+ context-dependent impairment of RP induction
How does [Ca2+]i increase negatively regulate the RP induction in a context-dependent manner? The signaling cascades regulating RP have two Ca2+-activated inhibitory pathways to CaMKII via indirect activation of PP-1: one is through PDE1 and the other is through calcineurin (see Fig. 1a). Reduction of PKA activity caused by PDE1-mediated hydrolysis of cAMP leads to a decrease in phosphorylation of DARPP-32, while calcineurin directly dephosphorylates DARPP-32. Dephosphorylated DARPP-32 no longer inhibits PP-1 activity. The involvement of these feedforward inhibitory pathways was examined by altering the amount of each protein in the model. A decrease of the amount of PDE1 drastically diminished the suppressive effect of the strong-weak sequence of [Ca2+]i increase on the CaMKII activity, compared to a decrease of calcineurin (Fig. 6a–c). Thus, our simulation suggested that the context-dependent negative regulation of RP is predominantly mediated by PDE1.

Predominant role of PDE1 in the temporal context-dependent CaMKII inhibition and RP suppression.
(a, b) Simulated time courses of amount of active CaMKII in response to the strong-weak sequence of [Ca2+]i increase under the condition of decreased amount (as indicated by color) of PDE1 (a) or calcineurin (b) in the model. (c) Relation between the amount of active CaMKII at 30 minutes and the amount of PDE1 or calcineurin in the model. (d) Time courses of GABA response amplitudes before and after the strong-weak sequence of conditioning depolarization with or without a selective PDE1 inhibitor, 8-MM-IBMX (20 μM), and/or a specific calcineurin inhibitor, FK506 (400 nM). n = 5 for each. The time course of RP induced by the strong alone conditioning depolarization is also shown for comparison. (e, f) Representative immunofluorescent images (e) and normalized intensity (f) of active CaMKII phosphorylated at Thr286 (magenta) in PCs before (control) or 30 minutes after the conditioning treatments with high K+-containing external solution. Green color shows the calbindin signal used as a marker for PCs. Strong depolarization treatment with 50 mM K+ was applied for 10 seconds, without (strong) or with (strong-weak) subsequent weak depolarization treatment with 10 mM K+-containing solution for 5 minutes in the presence or absence of FK506 (5 μM) and/or 8-MM-IBMX (20 μM). n = 22 cells for each condition. ***: p < 0.001 by Dunnet T3 test.
The model suggestion was then tested experimentally. When PDE1 activity was decreased by a selective inhibitor (8-MM-IBMX, 20 μM), the suppression of RP induction by the strong-weak conditioning was abolished (153 ± 9 %, p = 0.002) (Fig. 6d). On the other hand, inhibition of calcineurin by FK506 (400 nM) did not affect the Ca2+-context-dependent RP impairment (107 ± 4 %, p = 0.18) (Fig. 6d). When both PDE1 and calcineurin were inhibited simultaneously, a marginal, but not significant additive effect was observed (180 ± 16 %, p = 0.19 compared with 8-MM-IBMX alone). Neither 8-MM-IBMX nor FK506 by itself affected depolarization-induced [Ca2+]i increase (Supplementary Fig. S2). Thus, PDE1 is the predominant mediator of the Ca2+-context-dependent RP impairment.
Negative regulation of CaMKII by weak depolarization through PDE1
We next studied whether CaMKII activity is indeed affected by the temporal pattern of [Ca2+]i increase. The CaMKII activity was monitored by immunocytochemistry for CaMKII autophosphorylated at the Thr286 residue. When cultured PCs were depolarized by treatment with 50 mM K+-containing external solution for 10 seconds, CaMKII was persistently activated for longer than 30 minutes (190 ± 14 %) (Fig. 6e, f). In parallel to the simulation results, the CaMKII activation by the 50 mM K+ treatment was cancelled by the following weaker depolarization treatment with 10 mM K+ containing solution for 5 minutes (102 ± 13 %) (Fig. 6e, f). Thus, the CaMKII activation was negatively regulated by the weak depolarization after the strong one. The weak depolarization-mediated reversal of CaMKII activation was abolished by a PDE1 inhibitor, 8-MM-IBMX (232 ± 18 %), but not by a calcineurin inhibitor, FK506 (128 ± 13 %) (Fig. 6e, f). Neither 8-MM-IBMX nor FK506 by itself affected the basal or the 50 mM K+-mediated augmented immunofluorescence for phospho-Thr286 CaMKII (Supplementary Fig. S3). To summarize, the CaMKII activity is regulated by the context of stimuli through the PDE1-mediated feedforward inhibitory pathway.
