
Abstract
Arynes are highly reactive and versatile intermediates for the functionalization of aromatic rings that are often generated using strong bases or fluoride sources, which, in some cases, can limit functional group tolerance. Here we demonstrate that triaryloxonium ions can be transformed into arynes through treatment with solid potassium phosphate at room temperature. A substantial range of functional group-bearing arynes, including 4,5-pyrimidynes, may be generated and trapped using cycloaddition reactions with high yields. Other arynophiles including nitrones, alkenes and azides are compatible with these conditions. Quantum computation in conjunction with an intramolecular kinetic isotope study is consistent with an elimination, unimolecular, conjugate base-like mechanism of elimination to form the aryne. These investigations demonstrate that the oxonium ion is a powerful electron-withdrawing group and a particularly effective leaving group. We anticipate that this study will stimulate further investigations into the synthetic utility of aryl oxonium ions.

Similar content being viewed by others
Main
Arynes1 possess particularly rich history and varied chemistry, which has led to their broad application as reactive intermediates in synthesis2. The emerging field of arynes was initially dominated by a focus on structure and mechanism, leading to the original formulation of benzyne as a diradical intermediate3. Later, a dipolar form for benzyne was proposed4 and finally, by virtue of a classical 14C-labelling experiment, the bent acetylene-type structure, now regarded as correct, was proposed5. The practical use of arynes in synthesis had to await the arrival of accessible and stable precursors (Fig. 1a)6. These include haloarenes that generate benzynes upon treatment with organometallic bases7 and aryl ortho-diazonium carboxylates that offer reliable access to arynes despite their shock sensitivity8. Other eliminative approaches include transition metal-mediated functionalizations that can be employed in mild and chemoselective approaches to metal-bound arynes9,10. An alternative method for benzyne generation exploits the hexadehydro Diels–Alder reaction of tethered triynes11,12,13, which provides reagent-free (thermal) access to polycyclic arynes. However, among contemporary precursors, Kobayashi’s ortho-silyl triflate14,15 is largely responsible for the rehabilitation of arynes as viable intermediates in synthetic chemistry as a consequence of its ability to generate a low steady-state concentration of aryne upon treatment with a sparingly soluble fluoride source16. This has also led to a resurgence in fluoride-free17 methods for aryne generation, often from monosubstituted precursors18,19,20. Many of these methods use activated leaving groups such as λ3-iodanes to enable milder conditions for aryne generation21,22,23, but these often necessitate the use of strong bases such as lithium bis(triethylsilyl)amide or potassium tert-butoxide. There are relatively few reports of benzyne formation with weaker bases23,24 although notable progress has been made in the elimination of tricyclic λ3-chloranes and bromanes25,26,27.

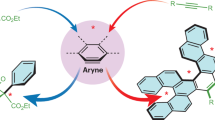
a, Previous strategies and methods for the synthesis of benzynes include the organometallic elimination of haloarenes, decomposition of diazo-anthranilic acid derivatives, the hexadehydro Diels–Alder reaction of tethered trienes, the oxidative insertion-mediated elimination of ortho-substituted leaving groups, the fluoride-mediated elimination of ortho-silyl triflates, the base-mediated elimination of λ3-iodanes and the base-mediated elimination of tricyclic λ3-chloranes and bromanes. b, In this work, a diverse range of triaryloxonium ions can be synthesized through an intramolecular O-arylation process. These oxonium ions can be used in mild and functional group-tolerant synthesis of arynes through a base-mediated elimination reaction. A range of arynophiles are effective trapping agents in this process. Hal, halogen; Ts, tosylate; Tf, triflate; Mes, mesityl; R, R1 = alkyl and/or aryl group.
We reasoned that a monosubstituted arene bearing a particularly good leaving group (that could also function as a powerful electron-withdrawing group) could enable the formation of arynes using mild and functional group-compatible conditions. The leaving group ability of onium salts from nitrogen, oxygen, phosphorus and sulfur has been calculated to be the greatest for oxygen28. Hence, we decided to examine whether oxonium salts could function as aryne precursors. Alkyloxonium salts are exemplified by Meerwein’s salt, which although kinetically stable, is a powerful alkylating agent that reacts with many nucleophiles, including water29,30. More complex oxonium ions have been proposed as intermediates in natural product biosynthesis31, and aside from Mascal’s oxatriquinane oxonium ions32, are considered to be highly reactive. In contrast, triaryloxonium ions are resistant to the action of many nucleophiles and are kinetically stable isolable solids. An early report from Hellwinkel33 showed that the addition of phenyllithium to phenyl dibenzofuranium ion led to products consistent with the generation of an aryne. Tolstaya later showed that treatment of a symmetrical triaryloxonium ion with potassium acetate in refluxing benzene led to an aryne that could be trapped in situ with tetraphenylcyclopentadienone34.
In this article, we describe a mild and functional group-tolerant method for the generation and trapping of functionalized arynes. We reasoned that, with an appropriately designed oxonium precursor, we could generate arynes using weak inorganic bases without any special precautions to exclude moisture or oxygen. While this approach would demonstrate only modest atom economy in common with other eliminative aryne generation methods, this could be compensated by the mildness of the reaction conditions for onward reaction and trapping. An appropriate oxonium precursor should possess ortho-blocking groups to prevent uncontrolled elimination (Fig. 1b). This structure could be assembled via cross-coupling between two suitably activated precursors. A methyl group is employed as the ortho-blocking group due to its small size and the availability of precursors. We have demonstrated that helically chiral triaryloxonium ions can be generated by intramolecular O-arylation of a diphenylether derivative35 with a diazonium salt, and we rationalized that this strategy offered a viable route to suitable oxonium ions for aryne generation36.
