
Key Points
-
Provides general dental practitioners with an overview of the background, epidemiology and aetiology of amelogenesis imperfecta (AI).
-
Delineates the classification systems associated with AI.
-
Outlines the different characteristics of AI clinical variability.
-
Provides an understanding of how to diagnose AI.
Abstract
Amelogenesis imperfecta (AI) is an inherited disorder that is associated with mutations in five genes (AMEL; ENAM; MMP20; KLK4 and FAM83H) with a wide range of clinical presentations (phenotypes). It affects the structure and appearance of enamel of all teeth, both in the primary and secondary dentition. In this review paper, we look at the epidemiology, classification, aetiology, clinical description and diagnosis of AI. In the following three papers of this series, we aim to describe the role of paediatric dentists, orthodontists and restorative dentists in the clinical management of patients with AI.
Similar content being viewed by others
Background
Amelogenesis imperfecta (AI) is a term used to describe a group of hereditary conditions that affect the structure and appearance of dental enamel, often in conjunction with changes in other intra-oral and/or extra-oral tissues. The structure and clinical appearance of dental enamel of nearly all teeth are affected in both primary and secondary dentition and is characterised by hypomineralisation and/or hypoplasia with discolouration, sensitivity and fragility. The term amelogenesis imperfecta is also used to describe enamel phenotypes in other syndromes.
Dental enamel is an epithelial-derived tissue comprised of highly organised hydroxyapatite crystals that form in a defined extracellular space. It is acellular and has no intrinsic repair potential. It is formed by ameloblasts during tooth developmental stages after dentinogenesis. The entire process of amelogenesis is under genetic control. Mutations in genes responsible for amelogenesis result in diverse phenotypes showing a wide spectrum of characteristics. The aim of this series of review papers is to understand the epidemiology, genetics, classification, clinical presentation, diagnosis and management options. The condition is associated with high morbidity for the patients, and therefore early detection and careful planning and management in a multidisciplinary environment is a key ingredient to a successful outcome.
Amelogenesis
Enamel is derived from epithelial (ectodermal) origin while dentine, cementum, pulp and periodontal ligament are derived from mesenchymal (mesodermal) origin. Enamel formation begins from cells that are derived from the internal enamel epithelium of the dental organ after initiation of dentinogenesis. Amelogenesis has been described in mainly three functional stages: presecretory, secretory and maturation stages. In the presecretory stage ameloblasts acquire their characteristics and develop an extensive protein synthetic apparatus to secrete the organic matrix of enamel. In the secretory phase the ameloblasts secrete the entire thickness of the enamel. At this stage the hydroxyapatite crystals are separated by organic and water contents. In the maturation stage the ameloblasts allows inorganic ions to be secreted and exchanged for the water and organic contents, resulting in an increase in the inorganic content and the length and width of the enamel prisms. Dental enamel is made of highly organised hydroxyapatie crystals occupying 95% of the maxtrix volume. Ameloblasts control the secretion, formation and maturation of the enamel matrix using matrix molecules such as enamelin, amelogenin, ameloblastin, tuftelin, amelotin, dentine sialophophoprotein and matrix enzymes such as kallikrein-4 and matrix metalloproteinaise-20.
The diversity of enamel malformations observed in AI is believed to reflect the differences in the timing during amelogenesis, when the disruptions occur. Flaws incorporated during formation of the dentino-enamel junction can result in an enamel layer that shears easily from the underlying dentine. Secretory stage defects result in insufficient crystal elongation and leave the enamel layer pathologically thin, or hypoplastic. Maturation stage defects, such as those that might occur if the enamel matrix is not properly degraded and reabsorbed, produce an enamel layer that is of normal thickness, but is pathologically soft.
Classification
Various classification systems have been described in the literature, dating back to as early as the 1940s. AI presents with various phenotypes and makes appearance invalid as a criterion for sorting. With advances over time and a better understanding of the genetics of AI, genotypic information can be used to distinguish between different forms. Molecular genetic tools will allow more precise diagnosis and classification in the future. A recent paper by Aldred et al. discusses previous systems used and proposes a new model for classifying AI based on four broad areas.5 These are described below:
-
Mode of inheritance: autosomal dominant, autosomal recessive, X-linked, isolated case
-
Molecular basis: chromosomal localisation/locus/mutation
-
Biochemical outcome: putative result of mutation when known
-
Phenotype: hypoplastic, hypocalcified, hypomaturation, hypomaturation-hypoplastic with taurodontism.