CaMKII autophosphorylation at Thr305/306 as molecular memory of [Ca2+]i increase history
How does a PC discern the [Ca2+]i context to dynamically control the RP induction? We first expected that PDE1 might be activated more effectively by the strong-weak sequence of [Ca2+]i increase than by the weak-strong one. However, simulation showed that the strong-weak [Ca2+]i increase caused slightly smaller PDE1 activation (Supplementary Fig. S4). Thus, the PDE1 pathway did not seem responsible for the discrimination of the temporal patterns of [Ca2+]i increase.
As an alternative candidate, we focused on the feedback inhibition of CaMKII through autophosphorylation at Thr305/306 (Thr306/307 in βCaMKII). Autophosphorylation at Thr305/306 slowly occurs after unbinding of Ca2+/CaM and then suppresses reactivation of the enzyme by inhibiting re-association with Ca2+/CaM29. Phosphorylation at Thr305/306 was shown essential for stimulation context-dependent suppression of the following induction of hippocampal LTP, called metaplasticity30,31. To examine whether the phosphorylation at Thr305/306 is required for the discrimination of the temporal order of [Ca2+]i increases, we first abolished the negative effect of the phospho-Thr305/306 on the CaMKII activity in the model. The affinity of phospho-Thr305/306 CaMKII for Ca2+/CaM was changed to equal to that of non-phosphorylated CaMKII. Here the amount of PP-1 was also slightly increased (∼ 8 %) to balance the dephosphorylation by PP-1 and the autophosphorylation by CaMKII. Elimination of the negative feedback regulation dampened the discrimination of strong-weak and weak-strong sequences of [Ca2+]i increase for the control of CaMKII activation (Fig. 7a). Thus, our model simulation suggested that the autophosphorylation at Thr305/306 works as a negative memory element for the RP induction by storing the history of [Ca2+]i increase.

CaMKII autophosphorylation at Thr305/306 detects the temporal context of [Ca2+]i increase.
(a) Simulated time courses of active CaMKII in response to a strong-weak or weak-strong sequence of [Ca2+]i increase in a modified model which lacks the negative feedback regulation mediated by phospho-Thr305/306. The duration of weak [Ca2+]i increase following the strong one was 80 or 160 sec. (b) Simulated time courses of active CaMKII in response to a strong or weak [Ca2+]i increase. Phospho-Thr305/306 CaMKII was arbitrarily increased by 50 % in both cases (pT305/306) at the onset of [Ca2+]i increase. (c) Relation between the amount of active CaMKII at 30 minutes after a strong or weak [Ca2+]i increase and the amount of arbitrarily increased phospho-Thr305/306. (d) Immunofluorescent images of α and β CaMKII in a PC with or without transfection of wild-type or T305/306A mutated CaMKII. (e, f) Time courses of GABA response amplitudes before and after the strong-weak sequence of conditioning depolarization recorded from a PC with or without transfection of wild-type (e) or T305/306A (f) CaMKII. The duration of weak depolarization was 80 or 160 sec resulting from altering the number of depolarization pulses (40 or 80 times). n = 5 for each.
We next examined whether the autophosphorylation at Thr305/306 by itself suppresses the CaMKII activation by [Ca2+]i increase. The amount of phospho-Thr305/306 CaMKII was arbitrarily increased at the onset of [Ca2+]i increase in the original model. Replacement of 50 % of inactive CaMKII by Thr305/306-phosphorylated CaMKII impaired the sustained activation of CaMKII in response to the weak [Ca2+]i increase but not to the strong one (Fig. 7b). As summarized in Fig. 7c, the CaMKII activation caused by the weak [Ca2+]i increase was more sensitive to the negative effect of Thr305/306 phosphorylation. Taken together, our model simulation predicted that the autophosphorylation at Thr305/306 mediates temporal discrimination of the strong and weak [Ca2+]i increase for the context-dependent regulation.
The model prediction was tested by electrophysiological experiments on PCs. We aimed to weaken the phospho-Thr305/306-mediated feedback inhibition by overexpression of mutant CaMKII in which Thr305/306 residues were replaced by alanine. As shown in Fig. 7d, transfection of plasmids encoding α and β subunits of CaMKII with or without T305/306A mutations increased both subunits to approximately similar extents. Overexpression of wild-type CaMKII did not affect the RP induction by the strong conditioning depolarization (158 ± 11 %, p = 0.55 compared with no transfection) (Fig. 7e). In addition, the strong-weak sequence of depolarization impaired the RP induction (105 ± 7 %, p = 0.007), while that with the reversed temporal order resulted in the RP establishment (158 ± 12 %, p = 0.98) (Fig. 7e). These results indicated that the [Ca2+]i context–dependent dynamic regulation of RP remained intact even when the total amount of CaMKII was increased.