Results
The 2-bromobiaryl ethers (such as 1a–1v) can be synthesized by a base-mediated SNAr reaction between an electron-deficient fluoroarene and a phenol derivative; this reaction is relatively tolerant of different functional groups and can be performed where the bromine atom is part of either the nucleophilic or electrophilic species. These compounds can also be manipulated to introduce other functional groups (for full details of oxonium ion synthesis, see Supplementary Section 2). Aniline-containing pinacol borane 2 can be efficiently cross-coupled with aryl bromides in the presence of a catalytic quantity of 1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) to yield anilines 3a–3v. Treatment with tert-butylnitrite in the presence of tetrafluoroboric acid led to an intermediate diazonium salt, which upon gentle warming, underwent intramolecular O-arylation to yield oxonium tetrafluoroborates 4a–4v (Fig. 2). Aldehyde and ketone functional groups were incompatible with the cross-coupling strategy due to the presence of an aniline, and hence an alternative procedure was utilized to generate oxonium ions 4d and 4e (route not shown; Supplementary Section 3.5). Using these methods, we were able to synthesize a wide range of triaryloxonium tetrafluoroborates. These are generally crystalline materials that can be stored indefinitely at ambient temperature without special precautions; there is little observable difference in reactivity or stability between oxonium ions with a para-acetamide or para-methyl group. The structure of compounds 4a, 4j, 4k and 4l was confirmed by single crystal X-ray diffraction, which in all cases shows that these compounds are pyramidal around the trivalent oxygen, with the exocyclic O-aryl substituent oriented almost perpendicular to the plane of the dibenzofuran ring system. These structural features are consistent with previous solid-state investigations of related materials37.

a, Triaryloxonium ions can be generated from bromobiaryl ethers via cross-coupling, diazotization and O-arylation procedures. Reagents and conditions: (i) Pd(dppf)Cl2˙CH2Cl2 (5 mol%), Na2CO3 (4 eq.), PhMe/H2O (1:1, vol/vol), 95 °C, 24 h and (ii) HBF4 (48% aq., 5 eq.), t-BuONO (5 eq.), CH2Cl2/IPA (1:1), 0 °C, 1 h then 30 °C, 48 h. Yields are reported for isolated and purified materials for each of the two steps pictured. For oxonium ions in which R1 = NHAc, yields are reported from a precursor R1 = NO2 and include a reduction and N-acylation step (not pictured; for full details, see Supplementary Information). b, Crystal and molecular structure of dibenzofuran-derived oxonium salts from single crystal X-ray diffraction studies. Tetrafluoroborate counterion is omitted for clarity. Yields for salts 4d and 4e reflect a different synthetic method; for details, see Supplementary Information. Pin, pinacol; dppf, 1,1′-ferrocenediyl-bis(diphenylphosphine).
With a range of oxonium salts in hand, we examined conditions for elimination of para-nitro oxonium 4a in the presence of furan as an exemplar arynophile. Homogeneous bases such as 2,6-lutidine and diisopropylethylamine were mostly ineffective. However, solid inorganic bases such as potassium carbonate at room temperature (r.t.) led to the formation of bicyclic product 5a, while treatment with sodium hydrogen carbonate led to recovery of oxonium ion. Potassium phosphate led to complete consumption of oxonium ion and a high yield of the furan trapping product 5a, and hence we focused on the use of this base to enable aryne generation (Supplementary Section 3.7). Our optimized conditions employ 5 equivalents of solid potassium phosphate in the presence of 5 equivalents of the trapping agent in acetonitrile. This process does not require any special precautions beyond rapid stirring, is not carried out under an inert atmosphere and does not require dry solvents or reagents (Table 1). Under our optimized conditions, p-nitro oxonium ion 4a led to the formation of tricycle 5a in 97% isolated yield. This increased to 99% when the reaction was carried out on a 1 mmol scale. This aryne generation and trapping process is tolerant of amides (to afford 5b in 91% yield) and esters (yielding 5c in 94% yield). Ketone- and aldehyde-containing oxonium salts can also be transformed into arynes and trapped with furan to yield 5d (96% yield) and 5e (93% yield), respectively. Oxonium ions bearing other electron-withdrawing groups such as p-chloro (affording 5f, 94%) and p-methylsulfone 5g (94%) are also effective in this transformation. Free alcohols do not have any deleterious effect on the aryne generation and trapping reaction. Oxonium 4h affords p-hydroxymethyl tricycle 5h in 96% yield. Oxonium 4i bearing a para-aryl group is also successfully converted to 5i (86% yield), and simple unsubstituted benzyne can also be generated from 4j, yielding 5j in 81% yield. Ortho-nitro23,38,39 oxonium ions smoothly generate the corresponding aryne, which upon trapping generates 5k in 91% yield; this reactivity is complementary to that of the corresponding Kobayashi silyl triflate aryne precursor, which preferentially undergoes the thia–Fries rearrangement40. Other ortho-substituents on the oxonium ion that successfully yield bicyclic products include o-cyano (5l, 97%), o-methyl (5m, 95% yield), o-methoxy (5n, 98%) and 1-naphthyl (5o, 97% yield). Meta-substituted oxonium ions also eliminate smoothly to generate arynes, and regioselectivity is dictated by the steric and/or electronic nature of the substituent group.41 Meta-fluoro oxonium 4p generates 5p upon treatment with base and reaction with furan (80% yield); other electron-withdrawing groups such as trifluoromethyl proceed with the same regioselectivity (to give 5q, 84% yield). This regioselectivity is reversed with a relatively large group such as tert-butyl, yielding 5r in 87% yield. Para-alkenes bearing electron-withdrawing groups are tolerated without incident (5s, 86% yield) as are more functionalized alkyl groups (to afford 5t), albeit in reduced yield.
