
Abstract
Study design:
This work is an experimental and prospective study in adult, female, Long–Evans rats.
Objectives:
The aim of this study was to probe the effect of metabolic inhibition after an acute traumatic spinal cord injury (TSCI) using a standardized contusion model (NYU impactor) to know whether the metabolic inhibition is a ‘secondary mechanism of injury’ or a mechanism of protection.
Setting:
All experimental procedures were carried out in the Mexico City.
Methods:
Animals were divided into five groups: one sham and four with TSCI, including no treatment, rotenone (inhibitor of mitochondrial complex I), sodium azide (inhibitor of mitochondrial complex IV) and pyrophosphate of thiamine or non-degradable cocarboxylase as a metabolic reactivator.
Results:
After TSCI, the metabolic inhibition with sodium azide treatment diminished the lipid peroxidation process (malondialdehyde levels by spectrophotometric procedures) and the damage to the spinal cord tissue (morphometric analysis), and increased the activity of creatine kinase and lactate dehydrogenase enzymes (P<0.05) (measured by spectrophotometric procedures 24 h after TSCI as well as after the functional recovery of the hind limb (evaluated weekly for 2 months by the BBB (Basso, Beattie and Bresnahan) scale)) when compared with the TSCI group without treatment.
Conclusion:
The results show that the partial and transitory inhibition of the aerobic metabolism after an acute TSCI could be a self-protection mechanism instead of being a ‘secondary mechanism of injury’.
Similar content being viewed by others


Introduction
In spite of preventive efforts, traumatic spinal cord injury (TSCI) continues to be a significant public health problem, with high rates of morbidity and mortality1 and, currently, with no cure.
TSCI can result in irreversible autonomic, sensorial and motor dysfunctions through not only the mechanical processes at the time of the traumatic event but also through a variety of ‘secondary mechanisms of injury’. These mechanisms contribute to cell dysfunction, and death of neuronal, glial and endothelial cells through events, such as ischemia, edema, ionic imbalances, glutamate-mediated excitotoxicity, mitochondrial disfunction, metabolic inhibition, oxidative damage (formation of reactive oxygen species) and lipid peroxidation.2
The mitochondrial electron transport chain is the main source of reactive oxygen species during normal metabolism,3 but this rate of reactive oxygen species generation may be altered by different physiological or pathological conditions, including hypoxia,4 ischemia5 and chemical inhibition of mitochondrial respiration.6 The molecular sites of mitochondrial reactive oxygen species production are not well established, but are generally thought to be located in complexes I and III of the electron transport chain.7 The mitochondrial disfunction causes the switch from aerobic to anaerobic metabolism, accumulation of lactate (which generates an acid environment), loss of ATP (adenosine triphosphate) production and formation of reactive oxygen species that contribute to cell death by shooting the lipid peroxidation process.8
Although hypoxia, hypothermia, acidosis and metabolic inhibition have traditionally been considered ‘secondary mechanisms of injury’ after a TSCI, they have shown a beneficial effect in other pathological conditions. For example, hibernation, which implies conditions, such as hypoxia and metabolic inhibition, allows the organism to survive for a long time without food and tolerate the inclement conditions of the season.9 Another example is acidosis, which might inhibit calcium overload and/or degradative enzyme activation, or block sodium entry due to sodium/hydrogen exchange.10 Hypothermia (34 °C) could also be a protective mechanism that decreases energetic metabolism to avoid reactive oxygen species formation and catalytic enzyme activation.11
In spite of the great advances in the physiology of TSCI, the mechanisms of damage that activate the organism after the injury have not been completely deciphered. We think that some of these ‘mechanisms of secondary damage’ could in fact be acting as ‘mechanisms of protection’ or defense. Therefore, the goal of this work is to determine whether metabolic inhibition could be acting as a ‘mechanism of protection’ after an acute TSCI.
Materials and methods
Experiments were carried out on 30 male (280–320 g) and 30 female adult Long–Evans rats (200–250 g). The experimental protocol was approved by the Institutional Review Board of the National Medical Center, Century XXI, IMSS, and all procedures were in accordance with the Mexican General Law in Health, concerning the care and use of laboratory animals (Ley Mexicana General de Salud).12 Rats were divided into five groups: one sham and four with TSCI, including no treatment, rotenone (inhibitor of mitochondrial complex I), sodium azide (inhibitor of mitochondrial complex IV), and pyrophosphate of thiamine or non-degradable cocarboxylase (NDC) as a metabolic reactivator. Each group included 14 rats, seven for biochemical analysis (the rats were killed by decapitation 24 h after TSCI) and seven for histological, morphological and neurological assessments (the rats were perfused via the ascending aorta with 10% formaldehyde 2 months after TSCI).