Aetiology
AI is caused by mutations or altered expression in five genes: AMEL (amelogenin), ENAM (enamelin), MMP20 (matrix metalloproteinaise-20), KLK4 (kallikrein-4) and FAM83H.6,7,8,9,10,11,12,13,14,15,16 Table 1 describes the phenotypes associated with the mutations or variable expression associated with these genes. Unlike other genes that cause AI, FAM83H does not encode an extracellular matrix protein. Its location inside the cell is completely unknown, as is its function.
Sporadic cases of AI may represent new mutations, illustrative of variable expressions of the genes. In such cases, careful assessment of other family members is important to exclude non-genetic causes.17
Clinical description
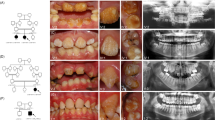
The phenotype (clinical appearance) of AI shows a spectrum of clinical variability (Fig. 1) and this depends on the pattern of inheritance, the mutation involved, expression of matrix proteins and biochemical changes associated with the mutations.

Some of these show composites/crowns placed by the patients' practitioners to mask sensitivity or for aesthetic reasons
Characterisitcs of hypoplastic AI
-
Enamel of reduced thickness due to a defect in the formation of normal matrix
-
Pitting and grooves
-
Hard and translucent enamel
-
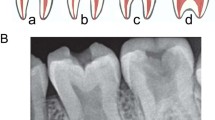
Radiographically, the enamel contrasts normally from dentine.
Characteristics of hypocalcified AI
-
Defect in enamel calcification
-
Enamel of normal thickness
-
Weak in structure
-
Appears opaque or chalky
-
Teeth become stained and rapidly wear down
-
Radiographically, enamel is less radio-opaque than dentine.
Characterisitcs of hypomaturation AI
-
Enamel of normal thickness but mottled in appearance
-
Slightly softer than normal and vulnerable to tooth wear, but not as severe as the hypocalcified type
-
Radiographically, similar radiodensity as dentine.
Characteristics of hypomaturation-hypoplasia with taurodontism
-
Mixed hypomaturation and hypoplasia appearance
-
Taurodontism: body and pulp chamber enlarged, and the floor of pulp chamber and furcation is moved apically down the root.
A review paper in the literature reported other clinical features of AI such as delay in dental eruption, microdontia, deviant crown and morphology, root resorption, short roots, enlarged pulp chamber, pulp stones, dens in dente, tooth agenesis, crowding of teeth, gingival enlargement, gingivitis and periodontitis.18 Additionally there are other reported skeletal abnormalities such as overbite, overjet and cross bites in this review paper.18
Diagnosis
It is important to understand the different phenotypes and associated inheritance patterns related to AI to help narrow down the search for a candidate gene in order to establish a definitive molecular aetiology.
Factors to consider during diagnosis include family history, pedigree plotting (a diagram of a family health history tree), clinical observations and radiographic assessment. Further laboratory-based genetic diagnosis can be done, but this is more useful as a research tool.
It is important to identify other causes of defects in enamel, both extrinsic and intrinsic, to exclude these during diagnosis of AI. These include past medical history (tertracycline staining), dental fluorosis, enamel hypoplasia, trauma, or molar-incisor hypomineralisation (MIH).
Discussion
The appearance of teeth and problems associated with the enamel (such as sensitivity, staining and roughness) can be of major psychological and functional concern to the patient. The aim of any treatment plan is early diagnosis, pain management, prevention, stabilisation, restoration of any defects and regular maintenance.
The management of teeth begins in the primary dentition, followed by the secondary dentition as the teeth erupt. Children with AI may also present with malocclusion and it is therefore essential that a multidisciplinary management involving paediatric dentists, orthodontists, oral surgeons, oral and maxillofacial surgeons and restorative dentists is sought to maintain pro-active care for the patient. At all stages, however, it is important to reinforce and maintain good oral hygiene.
In the next three papers of this series, we aim to describe the role of each of the specialties: paediatric dentistry, orthodontic dentistry and restorative dentistry and their role in management of AI.
