
Abstract
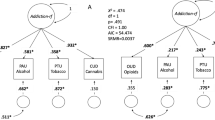
Genetic liability to substance use disorders can be parsed into loci that confer general or substance-specific addiction risk. We report a multivariate genome-wide association meta-analysis that disaggregates general and substance-specific loci from published summary statistics of problematic alcohol use, problematic tobacco use, cannabis use disorder and opioid use disorder in a sample of 1,025,550 individuals of European descent and 92,630 individuals of African descent. Nineteen independent single-nucleotide polymorphisms were genome-wide significant (P < 5 × 10–8) for the general addiction risk factor (addiction-rf), which showed high polygenicity. Across ancestries, PDE4B was significant (among other genes), suggesting dopamine regulation as a cross-substance vulnerability. An addiction-rf polygenic risk score was associated with substance use disorders, psychopathologies, somatic conditions and environments associated with the onset of addictions. Substance-specific loci (9 for alcohol, 32 for tobacco, 5 for cannabis and 1 for opioids) included metabolic and receptor genes. These findings provide insight into genetic risk loci for substance use disorders that could be leveraged as treatment targets.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$59.00 per year
only $4.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

Data availability
The MVP summary statistics were obtained via an approved dbGaP application (phs001672.v4.p1). For details on the MVP, see https://www.research.va.gov/mvp/ and ref. 76. This research is based on data from the MVP, Office of Research and Development, Veterans Health Administration, and was supported by the Veterans Administration Cooperative Studies Program award G002.
Publicly available data were also downloaded from the psychiatric genomics consortium (https://www.med.unc.edu/pgc/) and the GSCAN consortium (https://conservancy.umn.edu/handle/11299/201564).
The datasets used for the BioVU analyses described were obtained from Vanderbilt University Medical Center’s biorepository, which is supported by numerous sources: institutional funding, private agencies and federal grants. These include the National Institutes of Health-funded Shared Instrumentation grant S10RR025141; and Clinical and Translational Science Awards (CTSA) grants UL1TR002243, UL1TR000445 and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962 and R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/.
Data from Yale–Penn 1 are available through dbGAP accession no phs000425.v1.p1 including 1,889 African American subjects and 1,020 European-American subjects. Yale–Penn 1 data are also available through dbGAP accession no phs000952.v1.p1 including 1,531 African American subjects and 1,339 self-reported European-American subjects. Summary statistics for all Yale–Penn data are available on request to J.G. (joel.gelernter@yale.edu).
References
Degenhardt, L. et al. The impact of cohort substance use upon likelihood of transitioning through stages of alcohol and cannabis use and use disorder: findings from the Australian National Survey on Mental Health and Wellbeing. Drug Alcohol Rev. 37, 546–556 (2018).
Peacock, A. et al. Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction 113, 1905–1926 (2018).
Reitsma, M. B. et al. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and initiation among young people in 204 countries and territories, 1990–2019. Lancet Public Health 6, e472–e481 (2021).
Odds of dying. Injury Facts https://injuryfacts.nsc.org/all-injuries/preventable-death-overview/odds-of-dying/ (accessed 3 December 2021).
Vanyukov, M. M. An eternal epidemic: 1. Why substance use problems persist. Preprint at PsyArXiv https://psyarxiv.com/tkm5u/ (2021).
Deak, J. D. & Johnson, E. C. Genetics of substance use disorders: a review. Psychol. Med. 51, 2189–2200 (2021).
Gelernter, J. & Polimanti, R. Genetics of substance use disorders in the era of big data. Nat. Rev. Genet. 22, 712–729 (2021).
Zhou, H. et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat. Neurosci. 23, 809–818 (2020).
Mallard, T. T. et al. Item-level genome-wide association study of the alcohol use disorders identification test in three population-based cohorts. Am. J. Psychiatry 179, 58–70 (2021).
Walters, R. K. et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 21, 1656–1669 (2018).