The spinal cord contusions were carried out with a well-characterized technique using the NYU (New York University) weight-drop device.13 Rats were anesthetized with ketamine (75 mg kg−1) and xylazine (12.5 mg kg−1). Then, a dorsal laminectomy at the thoracic 9 level (T9) was carried out exposing the spinal cord. TSCI was induced after placing the animals in the stereotaxic system; a 10 g rod was dropped onto the spinal cord from a distance of 25 mm, inflicting a moderate contusion injury that results in hind limb locomotor deficit. Animals from the sham group received a dorsal laminectomy without a TSCI.
To select the best metabolic inhibitor for this study, a preliminary analysis was made using rats randomly assigned to receive treatment by intraperitoneal injection at 5 min, and at 2, 6 and 20 h after TSCI. Rats received rotenone (Sigma M1018, Sigma-Aldrich, Toluca, Mexico) at 3 mg kg−1 of body weight, sodium azide (Sigma-Aldrich) at 5 mg kg−1 of body weight, 3-nitropropionic acid (Sigma-Aldrich) at 25 mg kg−1 of body weight, antimicina A (Sigma-Aldrich) at 0.5 mg kg−1 of body weight and cyanide (Sigma-Aldrich) at 3 mg kg−1 of body weight. Once the metabolic inhibitors were selected, the experiments were conducted (Figure 1).

Lipid peroxidation 24 h after traumatic spinal cord injury (TSCI) in animals undergoing different treatments. Each group is represented by means±s.d. (n=7). Parametric ANOVA (analysis of variance) followed by Tukey's DVS test. Significant difference with P<0.05. ANT A, antimicina A; AZIDE, sodium azide; CYANIDE, cyanide; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy; 3-NPA, 3-nitropropionic acid.
The injured rats were randomly assigned to receive treatment by intraperitoneal injection at 5 min, and at 2, 6 and 20 h after TSCI. Rats received rotenone at 3 mg kg−1 of body weight, sodium azide at 5 mg kg−1 of body weight, as a single dose, and increasing doses of NDC (Manuelle Laboratories SA, Mexico City, México) at 50, 80, 100 and 150 mg kg−1 of body weight.
Rotenone and sodium azide were used as metabolic inhibitors, and NDC enzyme was used as an energy reactivator. After drug administration, each rat was placed in a cage with free access to food and water.
For biochemical assays, 24 h after TSCI, 2 ml of blood samples were taken from each animal and centrifuged at 3000 rpm for 15 min to isolate plasma. In parallel, the spinal cord was removed from T8 to T10 levels and placed free from meninges in physiological solution. The spinal cord was weighed and then homogenized in 20 mM Tris buffer in a 3/7 (w/v) proportion.
To evaluate the lipid peroxidation process, the bases of Schiff (an end product of this process) were quantified through a spectrofluorimetric technique, in which a volume of 4 ml of the homogenate was taken and 1 ml of a mixture of chloroform–methanol (2:1) was added. The mixture was shaken for 5 s and left in ice for 30 min. The methanolic phase was removed, and 900 μl of the solution was taken along with 100 μl of methanol. Finally, the reading was taken in a fluorometer with an excitation of 370 nm and an emission of 430 nm wavelength.
The lactate dehydrogenase (LDH) activity was evaluated by an enzymatic method using a specific kit (Sigma no. 735-10, Sigma-Aldrich). 10 μl of the homogenate of the spinal cord was used, and it was incubated with the reagents for 5 min at a temperature of 20–25 °C. The absorbance was measured in a Beckman Du-650 spectrophotometer (Beckman Instruments, Inc., CA, USA) at 540 nm wavelength. Results are directly proportional to the activity of the LDH in the sample.