References
Altug-Atac A T, Erdem D . Prevalence and distribution of dental anomalies in orthodontic patients. Am J Orthod Dentofacial Orthop 2007; 131: 510–514.
Bäckman B, Holm A K . Amelogenesis imperfecta: prevalence and incidence in a northern Swedish county. Community Dent Oral Epidemiol 1986; 14: 43–47.
Sedano H O . Congenital oral anomalies in Argentinian children. Community Dent Oral Epidemiol 1975; 3: 61–63.
Chosack A, Eidelman E, Wisotski I, Cohen T . Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol 1979; 47: 148–156.
Aldred M J, Savarirayan R, Crawford P J . Amelogenesis imperfecta: a classification and catalogue for the 21st century. Oral Dis 2003; 9: 19–23.
Wright J T, Hart T C, Hart P S et al. Human and mouse enamel phenotypes resulting from mutation or altered expression of AMEL, ENAM, MMP20 and KLK4. Cells Tissues Organs 2009; 189: 224–229.
Mårdh C K, Bäckman B, Holmgren G, Hu J C, Simmer J P, Forsman-Semb K. A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2). Hum Mol Genet 2002; 11: 1069–1074.
Hu J C, Hu Y, Smith C E et al. Enamel defects and ameloblast-specific expression in Enam knock-out/lacz knock-in mice. J Biol Chem 2008; 283: 10858–10871.
Gibson C W, Yuan Z A, Hall B et al. Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 2001; 276: 31871–31875.
Wright J T, Hart P S, Aldred M J et al. Relationship of phenotype and genotype in X-linked amelogenesis imperfecta. Connect Tissue Res 2003; 1: 72–78.
Hart P S, Hart T C, Michalec M D et al. Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 2004; 41: 545–549.
Kim J W, Simmer J P, Hart T C et al. MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 2005; 42: 271–275.
Simmer J P, Hu Y, Lertlam R, Yamakoshi Y, Hu J C . Hypomaturation enamel defects in Klk4 knockout/lacz knockin mice. J Biol Chem 2009; 284: 19110–19121.
Santos M C, Hart P S, Ramaswami M, Kanno C M, Hart T C, Line S R . Exclusion of known gene for enamel development in two Brazilian families with amelogenesis imperfecta. Head Face Med 2007; 3: 8.
Lee S K, Hu J C, Bartlett J D et al. Mutational spectrum of FAM83H: the C-terminal portion is required for tooth enamel calcification. Hum Mutat 2008; 8: E95–E99.
Wright J T, Frazier-Bowers S, Simmons D et al. Phenotypic variation in FAM83H-associated amelogenesis imperfecta. J Dent Res 2009; 88: 356–360.
Crawford P J, Aldred M, Bloch-Zupan A. Amelogenesis imperfecta. Orphanet J Rare Dis 2007; 2: 17.
Poulsen S, Gjørup H, Haubek D et al. Amelogenesis imperfecta – a systematic literature review of associated dental and oro-facial abnormalities and their impact on patients. Acta Odontol Scand 2008; 66: 193–199.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Refereed paper
Rights and permissions
About this article
Cite this article
Gadhia, K., McDonald, S., Arkutu, N. et al. Amelogenesis imperfecta: an introduction. Br Dent J 212, 377–379 (2012). https://doi.org/10.1038/sj.bdj.2012.314
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bdj.2012.314
This article is cited by
-
Tooth ultrastructure changes induced by a nonsense mutation in the FAM83H gene: insights into the diversity of amelogenesis imperfecta
Clinical Oral Investigations (2023)
-
Identification of the C-terminal region in Amelogenesis Imperfecta causative protein WDR72 required for Golgi localization
Scientific Reports (2022)
-
Patient-reported outcome measures in individuals with amelogenesis imperfecta: a systematic review
European Archives of Paediatric Dentistry (2022)
-
White enamel lesions in children: considering caries, chronological and congenital causes and their consequent care
British Dental Journal (2021)
-
Insights into the palaeobiology of an early Homo infant: multidisciplinary investigation of the GAR IVE hemi-mandible, Melka Kunture, Ethiopia
Scientific Reports (2021)