We can also apply these conditions to the synthesis of modified natural products and drugs. A capsaicin-derived oxonium ion was generated, which yielded tricycle 5u in 31% yield upon trapping with furan. This approach was also successful in the generation of an aryne derived from a diflusinal derivative, which yielded 5v in 88% yield. There are relatively few substituents that are unsuccessful in this process. These include oxonium ions bearing nucleophilic groups such as p-SMe, and groups such as p-CH2CO2Et that may be preferentially deprotonated in the benzylic rather than the ortho-position.
We also considered whether we could use this strategy to access hetarynes. 4,5-Pyrimidynes are a class of aryne that are notably more challenging to generate than modified benzynes42,43 and there are few reports of their generation and reactivity44,45,46,47. Pyrimidine oxonium 6 was assembled via a Suzuki coupling route similar to that employed for other oxonium ions (Supplementary Section 3.8) and its structure confirmed by single crystal X-ray diffraction. We reasoned that the electron-donating methoxy group adjacent to the oxonium oxygen could lead to enhanced trapping reactivity, as demonstrated in 2,3-pyridynes48. Treatment of this oxonium ion with solid potassium phosphate at r.t. in the presence of furan as trapping agent led to tricyclic product 7 in 39% yield. Trapping with ethanol is also effective, leading to 8 in 38% yield. In both of these reactions, a high concentration of trapping agent led to substantially increased yields. Employing pyrazole as the trapping agent led to formal insertion of the aryne into the N–H bond, affording 9 as a single regioisomer in 55% yield. This regioselectivity is consistent with nucleophilic attack distal to the electron-withdrawing oxygen substituent49. We observed similar reactivity in the addition of N-methylaniline to give 10, albeit in a moderate 24% yield. These results indicated that trapping reactions beyond cycloadditions with furan could be viable, and we explored the compatibility of a range of different trapping agents under our base-mediated conditions for aryne generation (Table 2).
The 1-nitrobenzyne, generated from oxonium ion 4k, reacts with 6,6-dimethylfulvene to afford tricycle 11 in 59% yield, which is a precursor to the anti-fungal agent isopyrazam50. Formal (3 + 2) cycloaddition is also an effective trapping manifold. Tricycle 12 is isolated in 71% yield through reaction with a diarylnitrone. Dihydropyran reacts with in situ-generated 1-nitrobenzyne via a formal (2 + 2) cycloaddition to afford 13 in 92% yield. Benzyne itself, generated from oxonium 4j, can trap benzyl azide to afford triazene 14 in 84% yield, and 3-methyl-2-butenal affords fused bicycle 15 (92% yield) via a cascade of rearrangements upon reaction with 1-nitrobenzyne (from 4k). The reaction of 4k with potassium phosphate base in the presence of 1,3-dimethyl-2-imidazolidinone affords benzodiazepine 16 in 37% yield. Nucleophiles such as pyrazole and N-methylaniline both yield regioselective addition products 17 (87% yield) and 18 (74% yield), respectively, in the presence of oxonium 4k. This oxonium ion also engages in an ene reaction with cyclohexene in the presence of potassium phosphate to yield 19. More complex structures are accessible: the reaction of in situ-generated benzyne with tethered bisbiaryl furan led to exo,exo-polycycle 20 in 47% yield51. Tryptycene 21 was formed in 83% yield from cycloaddition between anthracene and the aryne generated from precursor 4a.
To probe the mechanism of the formation of arynes from oxonium ions, we synthesized ortho-deuterated fluoroarene 22 (80% deuterium), which was transformed into oxonium 23 in three steps without any loss of deuterium (Fig. 3). We reasoned that this monodeuterated intermediate could allow us to perform an intramolecular competition experiment by measuring the selectivity of proton versus deuteron removal52. Subjecting 23 to our standard aryne-generating conditions in the presence of furan as trapping agent afforded tricycle 24 in 98% isolated yield, in which 50% of the deuterium label was retained. This allows us to calculate the kinetic isotope effect (KIE) kH/kD = 1.67, where kH is the rate constant of the reaction involving removal of hydrogen and kD is the rate constant of the reaction involving removal of deuterium.

Generating an aryne from monodeuterated oxonium 23 allows determination of an intramolecular KIE. Yields refer to isolated and purified material. Reagents and conditions: oxonium ion (0.3 mmol), K3PO4 (s, 1.5 mmol), MeCN [oxonium = 0.05 M], furan (1.5 mmol), 16 h, r.t., stirring rate >600 rpm.