Creatine kinase catalyzes the reversible conversion of creatine and ATP to creatine phosphate and ADP (adenosine diphosphate). This technique is based on an important reduction from NADP to NADPH, which increases the absorbance at 340 nm and is proportional to the creatine kinase activity. One unit is defined as the amount of enzyme that will catalyze the conversion of 1.0 μmol of substrate (creatine phosphate) per minute at 25 or 37 °C. Sigma kit no. 45-UV (Sigma-Aldrich) was used to evaluate this process in the injured spinal cord. Using 100 μl of plasma per sample, 3 ml of work solution was added and mixed by inversion. The samples were incubated at room temperature. The absorbance was measured in duplicates 5 and 10 min later at 340 nm using the Beckman Du-650 spectrophotometer (Beckman Instruments, Inc.). The corresponding calculations were made.
Hind limb motor score was graded using the 0–21 point scoring system BBB (Basso, Beattie and Bresnahan motor score), which categorizes combinations of rat hind limb movements, trunk position, stability, stepping, coordination, paw placement, toe clearance and tail position.13 Hind limb motor function was recorded 24 h after the surgery and then after every week for the next 2 months. At the end of follow-up, rats were deeply anesthetized with ketamine and chlorhydrate of xylazine and transcardially perfused with 100 ml of heparinized saline solution followed by 500 ml of 10% formaldehyde in phosphate buffer (pH 7.4) at 30 ml min−1. Then, the spinal cords were cut from T8 to T10 and embedded in paraffin blocks. Transverse paraffin sections were cut at a thickness of 6 μm. The tissue was stained with hematoxylin and eosin. After examination of serial sections from the injury site, the section containing the largest lesion area from each rat was selected and analyzed, as well as one slide preceding and following the selected one. The amount of preserved tissue was evaluated with an image analysis system (software image database Leica).
Results
A significant difference in the number of Schiff bases was observed between the TSCI, rotenone and sodium azide groups (P<0.05), whereas the 3-nitropropionic acid and cyanide groups showed a slight tendency to increase the lipid peroxidation process.
On the other hand, a significant difference (P<0.05) was observed when comparing sodium azide (the best metabolic inhibitor) against the metabolic reactivator NDC and the TSCI non-treated group (Figure 2).

Lipid peroxidation 24 h after traumatic spinal cord injury in animals undergoing different treatments. Each group is represented by means±s.d. (n=7). Parametric ANOVA (analysis of variance) followed by Tukey's DVS test. Significant difference with P<0.05. AZIDE, sodium azide; NDC, non-degradable cocarboxylase; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.
A significant difference between the sham group, and the sodium azide and NDC groups (P<0.05) was observed when evaluating the enzymatic activity of creatine kinase, which was higher in the sodium azide- and NDC-treated rats than in the other groups (Figure 3).

Creatine kinase (CK) levels 24 h after traumatic spinal cord injury in animals undergoing different treatments. Each group is represented by means±s.d. (n=7). Kruskal–Wallis test followed by Bonferroni test. Significant difference with P<0.05. AZIDE, sodium azide; NDC, non-degradable cocarboxylase; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.
A significant difference (P<0.05) in DHL activity was shown between the sham group, and TSCI and sodium azide groups, as well as between rotenone group and sodium azide- and NDC-treated rats (Figure 4).

Dehydrogenase lactate (DHL) levels 24 h after traumatic spinal cord injury in animals undergoing different treatments. Each group is represented by means±s.d. (n=7). Parametric ANOVA (analysis of variance) followed by Tukey's DVS test. Significant difference with P<0.05. AZIDE, sodium azide; NDC, non-degradable cocarboxylase; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.
At the second week of follow-up, rats treated with sodium azide showed an important functional recovery when they reached a qualification of nine on the BBB motor scale, which means that the animals supported their hind limbs. In the sixth week, they showed a greater recovery (which they kept during the remainder of the study), showing a significant difference (P<0.05) compared with TSCI, rotenone and NDC groups, which had qualifications below nine (no weight supported plantar steps) on the BBB motor scale (Figure 5).

The mean motor scores of the hind limbs in the different treatment groups after traumatic spinal cord injury using the Basso, Beattie and Bresnahan (BBB) motor score. Each group is represented by means±s.d. (n=7). ANOVA (analysis of variance) of repeating measures. Significant difference with P<0.05. AZIDE, sodium azide; NDC, non-degradable cocarboxylase; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.