In contrast, overexpression of T305/306A CaMKII abolished the RP cancellation by the weak depolarization (40 pulses for 40 msec) following the strong one (149 ± 12 %, p = 0.02 compared with wild-type), resulting in a similar extent of RP establishment to that caused by the weak-strong sequence of depolarization (165 ± 8 %, p = 0.69) (Fig. 7f). On the other hand, the weak conditioning depolarization with longer duration (80 pulses for 40 msec) attenuated the RP triggered by the strong depolarization irrespective of the temporal order (strong-weak, 108 ± 7 %; weak-strong, 115 ± 10 %) (Fig. 7f). Thus, mutant CaMKII, which is insensitive to the negative feedback regulation, deprived a PC of the ability to distinguish the temporal sequence of [Ca2+]i increase. Taken together, autophosphorylation at Thr305/306 works as a memory element of the history of [Ca2+]i increase for the dynamic control of RP.
Temporal profiles of negative signals for RP induction
As described above, the CaMKII autophosphorylation at Thr305/306 triggered by the strong [Ca2+]i increase converts the effect of a forthcoming [Ca2+]i increase on RP from positive to negative. This dynamic change of the impact of [Ca2+]i increase would make the PDE1-mediated feedforward inhibition of CaMKII more effective upon the second [Ca2+]i increase, resulting in cancelation of the RP establishment. If this is the case, as phosphorylation at Thr305/306 declines with time after the strong conditioning, the negative effect of a second [Ca2+]i increase would attenuate. In support of this idea, simulation showed that the suppressive effect of the strong-weak sequence of [Ca2+]i increase on CaMKII activity decreased as the timing of weak [Ca2+]i increase was delayed, exhibiting a temporal profile similar to the time course of the reduction of Thr305/306 autophosphorylation after the strong depolarization (Fig. 8a, c). Accordingly, electrophysiological recordings demonstrated that the context-dependent RP impairment was weakened as the interval between strong and weak depolarizations was increased (1 min, 103 ± 14 %; 2 min, 140 ± 19 %; 5 min, 184 ± 13 %) (Fig. 8b, d). Thus, we conclude that the temporal window for the RP cancellation by weak [Ca2+]i increase is limited to 1–2 minutes after the preceding [Ca2+]i increase, due to the attenuation of the negative feedback signal stored by the CaMKII autophosphorylation at Thr305/306.

Temporal profiles of the context-dependent negative regulation of RP.
(a, b) Time courses of simulated active CaMKII amount (a) and electrophysiologically recorded GABA response amplitudes (b), before and after the strong-weak conditioning with various intervals (0, 1, 2, or 5 minutes). For comparison, the CaMKII activity (a) and RP (b) brought about by the strong depolarization alone are also plotted. n = 5 for each. (c, d) Relation between the simulated CaMKII activity (c, green) or the extent of recorded RP (d) at 30 minutes and the interval between the strong and the weak conditionings. For comparison, the time course of simulated phospho-Thr305/306 CaMKII amount after the strong [Ca2+]i increase is overlaid upside down in (c, blue). (e, f) Time courses of simulated active CaMKII amount (e) and the recorded GABA response amplitudes (f) before and after the strong-weak conditioning followed by the additional strong depolarization applied at various timings (0, 5, 10, 20, or 60 seconds after the end of weak stimulation). For comparison, the time courses resulting from the strong depolarization alone or from the strong-weak sequence of depolarization are also plotted. n = 5 for each. (g, h) Relation between the simulated CaMKII activity (g) or the extent of recorded RP (h) at 30 minutes and the timing of the second strong depolarization are shown with green color. The simulated time course of PDE1 activity caused by the strong-weak sequence of [Ca2+]i increase is overlaid upside down in (g, blue).
We also considered that as the PDE1 activity in response to the strong-weak [Ca2+]i increase returns to the basal level, the attempt to induce RP again would become successful. To test this idea, a second strong [Ca2+]i increase was applied after the strong-weak sequence of [Ca2+]i increase with various intervals in the simulation model. Although CaMKII could not be activated by a second strong [Ca2+]i increase immediately after the strong-weak sequence (0.024 μM at 30 minutes), the suppressive effect disappeared within 10–20 seconds (5 sec, 0.032 μM; 10 sec, 0.157 μM; 20 sec, 1.59 μM; 60 sec, 4.67 μM) (Fig. 8e, g). This relatively rapid disappearance of suppressive effect nicely fit to the time course of attenuation of PDE1 activity after the strong-weak [Ca2+]i increase (Fig. 8g). Consistently, whole-cell recordings also showed a similar time course of recovery of RP induction by the second strong depolarization (0 sec, 107 ± 6 %; 5 sec, 101 ± 8 %; 10 sec, 114 ± 13 %; 20 sec, 144 ± 16 %; 60 sec, 161 ± 13 %) (Fig. 8f, h). Thus, the lifetime of the context-dependent negative effect on RP seems to be determined by the time course of PDE1 activation. Taking all these results together, the temporal profiles of the [Ca2+]i context-dependent RP regulation is defined by two signaling elements, the CaMKII autophosphorylation at Thr305/306 and the PDE1-mediated feed-forward inhibition of CaMKII.