We used quantum chemical calculations to investigate the mechanism of benzyne formation from an oxonium ion with inorganic phosphate at the ωB97X-D/6-31 + G(d,p) level of theory with solvation model density solvation (CH3CN) (refs. 53,54,55,56).
The computed potential energy surface reveals a formally stepwise elimination, unimolecular, conjugate base (E1cB) elimination mechanism with respect to C–H abstraction and C–O bond cleavage steps (Fig. 4a). Deprotonation at the ortho-position by K3PO4 takes place via transition structure (TS) TS1 (ΔG‡ = 9.9 kcal mol−1) and leads to the formation of a stable but shallow intermediate on the potential energy surface, IM2 (Fig. 4b). Relatively little lengthening of the exocyclic C–O bond is observed in this 1,3-zwitterionic intermediate compared with the parent oxonium ion (0.01 Å) (refs. 57,58). Subsequently, C–O bond cleavage proceeds via TS2 with a low barrier (ΔG‡ = 2.3 kcal mol−1) relative to intermediate IM2, resulting in the exergonic and irreversible formation of benzyne and dibenzofuran products IM3. We also computed the Diels–Alder cycloaddition of this benzyne intermediate with furan, which occurs via TS3 (ΔG‡ = 7.1 kcal mol−1) in a highly exergonic fashion. These data are consistent with the rapid and irreversible consumption of benzyne once formed. The stepwise formation of benzyne from an oxonium ion (Fig. 4c) stands in contrast to the mechanism of benzyne formation from a diaryliodonium ion. We computationally investigated base-mediated aryne generation from an exemplar diphenyliodonium ion at the same level of theory; this demonstrated that C–H abstraction and C–I cleavage occur in a concerted asynchronous single step without an intervening intermediate (for further details, see Supplementary Section 5)21,26,59. This is partly due to the greater strength of the (exocyclic) C–O bond in an oxonium ion versus the C–I bond in an iodonium ion; however, electronic effects are also apparent. While the formal positive charge in the Lewis structures of both -onium ions is placed at the heteroatom, polarization in the oxonium ion differs profoundly from the halonium ion. For example, computed natural charges illustrate that the oxygen atom retains substantial negative charge in oxonium ion IM1 (−0.36, cf. −0.48 in neutral 2,4,6,8-tetramethyldibenzofuran), while the neighbouring carbon atom (C1) has a partial positive charge of 0.28. This dipole provides electrostatic stabilization to the adjacent developing negative charge during ortho-C–H abstraction, which reaches −0.40 in IM2. In contrast, for the iodonium ion, a large positive charge is coincident with the position of the formal charge at the iodine atom (1.11). Natural bond orbital (NBO) analysis shows that IM2 is further stabilized through delocalization of the in-plane C6 sp2 lone pair into the exocyclic C–O σ* orbital (by 26.4 kcal mol−1). Finally, we compared the computationally predicted kH/kD isotope effect based on our Gibbs energy profile against the experiment. While the TS for C–O bond cleavage is formally rate limiting, it is only 0.7 kcal mol−1 higher in energy than the C–H abstraction TS, such that the KIE value is influenced by both TS1 and TS2. Quantitatively, the computed value of 1.93 is in close agreement with the observed KIE of 1.67 (ref. 60).

a, Mechanism of base-mediated aryne formation from oxonium ions via an E1cB elimination reaction and subsequent trapping with furan. b, ωB97X-D/6-31 + G(d,p) calculated Gibbs free energy profile (kcal mol−1). c, (i) Optimized geometries of key TSs and intermediates (selected distances are indicated (Å), energies in kcal mol−1); (ii) polarization (natural charges) in oxonium IM1, zwitterion IM2 and an exemplar diphenyliodonium ion; and (iii) NBO plot showing n–σ* electron delocalization in IM2. R, universal gas constant; T, temperature; ΔEdeloc, change in delocalization energy.
Conclusion
Triaryloxonium ions are stable and accessible compounds that can be transformed into arynes via a practically simple and mild base-mediated elimination. The functional group tolerance of this process enables efficient trapping of a wide range of functionalized arynes, including those bearing alcohols, amides and ortho-electron-withdrawing groups. Extension to the synthesis and trapping of 2,3-pyrimidynes is also successful. We anticipate that triaryloxonium salts may find application in the generation and reactions of other functionalized arynes.
Methods
Typical procedures for the preparation and reactions of triaryloxonium tetrafluoroborate salts are (i) Suzuki cross-coupling, (ii) diazotization and intramolecular O-arylation and (iii) aryne formation and trapping with furan. These procedures are exemplified for the formation and elimination of p-nitro oxonium tetrafluoroborate 4a and trapping to form 5a. Selected nuclear magnetic resonance (NMR) data are provided.