The amount of preserved tissue in all of the experimental groups showed a significant difference (P<0.05) with respect to the sham group. The group that showed the highest amount of preserved tissue was the one treated with rotenone; however, significantly differences were found only among NDC and Azide experimental groups (Figures 6 and 7).

Preserved tissue percentage in traverse courts of spinal cord of rats 2 months after spinal cord injury in animals undergoing different treatments. Each group is represented by means±s.d. (n=7). Kruskal–Wallis test followed by Bonferroni test. Significant difference with P<0.05. AZIDE, sodium azide; NDC, non-degradable cocarboxylase; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.

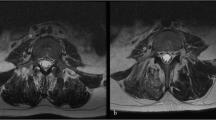
The cut traverse section of spinal cord in epicenter of injury or in corresponding area in the control group of rats 2 months after traumatic spinal cord injury and a corresponding treatment. The dye used was hematoxylin-eosin. AZIDE, sodium azide; ROTENONE, rotenone; TSCI, traumatic spinal cord injury without treatment; SHAM, laminectomy.
Discussion
For more than two decades, the goal of many researchers has been to limit the damage generated by the ‘secondary mechanisms of injury’ after a TSCI. Nevertheless, in a great number of studies, treatments have been far from improving the condition of the injured individual. Moreover, some of these therapeutic strategies show no change in, or heighten, the response to the injury.14
This study showed that metabolic inhibition in the acute phase of TSCI diminishes the lipid peroxidation process and increases the activity of DHL and creatine kinase enzymes, as well as the functional recovery of the hind limbs in rats. Many therapeutic strategies have maintained mitochondrial homeostasis with the purpose of attenuating the effects of an acute injury on the brain or on the spinal cord15 to decrease the formation of reactive oxygen species, which destroy the cellular components. However, it has been shown that rotenone (0.5 μg ml−1) decreases reactive oxygen species generation during a hypoxic phenomenon,16 which could avoid membrane damage, especially at the myelin sheath of the axons that keep them functional.
As the production of reactive oxygen species is very sensitive to the proton motive force, it can be strongly decreased by mild uncoupling, as this diminishes mitochondrial superoxide anion (O2•−) production, thus protecting against disease and oxidative damage at the expense of a small loss of energy.17 Moreover, it has been shown that the use of a metabolic inhibitor can be beneficial in certain pathological conditions, such as heart infarction, in which experimental precondition through metabolic inhibition with sodium cyanide significantly reduced the heart infarct size. Sodium cyanide uncouples oxidative phosphorylation, leading to a decrease in the cellular ATP levels; the more rapid depletion of high-energy phosphate leads to more rapid activation of mitochondrial KATP channels, which may be protective.18 These data support the idea that, although a complete or prolonged uncoupling of the mitochondrial chain can produce a series of harmful processes for the cellular function, partial or transitory uncoupling of the mitochondrial chain can protect against harmful processes such as excitotoxicity.6, 15 This may be explained by the fact that uncoupling of respiration from ADP phosphorylation increases mitochondrial respiratory rates, decreasing the lifetime of ubiquinone in the respiratory chain and, therefore, the probability of O2•− generation by electron donation from ubiquinone to O2. Another interesting hypothesis on why mitochondrial uncoupling reduces radical oxygen species generation proposes that the O2•− formed at the cytoplasmic side of the inner mitochondrial membrane during the coenzyme Q cycle may combine with protons and form perhydroxyl radicals. These radicals can readily diffuse through the inner membrane to regenerate O2•− in the more alkaline mitochondrial matrix. This process would decrease the concentration of O2•− at the cytoplasmic face of the inner mitochondrial membrane, increasing O2•− generation in the mitochondrial matrix, and is stimulated by the presence of a proton gradient. This model also explains how O2•− generated on the cytoplasmic face of the inner mitochondrial membrane diffuses through the membrane to be converted to H2O2 by Mn-superoxide dismutase in the mitochondrial matrix. Nevertheless, it is probable that only a fraction of O2•− generated in the intermembrane space combines with protons, whereas part of the O2•− combines with cytochrome C or promotes membrane lipid oxidation.6
Furthermore, metabolic inhibition increases the enzymatic activity of the LDH, which generates an acid environment (pH=6.2) that exerts a neuroprotector mechanism against the damage caused by the excitotoxicity in experimental models of focal ischemia.19 Two reasons for this phenomenon include: (1) acid pH inactivates the receptors type N-Methyl-D-Aspartate and limits the entrance of excessive amounts of calcium into the cell and (2) ATP production decreases, which is necessary to maintain the stable structure of the N-Methyl-D-Aspartate. As the calcium binding to the negatively charged phosphatidylserine results in a bilayer destabilizing effect of phosphatidylethanolamine and the lower pH greatly diminishes this binding, lower pH results in an attenuation of cell lyses due to the calcium overload inhibition or the degradative enzymes inactivation.20
The results show that the partial and transitory inhibition of the aerobic metabolism after an acute TSCI could be a self-protection mechanism instead of being a ‘secondary mechanism of injury’.