Discussion
Taking advantage of the quantitative and predictive nature of systems biological model simulation8,18,24,32,33,34,35,36,37 coupled with validation by electrophysiological experiments, the present study revealed the [Ca2+]i context-dependent dynamic regulation of RP, a form of LTP at inhibitory synapses on a cerebellar PC. In spite of the temporal integrative RP induction by [Ca2+]i, coupling of two patterns of RP-inducing [Ca2+]i increase suppressed RP depending on the temporal order. Weak [Ca2+]i increase with medium intensity and long duration negatively regulated the ongoing RP induction process triggered by a preceding strong [Ca2+]i increase. To the best of our knowledge, the temporal context-dependent conversion of plasticity signals from positive to negative is a novel regulatory mechanism of synaptic plasticity.
Our model focused on the CaMKII activity and the resultant GABAAR phosphorylation level as readout of RP and provided several predictions, which were verified with electrophysiological experiments. However, the precise mechanism how CaMKII establishes RP remains unknown. Increase of the surface GABAAR number or modulation of the channel properties might be caused by CaMKII-mediated GABAAR phosphorylation, such as β3 (S383), β2 (S410) and γ2 subunits (S348/T350)27,28. In addition to the direct phosphorylation of GABAARs, CaMKII contributes to the RP establishment through modulation of GABARAP, an intracellular GABAAR-associated protein23.
The temporal pattern of [Ca2+]i increase is processed in the signaling cascades relatively slowly within seconds to a few minutes. More rapid context-dependent synaptic modification would be accomplished by STDP, in which LTP or LTD is induced by different intensities of [Ca2+]i increase based on the temporal order of pre- and postsynaptic neuronal firing within tens of milliseconds16,17,18. On the other hand, longer context (minutes to hours) might be reflected by metaplasticity, in which neuronal activity alters the probability of future induction of synaptic plasticity by modulating intracellular signaling cascades30. Taken together, wide ranges of context in neuronal activity might be imprinted in neuronal networks.
Synaptic plasticity once induced can be modulated by the subsequent neuronal activity. For instance, LTP at hippocampal excitatory synapses is cancelled by the LTD-inducing weak stimuli and vice versa, called depotentiation and dedepression, respectively38,39. Because the direction of synaptic modification caused by a particular stimulation pattern does not change in these cases, they are essentially different from the context-dependent RP regulation shown here. On the other hand, at the retinotectal excitatory synapses in a Xenopus larvae tadpole, LTP induced by a pattern of visual stimulus attenuates in response to subsequent neuronal activity which is not correlated with that initially triggered LTP40. Thus, the LTP consolidation seems to require neuronal inactivity for a while after the induction, which is reminiscent of the requirement of rapid recovery of [Ca2+]i after the strong increase for the RP consolidation.
The context-dependent RP regulation involved the feedforward inhibition of CaMKII through PDE1, but not through calcineurin. This difference might be ascribed to the much higher affinity of PDE1 for Ca2+/CaM (Kd: ∼ 0.1 nM) compared with that of calcineurin (Kd: ∼ 6 nM)41. Considering that PDE1 determines the [Ca2+]i threshold required for the RP induction24, it becomes effective at low [Ca2+]i and would control the RP establishment by monitoring both the intensity and duration of [Ca2+]i signals. In contrast, calcineurin might be effectively activated upon relatively stronger [Ca2+]i increase and then negatively regulate the RP induction in cooperation with the additional inputs including GABABRs21. On the other hand, decoding of the temporal context of [Ca2+]i increase was mediated by the negative feedback autophosphorylation of CaMKII at Thr305/306. Mutant mice lacking the autophosphorylation at Thr305/306 exhibit altered synaptic plasticity, impaired metaplasticity and deficits in context-dependent learning31,42,43. Notably, model mice of Angelman syndrome, a severe neurological disorder, somewhat shows increased phosphorylation at Thr305/30644 and the deficits in hippocampal synaptic plasticity and learning, epilepsy and motor dysfunction were shown to be rescued by genetic reduction of the Thr305/306 autophosphorylation45.