Suzuki cross-coupling
The 1-bromo-3,5-dimethyl-2-(4-nitrophenoxy)benzene 1a, (2.61 g, 8.00 mmol, 1 eq.) and a solution of 2,4-dimethyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline 2, (12.00 mmol, 1.5 eq.) in PhMe (48 ml) were sequentially added to a stirring solution of Pd(dppf)Cl2·CH2Cl2 (327 mg, 0.4 mmol, 5 mol%) and Na2CO3 (3.39 g, 32.00 mmol, 4 eq.) in PhMe (20 ml) and water (80 ml), under N2 at r.t. Argon was bubbled through the solution for 5 min, and the reaction mixture was heated to 95 °C. After 24 h, the reaction mixture was extracted (CH2Cl2, 3× 80 ml), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified by flash column chromatography (EtOAc:pentane, 7:93, Rf = 0.3). The crude product was suspended in 2-propanol (20 ml), and water (40 ml) was added. The solid precipitate was collected to give 3,3′,5,5′-tetramethyl-2’-(4-nitrophenoxy)-[1,1’-biphenyl]-2-amine 3a as a bright yellow solid (1.86 g, 64%).
1H NMR (600 MHz, CDCl3): δH = 8.04–7.96 (m, 2H, H11), 7.14–7.10 (m, 1H, H3), 7.05 (t, J = 1.5 Hz, 1H, H5), 6.75–6.72 (m, 1H, H16), 6.70 (m, 2H, H10), 6.57 (d, J = 2.1 Hz, 1H, H18), 2.37 (s, 3H, H8), 2.17 (s, 3H, H7), 2.11 (s, 3H, H19), 2.07 (s, 3H, H20). NH2 not observed.
13C NMR (151 MHz, CDCl3): δC = 163.1 (C9), 147.6 (C1), 141.9 (C12), 138.7 (br, C14), 136.2 (CAr), 132.7 (C6), 131.8 (C3), 131.4 (C16), 130.7 (CAr), 130.5 (C5), 128.7 (C18), 127.3 (br, CAr), 125.5 (C11), 123.2 (br, C13), 122.8 (br, CAr), 115.3 (C10), 20.9 (C8), 20.2 (C20), 17.8 (C19), 16.4 (C7).
Diazotization and intramolecular O-arylation
t-BuONO (2.23 ml, 1.94 g, 19.35 mmol, 5 eq.) was added to a solution of 3,3′,5,5′- tetramethyl-2’-(4-nitrophenoxy)-[1,1’-biphenyl]-2-amine 3a (1.40 g, 3.87 mmol, 1 eq.) and HBF4 (2.50 ml, 3.53 g, 19.35 mmol, 48% aq., 5 eq.) in CH2Cl2:IPA (1:1, 16 ml) at 0 °C. After 1 h, the reaction mixture was diluted (CH2Cl2, 20 ml), washed (H2O, 8 ml) and warmed to 30 °C. After 36–48 h, the solvent was removed by a steady stream of N2 and Et2O (20 ml) was added, resulting in a solid precipitate. The Et2O layer was passed through Celite® (4 ml). The solid precipitate was washed, and the solvent passed through Celite® (Et2O, 4× 4 ml). The solid was dissolved in MeCN (30 ml), passed through Celite® and eluted (MeCN, 4× 20 ml). The solvent was removed in vacuo at r.t. to give oxonium ion 4a as a tan solid (1.10 g, 65%).
1H NMR (500 MHz, CD3CN): δH = 8.49–8.42 (m, 2H, H10), 8.04–7.98 (m, 2H, H11), 7.93–7.89 (m, 2H, H5), 7.34–7.30 (m, 2H, H3), 2.51 (s, 6H, H8), 2.13 (s, 6H, H7).
13C NMR (126 MHz, CD3CN): δC = 162.3 (C9), 161.4 (C1), 150.4 (C12), 143.4 (CAr), 135.5 (C3), 128.5 (C10), 124.7 (CAr), 124.2 (CAr), 123.8 (C11), 122.7 (C5), 21.1 (C8), 16.7 (C7).
19F NMR (471 MHz, CD3CN): δF = −151.8.
Aryne formation and trapping with furan
K3PO4 (1.06 g, 5.00 mmol, 5 eq.) was added to a stirring (>600 rpm) solution of 2,4,6,8-tetramethyl-5-(4-nitrophenyl)-5H-dibenzo[b,d]furan-5-ium tetrafluoroborate 4a (433 mg, 1.00 mmol, 1 eq.) and furan (0.37 ml, 340 mg, 5.00 mmol, 5 eq.) in MeCN (6 ml). After 16 h, the reaction was filtered through Celite® (5 ml), eluted (CH2Cl2, 4× 10 ml) and concentrated in vacuo. The crude residue was purified by flash column chromatography (Et2O:pentane, 15:85, Rf = 0.3) to give 6-nitro-1,4-dihydro-1,4-epoxynaphthalene 5a as a white solid (189 mg, 99%).
1H NMR (400 MHz, CDCl3): δH = 8.04 (d, J = 2.0 Hz, 1H, H7), 7.98 (dd, J = 7.8, 2.0 Hz, 1H, H9), 7.36 (d, J = 7.8 Hz, 1H, H10), 7.10 (dd, J = 5.5, 1.9 Hz, 1H, H3/4), 7.05 (dd, J = 5.5, 1.9 Hz, 1H, H3/4), 5.83–5.78 (m, 2H, H2, H5).
13C NMR (126 MHz, CDCl3): δC = 156.6 (C1), 151.4 (C6), 145.9 (C8), 143.6 (C3/4), 142.5 (C3/4), 122.5 (C9), 120.2 (C10), 115.4 (C7), 82.2 (C2/5), 82.2 (C2/5).