References
Sekhon HL, Fehlings GM . Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine 2001; 26: 2–12.
Dumont JR, Okonkwo OD, Verma S, Hurlbert JR, Boulos TP, Ellegala BD et al. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 2001; 24: 254–264.
Kroemer G, Reed JC . Mitochondrial control of cell death. Nat Med 2000; 6: 513–519.
LaManna JC, Chavez JC, Pichiule P . Structural and functional adaptation to hypoxia in the rat brain. J Exp Biol 2004; 207: 3163–3169.
Reiter RJ, Tan DX, Manchester LC, Tamura H . Melatonin defeats neurally-derived free radicals and reduces the associated neuromorphological and neurobehavioral damage. J Physiol Pharmacol 2007; 58 (Suppl 6): 5–22.
Kowaltowski AJ, Vercesi AE . Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 1999; 26: 463–471.
Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS et al. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 2004; 24: 7779–7788.
Hall ED, Springer EJ . Neuroprotection and acute spinal cord injury: a reappraisal. J Am Soc Exp Neuro Rx 2004; 1: 80–100.
Zhou F, Zhu X, Castenalli RJ, Stimmelmayr R, Perry G, Smith MA et al. Hibernation, a model of neuroprotection. Am J Pathol 2001; 158: 2145–2151.
Imaizumi T, Kocsis JD, Waxman SG . Anoxic injury in the rat spinal cord: pharmacological evidence for multiple steps in Ca(2+)-dependent injury of the dorsal columns. J Neurotrauma 1997; 14: 299–311.
Dimar JR, Shields CB, Zhang YP, Burke DA, Raque GH, Glassman SD . The role of directly applied hypothermia in spinal cord injury. Spine 2000; 25: 2294–2302.
Ley Mexicana General de Salud: Uso de animales en experimentación (Mexican General Law in Health: The Use of Animals in Experimentation). Porrúa: México, 1995, pp 430–431.
Basso DM, Beattie MS, Bresnahan JC . Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transaction. Exp Neurol 1996; 139: 244–256.
Hulbert RJ . Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg 2000; 93: 1–7.
Maragos WF, Korde AS . Mitochondrial uncoupling as a potential therapeutic target in acute central nervous system injury. J Neurochem 2004; 91: 257–262.
Schroedl C, McClintock DS, Budinger GR, Navdeep SC . Hypoxic but not anoxic stabilization of HIF-1α requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol 2002; 283: L922–L931.
Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, Miwa S et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 2004; 37: 755–767.
Irie H, Gao J, Gaudette GR, Cohen IS, Mathias RT, Saltman AE et al. Both metabolic inhibition and mitochondrial KATP channel opening are myoprotective and initiate a compensatory sarcolemmal outward membrane current. Circulation 2003; 108 (Suppl II): II-341–II-347.
Vornov JJ, Thomas AG, Jo D, Ajit GT, David J . Protective effects of extracellular acidosis and blockade of sodium/hydrogen ion exchange during recovery from metabolic inhibition in neuronal tissue culture. J Neurochem 1996; 67: 2379–2389.
Nicholls DG, Budd SL . Mitochondria and neuronal Survival. Physiologic Rev 2000; 80: 315–334.
Acknowledgements
Sergio Torres-Castillo was supported by CONACyT, scholarship no. 153029.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Torres, S., Salgado-Ceballos, H., Guizar-Sahagún, G. et al. Deleterious versus neuroprotective effect of metabolic inhibition after traumatic spinal cord injury. Spinal Cord 47, 745–750 (2009). https://doi.org/10.1038/sc.2009.27
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sc.2009.27