Kranzler, H. R. et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat. Commun. 10, 1499 (2019).
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244 (2019).
Hancock, D. B. et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol. Psychiatry 23, 1911–1919 (2018).
Quach, B. C. et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat. Commun. 11, 5562 (2020).
Johnson, E. C. et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 7, 1032–1045 (2020).
Zhou, H. et al. Association of OPRM1 functional coding variant with opioid use disorder: a genome-wide association study. JAMA Psychiatry 77, 1072–1080 (2020).
Kendler, K. S. et al. Recent advances in the genetic epidemiology and molecular genetics of substance use disorders. Nat. Neurosci. 15, 181–189 (2012).
Hatoum, A. S. et al. The addiction risk factor: a unitary genetic vulnerability characterizes substance use disorders and their associations with common correlates. Neuropsychopharmacology 47, 1739–1745 (2022).
Abdellaoui, A., Smit, D. J. A., van den Brink, W., Denys, D. & Verweij, K. J. H. Genomic relationships across psychiatric disorders including substance use disorders. Drug Alcohol Depend. 220, 108535 (2021).
Waldman, I. D., Poore, H. E., Luningham, J. M. & Yang, J. Testing structural models of psychopathology at the genomic level. World Psychiatry 19, 350–359 (2020).
Grotzinger, A. D. et al. Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic and molecular genetic levels of analysis. Nat. Genet. 54, 548–559 (2022).
Duan, Q. et al. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res. 42, W449–W460 (2014).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Bhattacharjee, S. et al. A subset-based approach improves power and interpretation for the combined analysis of genetic association studies of heterogeneous traits. Am. J. Hum. Genet. 90, 821–835 (2012).
Barbeira, A. N. et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 15, e1007889 (2019).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 e17 (2017).
Karlsson Linner, R. et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat. Neurosci. 24, 1367–1376 (2021).
O’Connor, L. J. & Price, A. L. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat. Genet. 50, 1728–1734 (2018).
Association, A. P. Diagnostic and Statistical Manual (DSM-IV) (American Psychiatric Press, 1994).
Roden, D. M. et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 84, 362–369 (2008).
Volkow, N. D. et al. The conception of the ABCD study: from substance use to a broad NIH collaboration. Dev. Cogn. Neurosci. 32, 4–7 (2018).
Wise, R. A. & Robble, M. A. Dopamine and addiction. Annu. Rev. Psychol. 71, 79–106 (2020).
Hatoum, A.S. et al. Genome-wide association study shows that executive functioning is influenced by gabaergic processes and is a neurocognitive genetic correlate of psychiatric disorders. Biol. Psychiatry 93, 59–70 (2022).
Snyder, G. L. & Vanover, K. E. PDE inhibitors for the treatment of schizophrenia. Adv Neurobiol 17, 385–409 (2017).
Olsen, C. M. & Liu, Q. S. Phosphodiesterase 4 inhibitors and drugs of abuse: current knowledge and therapeutic opportunities. Front. Biol. 11, 376–386 (2016).
Burnette, E. M., Ray, L. A., Irwin, M. R. & Grodin, E. N. Ibudilast attenuates alcohol cue-elicited frontostriatal functional connectivity in alcohol use disorder. Alcohol. Clin. Exp. Res. 45, 2017–2028 (2021).
Grodin, E. N. et al. Ibudilast, a neuroimmune modulator, reduces heavy drinking and alcohol cue-elicited neural activation: a randomized trial. Transl. Psychiatry 11, 355 (2021).
Li, M. J., Briones, M. S., Heinzerling, K. G., Kalmin, M. M. & Shoptaw, S. J. Ibudilast attenuates peripheral inflammatory effects of methamphetamine in patients with methamphetamine use disorder. Drug Alcohol Depend. 206, 107776 (2020).
Schoeler, T. et al. Novel biological insights into the common heritable liability to substance involvement: a multivariate genome-wide association study. Biol. Psychiatry 93, 524–535 (2022).