RP is heterosynaptically induced by repetitive excitatory inputs from a climbing fiber, which transmits an error signal detected by a sensory organ during motor control19. As each climbing fiber input causes large [Ca2+]i increase (∼μM) for tens of milliseconds, the frequency and number of climbing fiber inputs for a few minutes would be integrated and processed by the signaling mechanism for [Ca2+]i context-dependent RP regulation. Our simulation suggests that trains of brief [Ca2+]i increase (80 pulses to 2 μM for 20 msec at 1 Hz), imitating the basal firing of a climbing fiber in vivo, cancels the RP induction triggered by the preceding high frequency [Ca2+]i increases (see Supplementary Fig. S5). Thus, RP might be consolidated only when the error signals encoded by climbing fiber activity attenuates for a while after the intensive activity. RP at inhibitory synapses and LTD at excitatory synapses on a PC, a cellular basis for motor learning, correlate in many aspects, including the temporal integrative induction by [Ca2+]i and the involvement of several common molecules such as CaMKII, mGluR1 and GABABRs5,7,8,20,21,22,46,47,48,49,50. Interestingly, the leaky integrative induction of LTD (τ: 0.6∼1.5 sec)8 is quite similar to that of RP (τ: ∼ 1.7 sec). Thus, the concerted regulation of RP and LTD by the [Ca2+]i context might bring about temporally tuned motor control.
Methods
Culture
The method for preparing primary cultures of cerebellar neurons was similar to that in a previous study50. Whole-cell patch clamp recordings and immunocytochemistry were performed 2–3 weeks after preparation of the culture. Experimental procedures were performed in accordance with the guidelines regarding care and use of animals for experimental procedures of the National Institutes of Health, U.S.A. and Kyoto University and approved by the local committee for handling experimental animals in the Graduate School of Science, Kyoto University.
Electrophysiology
Methods used for electrophysiological experiments were similar to those in previous studies21,22,23,24. Briefly, whole-cell patch clamp recording from a cultured PC was performed with an amplifier (EPC9 or EPC10, HEKA, Germany) in a solution containing (in mM) 145 NaCl, 5 KOH, 2 CaCl2, 1 MgCl2, 10 Hepes and 10 glucose (pH 7.3) at room temperature (20∼24 °C). The solution contained 6-cyano-7-nitroquinoxaline-2, 3-dione disodium (CNQX, 10 μM, Tocris Cookson, UK), tetrodotoxin (TTX, 1 μM, Wako, Japan) and SCH50911 (10 μM, Tocris Cookson) to inhibit glutamatergic EPSCs, action potentials and GABABR activation, respectively. A patch pipette used to record from a PC was filled with an internal solution (pH 7.3, adjusted by CsOH) containing (in mM) 121 CsCl, 33 KCl, 1.4 ethylene glycol bis (β-aminoethylether) N,N,N',N'-tetraacetic acid (EGTA), 10 Hepes, 2 Mg-ATP and 0.2 Na-GTP. The membrane potential of a PC was held at −70 mV. Only recordings with input resistance of more than 100 MΩ and series resistance of less than 25 MΩ were accepted. To minimize the voltage clamp error, the GABA response amplitude at the beginning of experiments was set at around 200 pA. Series resistance and input resistance were monitored every 2 min and experiments were terminated when a change of more than 20 % was detected. The method for iontophoretic application of GABA was similar to that in previous studies21,22,23,24. A glass pipette containing 10 mM GABA was aimed at a proximal dendrite and 20-msec positive voltage pulses were applied every 20 seconds. 8-MM-IBMX (20 μM, Calbiochem, USA), was applied to the bath 5–10 minutes before recording. FK506 (400 nM, Calbiochem) was applied intracellularly through a patch pipette.
Immunocytochemistry
Cultured neurons were fixed with 4 % paraformaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.5 % Tween 20, then blocked with 2 % skim milk and finally labeled with primary and secondary antibodies. The following antibodies were used: a mouse monoclonal antibody (mAb) against calbindin D28 (1∶500, Swant Bellinzona, Switzerland), a mAb against αCaMKII (1∶500, Chemicon, USA), a mAb against βCaMKII (1∶500, Zymed Laboratories, Inc., USA), a rabbit polyclonal antibody (pAb) against active CaMKII (1∶500, Promega, USA) and Alexa 488- or 568-conjugated pAb against rabbit or mouse IgG (1∶400, Molecular Probes, USA). Fluorescent images were obtained with a confocal laser microscope (FV1000 imaging system, Olympus, Japan) and analyzed using ImageJ software (NIH, USA). The high K+-containing solution for conditioning treatment was prepared by replacing 50 or 10 mM Na+ with K+ in the normal external solution. Cultured cerebellar neurons were treated with the 50 mM K+- containing solution for 10 seconds in the presence of SCH50911 (10 μM), CNQX (10 μM), TTX (1 μM), with or without subsequent treatment with 10 mM K+-containing solution for 5 minutes. Then, neurons were washed with the normal external solution until fixation at 30 minutes after the onset of conditioning treatment. Cell-permeable inhibitor FK506 (5 μM) and/or 8-MM-IBMX (20 μM) was added to the external solution from > 5 minutes before the conditioning treatment. The averaged fluorescent signals for active CaMKII in the area positive for calbindin, a molecular marker of PCs, were compared.