Data availability
All data (experimental procedures, characterization data and cartesian coordinates for all density functional theory calculations) supporting the findings of this study are available within the article and its supplementary information. Crystallographic data for compounds 4a, 4j, 4k, 4l and 6 have been deposited with the Cambridge Crystallographic Data Centre under deposition numbers 2109776, 2109774, 2251371, 2109775 and 2251372, respectively. These data can be obtained free of charge from www.ccdc.cam.ac.uk/data_request/cif.
References
Wentrup, C. The benzyne story. Aust. J. Chem. 63, 979–986 (2010).
Takikawa, H., Nishii, A., Sakai, T. & Suzuki, K. Aryne-based strategy in the total synthesis of naturally occurring polycyclic compounds. Chem. Soc. Rev. 47, 8030–8056 (2018).
Bachmann, W. E. & Clarke, H. T. The mechanism of the Wurtz–Fittig reaction. J. Am. Chem. Soc. 49, 2089–2098 (1927).
Wittig, G. Phenyl-lithium, der schlüssel zu einer neuen chemie metallorganischer verbindungen. Naturwissenschaften 30, 696–703 (1942).
Roberts, J. D., Simmons, H. E., Carlsmith, L. A. & Vaughan, C. W. Rearrangement in the reaction of chlorobenzene-1-C14 with potassium amide. J. Am. Chem. Soc. 75, 3290–3291 (1953).
Yoshida, S. & Hosoya, T. The renaissance and bright future of synthetic aryne chemistry. Chem. Lett. 44, 1540–1560 (2015).
Ramírez, A., Bashore, C. G., Coe, J. W. & Collum, D. B. Formation of benzynes from 2,6-dihaloaryllithiums: mechanistic basis of the regioselectivity. J. Am. Chem. Soc. 126, 14700–14701 (2004).
Stiles, M., Miller, R. G. & Burckhard, U. Reactions of benzyne intermediates in non-basic media. J. Am. Chem. Soc. 85, 1792–1797 (1963).
Antonio, J., López, G. & Greaney, M. F. Use of 2-bromophenylboronic esters as benzyne precursors in the Pd-catalyzed synthesis of triphenylenes. Org. Lett. 16, 2338–2341 (2014).
Dong, C.-G. & Hu, Q.-S. Pd(OAc)2-Catalyzed domino reactions of 1-chloro-2-haloarenes and 2-haloaryl tosylates with hindered Grignard reagents via palladium-associated arynes. Org. Lett. 22, 5057–5060 (2006).
Miyawaki, K., Suzuki, R., Kawano, T. & Ueda, I. Cycloaromatization of a non-conjugated polyenyne system: synthesis of 5H-benzo[d]fluoreno[3,2-b]pyrans via diradicals generated from 1-[2-{4-(2-alkoxymethylphenyl)butan-1,3-dinyl}]phenylpentan-2,4-diyn-1-ols and trapping evidence for the 1,2-didehydrobenzene diradical. Tetrahedron Lett. 38, 3943–3946 (1997).
Holden, C. & Greaney, M. F. The hexadehydro-Diels–Alder reaction: a new chapter in aryne chemistry. Angew. Chem. Int. Ed. 53, 5746–5749 (2014).
Fleugel, L. L. & Hoye, T. R. Hexadehydro-Diels–Alder reaction: benzyne generation via cycloisomerization of tethered triynes. Chem. Rev. 121, 2413–2444 (2021).
Himeshima, Y., Sonoda, T. & Kobayashi, H. Fluoride-induced 1,2-elimination of o-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem. Lett. 12, 1211–1214 (1983).
Shi, J., Li, L. & Li, Y. O-Silylaryl triflates: a journey of Kobayashi aryne precursors. Chem. Rev. 121, 3892–4044 (2021).
Dubrovskiy, A. V., Markina, N. A. & Larock, R. C. Use of benzynes for the synthesis of heterocycles. Org. Biomol. Chem. 11, 191–218 (2013).
Idiris, F. I. M. & Jones, C. R. Recent advances in fluoride-free aryne generation from arene precursors. Org. Biomol. Chem. 15, 9044–9056 (2017).
García-López, J.-A. & Greaney, M. F. Synthesis of biaryls using aryne intermediates. Chem. Soc. Rev. 45, 6766–6798 (2016).
Kim, K. S., Ha, S. M., Kim, J. Y. & Kim, K. 5-Arylthianthreniumyl perchlorates as a benzyne precursor. J. Org. Chem. 64, 6483–6486 (1999).
Roberts, R. A., Metze, B. E., Nilova, A. & Stuart, D. R. Synthesis of arynes via formal dehydrogenation of arenes. J. Am. Chem. Soc. 145, 3306–3311 (2023).
Yoshimura, A., Saito, A. & Zhdankin, V. V. Iodonium salts as benzyne precursors. Chem. Eur. J. 24, 15156–15166 (2018).
Nilova, A. et al. Regioselective synthesis of 1,2,3,4-tetrasubstituted arenes by vicinal functionalization of arynes derived from aryl(mes)iodonium salts. Chem. Eur. J. 27, 7168–7175 (2021).