Sanchez-Roige, S., Palmer, A. A. & Clarke, T. K. Recent efforts to dissect the genetic basis of alcohol use and abuse. Biol. Psychiatry 87, 609–618 (2020).
Marees, A. T. et al. Genetic correlates of socio-economic status influence the pattern of shared heritability across mental health traits. Nat. Hum. Behav. 5, 1065–1073 (2021).
Khantzian, E. J. Addiction as a self-regulation disorder and the role of self-medication. Addiction 108, 668–669 (2013).
Thapar, A. & Rice, F. Family-based designs that disentangle inherited factors from pre- and postnatal environmental exposures: in vitro fertilization, discordant sibling pairs, maternal versus paternal comparisons, and adoption designs. Cold Spring Harb. Perspect. Med. 11, a038877 (2021).
Haan, E. et al. Prenatal smoking, alcohol and caffeine exposure and maternal reported ADHD symptoms in childhood: triangulation of evidence using negative control and polygenic risk score analyses. Addiction 117, 1458–1471 (2021).
Cornelis, M. C., Zaitlen, N., Hu, F. B., Kraft, P. & Price, A. L. Genetic and environmental components of family history in type 2 diabetes. Hum. Genet. 134, 259–267 (2015).
Wendt, F. R. et al. Multivariate genome-wide analysis of education, socioeconomic status and brain phenome. Nat. Hum. Behav. 5, 482–496 (2021).
Grotzinger, A. D. et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav. 3, 513–525 (2019).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
International HapMap, C. et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–58 (2010).
Grotzinger, A. D., de la Fuente, J., Privé, F., Nivard, M. G. & Tucker-Drob, E. M. Pervasive downward bias in estimates of liability-scale heritability in genome-wide association study meta-analysis: a simple solution. Biol. Psychiatry, 93, 29–36 (2023).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Rosseel, Y. Lavaan: an R package for structural equation modeling. J. Stat. Software 48, 1–36 (2012).
Hu, L. & Bentler, P. M. Cutoff criteria for fit indexes in covariance strcuture analysis: conventional criteria versus new alternatives. Struct. Equation Modell. 6, 1 (1999).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Han, B. & Eskin, E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 88, 586–598 (2011).
Zhang, Y., Qi, G., Park, J. H. & Chatterjee, N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet. 50, 1318–1326 (2018).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Psych, E. C. et al. The PsychENCODE project. Nat. Neurosci. 18, 1707–1712 (2015).
Roadmap Epigenomics, C. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Ernst, J. & Kellis, M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. R. Stat. Soc. B 82, 1273–1300 (2020).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825 (2018).
Gandal, M. J. et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127 (2018).
Liu, C. et al. Compound signature detection on LINCS L1000 big data. Mol. Biosyst. 11, 714–722 (2015).
Viechtbauer, W. Accounting for heterogeneity via random-effects models and moderator analyses in meta-analysis. Z. Psychol. 215, 104–121 (2007).
Sherva, R. et al. Genome-wide association study of cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry 73, 472–480 (2016).
Pierucci-Lagha, A. et al. Diagnostic reliability of the semi-structured assessment for drug dependence and alcoholism (SSADDA). Drug Alcohol Depend. 80, 303–312 (2005).
Das, S. et al. Next-generation genotype imputation service and methods. Nat. Genet. 48, 1284–1287 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Cuellar-Partida, G. et al. Complex-Traits Genetics Virtual Lab: A community-driven web platform for post-GWAS analyses. Preprint at bioRxiv https://doi.org/10.1101/518027 (2021).
Carroll, R. J., Bastarache, L. & Denny, J. C. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376 (2014).
Lisdahl, K. M. et al. Substance use patterns in 9-10 year olds: baseline findings from the adolescent brain cognitive development (ABCD) study. Drug Alcohol Depend. 227, 108946 (2021).