Ca2+ imaging
[Ca2+]i was measured with a Ca2+ imaging system (Aquacosmos, Hamamatsu Photonics, Japan) mounted on an upright microscope (BX50WI, Olympus) using fura-2 (50 μM, Invitrogen, USA). Fura-2 was loaded into a PC through a patch pipette and excited alternately at 340 nm and 380 nm for 120 msec. Each fluorescence image was recorded at 2 Hz and the fluorescence ratio (the fluorescence excited at 340 nm divided by that at 380 nm) was calculated.
DNA construction and transfection
cDNAs of mouse CaMKII α and β subunits were cloned by PCR of cDNA template obtained by reverse transcription of mRNAs prepared from mouse cerebellar culture. The mutant CaMKII T305/306A was produced by PCR-mediated introduction of mutations. cDNA of wild-type or mutant CaMKII was inserted into pCAGplay expression vector50 at the EcoRI/XhoI site. The expression plasmid (50 ng/μl total) encoding wild-type or mutant CaMKII α and β subunits together with that encoding EGFP was injected into the nucleus of a PC through a sharp glass pipette. The electrophysiological or immunocytochemical experiments were performed 1–2 days after the injection.
Simulation
A computational model of signaling cascades regulating RP we previously developed23 was used with some modifications as explained briefly below (for details, see Supplementary Note). First, according to the recent prevailing view about the mechanisms of CaMKII autophosphorylation at Thr286 and at Thr305/30651, the autophosphorylation was assumed to take place in an intra-holoenzyme manner alone, although we previously assumed both intra- and inter-holoenzyme reactions. Second, the basal activity of “PP2As”, which was separately assumed to balance the basal phosphorylation level of CaMKII and that of DARPP-32 in the previous model, was integrated into one PP2A. Third, although the mechanism of dephosphorylation of PDE1 still remains largely unknown, calcineurin-mediated dephosphorylation reported previously was explicitly included in the present version52. Fourth, some parameters defining molecular concentrations and molecular reactions were changed as summarized in Supplementary Note so that the temporal profile of context-dependent negative regulation of CaMKII in the model simulation better matches that obtained by the experiments shown in Fig. 5. It should be noted that the previous model also produced qualitatively similar results demonstrating the context-dependent regulation of CaMKII (data not shown).
In the model, biochemical reactions in the signaling cascades were represented either as binding-dissociation reactions or as enzymatic reactions. For example, a binding reaction in which A and B bind to form complex AB, is expressed as the following equation:

where kf and kb are the rate constants for the forward and backward processes. These rate constants are determined by the dissociation constant Kd and time constant τ. Kd is defined as kb / kf and τ reflects velocity of the reaction toward equilibrium. The reaction is represented as a differential equation:

The enzymatic reaction was expressed with the Michaelis-Menten formulation:

where S, E and P are substrate, enzyme and product, respectively. The Michaelis constant Km is defined as Km = (k−1 + kcat )/k1 . The maximum enzyme velocity Vmax is expressed as Vmax = kcat ·[E]total, where [E]total is the total concentration.
We performed the simulation using CellDesigner 4.153. Ordinary differential equations (ODEs) were numerically solved by the SOSlib (SBML ODE Solver Library). We started simulation experiments after the model reached equilibrium in the basal condition.
Statistics
Data are presented as mean ± s.e.m. Statistical significance was assessed by unpaired two-tailed Students' t-test or by one-way ANOVA followed by the post-hoc Dunnet T3 test.
References
Tulving, E. & Markowitsch, H. J. Episodic and declarative memory: role of the hippocampus. Hippocampus 8,198–204 (1998).
Bailey, C. H., Giustetto, M., Huang, Y. Y., Hawkins, R. D. & Kandel, E. R. Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat Rev Neurosci 1, 11–20 (2000).
Kandel, E. R. The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038 (2001).
Tang, Y. P. et al. Genetic enhancement of learning and memory in mice. Nature 401, 63–69 (1999).
Ito, M. Cerebellar long-term depression: characterization, signal transduction and functional roles. Physiol Rev 81, 1143–1195 (2001).
Gaiarsa, J. L., Caillard, O. & Ben-Ari, Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci 25, 564–570 (2002).