Metze, B. E., Roberts, R. A., Nilova, A. & Stuart, D. R. Deprotonation of arenes with weak base: assessing and expanding the functional group compatibility of aryne chemistry. Preprint at ChemRxiv https://doi.org/10.26434/chemrxiv-2023-bwkcd (2023).
Yuan, H., Yin, W. & Li, Y. 3-Sulfonyloxyaryl(mesityl)iodonium triflates as 1,2-benzdiyne precursors with activation via ortho-deprotonative elimination strategy. Nat. Commun. 14, 1841 (2023).
Lanze, M., Dherbassy, Q. & Wencel-Delord, J. Cyclic diaryl λ3-bromanes as original aryne precursors. Angew. Chem. Int. Ed. 60, 14852–14857 (2021).
Karandikhar, S. S. et al. Orbital analysis of bonding in diarylhalonium salts and relevance to periodic trends in structure and reactivity. Chem. Sci. 13, 6532–6540 (2022).
Lanzi, M., Rogge, T., Truong, T. S., Houk, K. N. & Wencel-Delord, J. Cyclic diaryl λ3-chloranes: reagents and their C–C and C–O couplings with phenols via aryne intermediates. J. Am. Chem. Soc. 145, 345–358 (2023).
Aggarwal, V. K., Harvey, J. N. & Robiette, R. On the importance of leaving group ability in reactions of ammonium, oxonium, phosphonium, and sulfonium ylides. Angew. Chem. Int. Ed. 44, 5468–5471 (2005).
Meerwein, H., Hinz, G., Hofmann, P., Kroning, E. & Pfeil, E. Über tertiäre oxoniumsalze I. J. Prakt. Chem. 147, 257–285 (1937).
Meerwein, H., Bettenberg, E., Gold, H., Pfeil, E. & Willfang, G. Über tertiäre oxoniumsalze II. J. Prakt. Chem. 154, 83–156 (1940).
Chan, H. S., Nguyen, Q. N. N., Paton, R. S. & Burton, J. W. Synthesis, characterization and reactivity of complex oxonium ions, proposed intermediates in natural product biosynthesis. J. Am. Chem. Soc. 141, 15951–15962 (2019).
Mascal, M., Hafezi, N., Meher, N. K. & Fettinger, J. C. Oxatriquinane and oxatriquinacene: extraordinary oxonium ions. J. Am. Chem. Soc. 130, 13532–13533 (2008).
Hellwinkel, D. & Seifert, H. Zur frage des pentakoordinierten Stickstoffs: reaktionen von (spiro)cyclischen tetraarylammonium-salzen mit nucleophilen. Justus Liebigs Ann. Chem. 762, 29–54 (1972).
Tolstaya, T. P., Tsariev, D. A. & Luzikov, Y. N. Reaction of triaryloxonium salts with bases via dehydroarenes. Tetrahedron Lett. 38, 4457–4458 (1997).
Nesmayanov, A. N. & Tolstaya, T. P. Salts of o, o’-diphenyloxonium ion. Russ. Chem. Bull. 8, 620–623 (1959).
Smith, O. et al. Control of stereogenic oxygen in a helically chiral soxonium ion. Nature 615, 430–435 (2023).
Lu, M. et al. Peraryl-X-onium ions of nitrogen and oxygen. Org. Chem. Front. 6, 2640–2646 (2019).
Gravel, D., Giasson, R. & Blanche, D. Photochemistry of the o-nitrobenzyl system in solution: effects of O-H distance and geometrical constraint on the hydrogen transfer mechanism in the excited state. Can. J. Chem. 69, 1193–1200 (1991).
Sapountzis, I., Lin, W., Fischer, M. & Knochel, P. Preparation of polyfunctional arynes via 2-magnesiated diaryl sulfonates. Angew. Chem. Int. Ed. 43, 4364–4366 (2004).
Hall, C., Henderson, J. L., Ernouf, G. & Greaney, M. F. Tandem thia–Fries rearrangement—cyclisation of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate benzyne precursors. Chem. Commun. 49, 7602–7604 (2013).
Goetz, A. E. et al. An efficient computational model to predict the synthetic utility of heterocyclic arynes. Angew. Chem. Int. Ed. 51, 2758–2762 (2012).
Kauffmann, T. & Wirthwein, R. Progress in the hetaryne field. Angew. Chem. Int. Ed. 10, 20–33 (1971).
Medina, J. M., Jackl, M. K., Susick, R. B. & Garg, N. K. Synthetic studies pertaining to the 2,3-pyridyne and 4,5-pyrimidyne. Tetrahedron 72, 3629–3634 (2016).
van der Plas, H. C., Smit, P. & Koudijs, A. Hetarynes XVI: evidence for the occurrence of 4-t-butyl-5,6-pyrimidyne in the amination of both 6-bromo- and 5-bromo-4-t-butylpyrimidine. Tetrahedron Lett. 9, 9–13 (1968).
Kauffman, T., Nürnberg, R. & Udluft, K. Hetarine, XIII. Cine-substitutionsreaktionen an 5-halogen-pyrimidinen mit EA- (über 4.5-dehydro-pyrimidin) und AEa-mechanismus. Chem. Ber. 102, 1177–1190 (1969).