Bates, D., Machler, M., Bolker, B. M. & Walker, S. C. Fitting linear mixed-effects models using lme4. J. Stat. Software 67, 1–48 (2015).
Gaziano, J. M. et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol. 70, 214–223 (2016).
Acknowledgements
The authors would like to thank Cold Harbor Labroatory for posting a preprint of this work on MedRxiv (https://www.medrxiv.org/). The authors thank Million Veteran Program (MVP) staff, researchers and volunteers, who have contributed to MVP, and especially participants who previously served their country in the military and now generously agreed to enroll in the study. Funding: K01 AA030083 (A.S.H.), T32 DA007261 (A.S.H.), DA54869 (A.A., J.G., H.E.), R01 DA54750 (A.A., R.B.), K02 DA32573 (A.A.), R21 AA027827 (R.B.), U01 DA055367 (R.B.), K01 DA51759 (E.C.J.), K23 MH121792 (N.R.K.), DP1 DA54394 (S.S.-R.), T32 MH014276 (G.A.P.), R01 AA027522 (A.E.), F31 AA029934 (S.E.P.), R01 MH120219 (E.M.T.-D., A.D.G.), RF1 AG073593 (E.M.T.-D., A.D.G.), P30 AG066614 (E.M.T.-D.), P2CHD042849 (E.M.T.-D.), R33 DA047527 (R.P., G.A.P.) and T32 AA028259 (J.D.D.)
Author information
Authors and Affiliations
Consortia
Contributions
A.S.H. designed the study and conducted analyses. S.M.C.C., E.C.J., S.B.H., J.D.D., G.A.P., M.V.J., S.S.-R., S.E.P., N.R.K., I.H. and D.A.A.B. conducted various analyses. A.A., R.B., H.J.E. and J.G. supervised the study. A.D.G. and E.M.T.-D. provided statistical guidance. A.E., H.R.K., R.P., L.K.D. and S.S.-R. guided interpretation of key findings. A.S.H., H.J.E., J.G., R.B. and A.A. drafted the manuscript. A.S.H., S.M.C.C., J.D.D., M.V.J., S.E.P., N.R.K. and I.H. organized the data. The consortium members provided insight into various aspects of analyses and interpretation and data for some of the discovery GWAS that were inputs to these analyses. All named authors reviewed, edited and approved the submission.
Corresponding author
Ethics declarations
Competing interests
H.R.K. is a member of advisory boards for Dicerna Pharmaceuticals, Sophrosyne Pharmaceuticals and Enthion Pharmaceuticals; a consultant to Sobrera Pharmaceuticals; and a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi and Otsuka. H.R.K. and J.G. hold US Patent 10900,082: ‘Genotype-guided dosing of opioid agonists’ issued on 26 January 2021. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Mental Health thanks Ditte Demontis and Eske Derks for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10 and methods.
Supplementary Tables
Supplementary Tables 1–22.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hatoum, A.S., Colbert, S.M.C., Johnson, E.C. et al. Multivariate genome-wide association meta-analysis of over 1 million subjects identifies loci underlying multiple substance use disorders. Nat. Mental Health 1, 210–223 (2023). https://doi.org/10.1038/s44220-023-00034-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44220-023-00034-y
This article is cited by
-
Pleiotropy and genetically inferred causality linking multisite chronic pain to substance use disorders
Molecular Psychiatry (2024)
-
Epigenetic alterations identify a confluence of genetic vulnerabilities tied to opioid overdose
Neuropsychopharmacology (2024)
-
Multi-ancestry meta-analysis of tobacco use disorder identifies 461 potential risk genes and reveals associations with multiple health outcomes
Nature Human Behaviour (2024)
-
Multi-trait genome-wide association analyses leveraging alcohol use disorder findings identify novel loci for smoking behaviors in the Million Veteran Program
Translational Psychiatry (2023)
-
Single nucleus transcriptomics of ventral midbrain identifies glial activation associated with chronic opioid use disorder
Nature Communications (2023)