Hansel, C., Linden, D. J. & D'Angelo, E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci 4, 467–475 (2001).
Tanaka, K. et al. Ca2+ requirements for cerebellar long-term synaptic depression: role for a postsynaptic leaky integrator. Neuron 54, 787–800 (2007).
Tanaka, K. & Augustine, G. J. A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron 59, 608–620 (2008).
Yang, S. N., Tang, Y. G. & Zucker, R. S. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol 81, 781–787 (1999).
Neveu, D. & Zucker, R. S. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron 16, 619–629 (1996).
Mizuno, T., Kanazawa, I. & Sakurai, M. Differential induction of LTP and LTD is not determined solely by instantaneous calcium concentration: an essential involvement of a temporal factor. Eur J Neurosci 14, 701–708 (2001).
Taniike, N., Lu, Y. F., Tomizawa, K. & Matsui, H. Critical differences in magnitude and duration of N-methyl D-aspartate(NMDA) receptor activation between long-term potentiation (LTP) and long-term depression (LTD) induction. Acta Med Okayama 62, 21–28 (2008).
Coesmans, M., Weber, J. T., De Zeeuw, C. I. & Hansel, C. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron 44, 691–700 (2004).
Wang, S. S., Denk, W. & Häusser, M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3, 1266–1273 (2000).
Bi, G. Q. & Poo, M. M. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength and postsynaptic cell type. J Neurosci 18, 10464–10472 (1998).
Dan, Y. & Poo, M. M. Spike timing-dependent plasticity of neural circuits. Neuron 44, 23–30 (2004).
Urakubo, H., Honda, M., Froemke, R. C. & Kuroda, S. Requirement of an allosteric kinetics of NMDA receptors for spike timing-dependent plasticity. J Neurosci 28, 3310–3323 (2008).
Kano, M., Rexhausen, U., Dreessen, J. & Konnerth, A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature 356, 601–604 (1992).
Kano, M., Kano, M., Fukunaga, K. & Konnerth, A. Ca2+-induced rebound potentiation of γ-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proc Natl Acad Sci USA 93, 13351–13356 (1996).
Kawaguchi, S. & Hirano, T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. J Neurosci 22, 3969–3976 (2002).
Kawaguchi, S. & Hirano, T. Suppression of inhibitory synaptic potentiation by presynaptic activity through postsynaptic GABAB receptors in a Purkinje neuron. Neuron 27, 339–347 (2000).
Kawaguchi, S. & Hirano, T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci 27, 6788–6799 (2007).
Kitagawa, Y., Hirano, T. & Kawaguchi, S. Prediction and validation of a mechanism to control the threshold for inhibitory synaptic plasticity. Mol. Syst. Biol., 5, 280 (2009).
Miller, S. G. & Kennedy, M. B. Regulation of brain type II Ca2+/calmodulin- dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell 44, 861–870 (1986).
Hashimoto, Y., Sharma, R. K. & Soderling, T. R. Regulation of Ca2+/calmodulin- dependent cyclic nucleotide phosphodiesterase by the autophosphorylated form of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 264, 10884–10887 (1989).
Houston, C. M., Hosie, A. M. & Smart, T. G. Distinct regulation of β2 and β3 subunit-containing cerebellar synaptic GABAA receptors by calcium/calmodulin- dependent protein kinase II. J Neurosci 28, 7574–7584 (2008).
Houston, C. M. & Smart, T. G. CaMK-II modulation of GABAA receptors expressed in HEK293, NG108-15 and rat cerebellar granule neurons. Eur J Neurosci 24, 2504–2514 (2006).
Patton, B. L., Miller, S. G. & Kennedy, M. B. Activation of type II calcium/calmodulin-dependent protein kinase by Ca2+/calmodulin is inhibited by autophosphorylation of threonine within the calmodulin-binding domain. J Biol Chem 265, 11204–11212 (1990).
Abraham, W. C. & Bear, M. F. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19, 126–130 (1996).
Zhang, L. et al. Hippocampal synaptic metaplasticity requires inhibitory autophosphorylation of Ca2+/calmodulin-dependent kinase II. J Neurosci 25, 7697–7707 (2005).
Bhalla, U. S. & Iyengar, R. Emergent properties of networks of biological signaling pathways. Science 283, 381–387 (1999).
Kim, M., Huang, T., Abel, T. & Blackwell, K. T. Temporal sensitivity of protein kinase A activation in late-phase long term potentiation. .PLoS Comput Biol. 6, e1000691 (2010).
Doi, T., Kuroda, S., Michikawa, T. & Kawato, M. Inositol,1,4,5-trisphosphate- dependent Ca2+ threshold dynamics detect spike timing in cerebellar Purkinje cells. J Neurosci 25, 950–961 (2005).