Christophe, D., Promel, R. & Maeck, M. Trapping of a 4,5-didehydropyrimidine with furan. Tetrahedron Lett. 45, 4435–4438 (1978).
Tiblemans, M. et al. Reactivity of 2-t-butyl-4,5-didehydropyrimidine and electronic structure of the parent hetaryne. Tetrahedron 48, 10575–10586 (1992).
Walters, M. A., Carter, P. H. & Banerjee, S. A practical 2,3-pyridyne precursor. Synth. Commun. 22, 2829–2837 (1992).
Medina, J. M., Mackey, J. L., Garg, N. K. & Houk, K. N. The role of aryne distortions, steric effects, and charges in regioselectivities of aryne reactions. J. Am. Chem. Soc. 136, 15798–15805 (2014).
Walter, H., Tobler, H., Gribkov, D. & Corsi, C. Sedaxane, Isopyrazam and Solatenol™: novel broad-spectrum fungicides inhibiting succinate dehydrogenase (SDH)—synthesis challenges and biological aspects. Chimia 69, 425–434 (2015).
Criado, A., Peña, D., Cobas, A. & Guitián, E. Domino Diels–Alder cycloadditions of arynes: new approach to elusive perylene derivatives. Chem. Eur. J. 16, 9736–9740 (2010).
Anderson, D. R., Faibish, N. C. & Beak, P. Complex-induced proximity effects in directed lithiations: analysis of intra- and intermolecular kinetic isotope effects in directed aryl and benzylic lithiations. J. Am. Chem. Soc. 121, 7553–7558 (1999).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
Hehre, W. J., Ditchfield, R. & Pople, J. A. Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular-orbital studies of organic-molecules. J. Chem. Phys. 56, 2257–2261 (1972).
Hariharan, P. C. & Pople, J. A. Influence of polarization functions on molecular-orbital hydrogenation energies. Theor. Chem. Acc. 28, 213–222 (1973).
Zhang, J. & Hoye, T. R. Divergent reactivity during the trapping of benzynes by glycidolanalogs: ring cleavage via pinacol-like rearrangements vs oxirane fragmentations. Org. Lett. 21, 2615–2619 (2019).
Arora, S., Zhang, J., Pogula, V. & Hoye, T. R. Reactions of thermally generated benzynes with six-membered N-heteroaromatics: pathway and product diversity. Chem. Sci. 10, 9069–9076 (2019).
Kitamura, T. et al. New and efficient hypervalent iodine-benzyne precursor, (phenyl)[O-(trimethylsilyl)phenyl]iodonium triflate: generation, trapping reaction, and nature of benzyne. J. Am. Chem. Soc. 121, 11674–11679 (1999).
Plata, R. E. & Singleton, D. A. A case study of the mechanism of alcohol-mediated Morita Baylis−Hillman reactions. The importance of experimental observations. J. Am. Chem. Soc. 137, 3811–3826 (2015).
Acknowledgements
The Engineering and Physical Sciences Research Council has provided financial support for studentships: to O.S. via the Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/ L015838/1) and to M.H. via a doctoral training partnership studentship (EP/T517811/1; project 2446204). R.S.P. acknowledges support from the National Science Foundation (CHE-1955876) and computational resources from the Rocky Mountain Advanced Computer Consortium Summit supercomputer supported by the National Science Foundation (ACI-1532235 and ACI-1532236), the University of Colorado Boulder and Colorado State University, and the Extreme Science and Engineering Discovery Environment through allocation TG-CHE180056. A CC-BY licence is applied to the author accepted manuscript arising from this submission, in accordance with Engineering and Physical Sciences Research Council’s open access conditions.
Author information
Authors and Affiliations
Contributions
O.S., M.J.H., R.S.P., J.W.B. and M.D.S. conceived and designed the study. O.S., V.T. and M.J.H. performed the synthetic experiments and analysed data for all compounds. O.S. performed the X-ray crystallography. A.S. and R.S.P. performed the computational study. O.S., M.J.H., A.S., R.S.P., J.W.B. and M.D.S. co-wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Torben Rogge and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Peter Seavill, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental details, Computational details, Sections 1–8, Figs. 1–3 and Tables 1–4.
Supplementary Data 1
Crystallographic data for compound 4a, CCDC 2109776.
Supplementary Data 2
Structure factors for compound 4a, CCDC 2109776.
Supplementary Data 3
Crystallographic data for compound 4j, CCDC 2109774.
Supplementary Data 4
Structure factors for compound 4j, CCDC 2109774.
Supplementary Data 5
Crystallographic data for compound 4k, CCDC 2251371.
Supplementary Data 6
Structure factors for compound 4k, CCDC 2251371.
Supplementary Data 7
Crystallographic data for compound 4l, CCDC 2109775.
Supplementary Data 8
Structure factors for compound 4l, CCDC 2109775.
Supplementary Data 9
Crystallographic data for compound 6, CCDC 2251372.
Supplementary Data 10
Structure factors for compound 6, CCDC 2251372.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smith, O., Hindson, M.J., Sreenithya, A. et al. Harnessing triaryloxonium ions for aryne generation. Nat. Synth 3, 58–66 (2024). https://doi.org/10.1038/s44160-023-00408-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44160-023-00408-1
This article is cited by
-
Oxonium ions for easy aryne chemistry
Nature Synthesis (2023)