Pettigrew, D. B., Smolen, P., Baxter, D. A. & Byrne, J. H. Dynamic properties of regulatory motifs associated with induction of three temporal domains of memory in aplysia. J Comput Neurosci 18, 163–181 (2005).
Kuroda, S., Schweighofer, N. & Kawato, M. Exploration of signal transduction pathways in cerebellar long-term depression by kinetic simulation. J Neurosci 21, 5693–5702 (2001).
Kotaleski, J. H. & Blackwell, K. T. Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci 11, 239–251 (2010).
Barrionuevo, G., Schottler, F. & Lynch, G. The effects of repetitive low frequency stimulation on control and “potentiated” synaptic responses in the hippocampus. Life Sci 27, 2385–2391 (1980).
Dudek, S. M. & Bear, M. F. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J Neurosci 13, 2910–2918 (1993).
Zhou, Q., Tao, H. W. & Poo, M. M. Reversal and stabilization of synaptic modifications in a developing visual system. Science 300, 1953–1957 (2003).
Meyer, T., Hanson, P. I., Stryer, L. & Schulman, H. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science 256, 1199–1202 (1992).
Elgersma, Y. et al. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity and learning. Neuron 36, 493–505 (2002).
Elgersma, Y., Sweatt, J. D. & Giese, K. P. Mouse genetic approaches to investigating calcium/calmodulin-dependent protein kinase II function in plasticity and cognition. J Neurosci 24, 8410–8415 (2004).
Weeber, E. J., Jiang, Y. H., Elgersma, Y., Varga, A. W., Carrasquillo, Y., Brown, S. E., Christian, J. M., Mirnikjoo, B., Silva, A., Beaudet, A. L., Sweatt, J. D. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci 23, 2634–2644 (2003).
van Woerden, G. M. et al. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10, 280–282 (2007).
Hansel, C. et al. αCaMKII is essential for cerebellar LTD and motor learning. Neuron 51, 835–843 (2006).
Kamikubo, Y. et al. Postsynaptic GABAB receptor signalling enhances LTD in mouse cerebellar Purkinje cells. J Physiol. 585, 549–563 (2007).
Tsuruno, S., Kawaguchi, S. & Hirano, T. Src-family protein tyrosine kinase negatively regulates cerebellar long-term depression. Neurosci Res 61, 329–332 (2008).
Sugiyama, Y., Kawaguchi, S. & Hirano, T. mGluR1-mediated facilitation of long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. Eur J Neurosci 27, 884–896 (2008).
Kawaguchi, S. & Hirano, T. Integrin α3β1 suppresses long-term potentiation at inhibitory synapses on the cerebellar Purkinje neuron. Mol Cell Neurosci 31, 416–426 (2006).
Bradshaw, J. M., Hudmon, A. & Schulman. . Chemical quenched flow kinetic studies indicate an intraholoenzyme autophosphorylation mechanism for Ca2+/calmodulin-dependent protein kinase II. H. .J Biol Chem 277, 20991–20998 (2002).
Sharma, R. K. & Wang, J. H. Calmodulin and Ca2+-dependent phosphorylation and dephosphorylation of 63-kDa subunit-containing bovine brain calmodulin-stimulated cyclic nucleotide phosphodiesterase isozyme. J Biol Chem. 261, 1322–1328 (1986).
Funahashi, A., Tanimura, N., Morohashi, M. & Kitano, H. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. BIOSILICO 1, 159–162 (2003).
Acknowledgements
We are grateful to Drs. Elizabeth Nakajima and Yoshiaki Tagawa for critical reading of the manuscript and helpful comments. This work was supported by grants from MEXT, Japan to S. K. and T. H. and from the Takeda Science Foundation to S. K. and by Global COE program A06 of MEXT, Japan, to Kyoto University.
Author information
Authors and Affiliations
Contributions
S. K. and T. H. designed the project and wrote the manuscript. S. K. conducted experiments and simulation. N. N. performed experiments examining the role of CaMKII autophosphorylation at Thr305/306.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Kawaguchi, Sy., Nagasaki, N. & Hirano, T. Dynamic impact of temporal context of Ca2+ signals on inhibitory synaptic plasticity. Sci Rep 1, 143 (2011). https://doi.org/10.1038/srep00143
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00143
This article is cited by
-
Regulation and Interaction of Multiple Types of Synaptic Plasticity in a Purkinje Neuron and Their Contribution to Motor Learning
The Cerebellum (2018)
-
Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP
Nature Communications (2014)
-
Regulation of Inhibitory Synaptic Plasticity in a Purkinje Neuron
The Cerebellum (2012)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
