
Abstract
Bacillus species are ubiquitous in nature and have tremendous application potential in agriculture, medicine, and industry. However, the individual species of this genus vary widely in both ecological niches and functional phenotypes, which, hence, requires accurate classification of these bacteria when selecting them for specific purposes. Although analysis of the 16S rRNA gene has been widely used to disseminate the taxonomy of most bacterial species, this gene fails proper classification of Bacillus species. To circumvent this restriction, we designed novel primers and optimized them to allow exact species resolution of Bacillus species in both synthetic and natural communities using high-throughput amplicon sequencing. The primers designed for the tuf gene were not only specific for the Bacillus genus but also sufficiently discriminated species both in silico and in vitro in a mixture of 11 distinct Bacillus species. Investigating the primers using a natural soil sample, 13 dominant species were detected including Bacillus badius, Bacillus velezensis, and Bacillus mycoides as primary members, neither of which could be distinguished with 16S rRNA sequencing. In conclusion, a set of high-throughput primers were developed which allows unprecedented species-level identification of Bacillus species and aids the description of the ecological distribution of Bacilli in various natural environment.
Similar content being viewed by others

Introduction
The Bacillus genus is a prolific and diverse prokaryotic genus consisting of more than 100 species with validly published names, widely distributed in soil, sediment, air, marine environment, and even human systems [1, 2]. Members of the Bacillus genus comprise important species with economic, medical, and sustainability values as well as pathogenic strains. As an example, the Bacillus cereus sensu lato (s.l.) group includes the human pathogen Bacillus anthracis, the food poisoning agent B. cereus, and insect biopesticide Bacillus thuringiensis [3]. On the other hand, other members, such as Bacillus subtilis, Bacillus amyloliquefaciens, and Bacillus velezensis are widely used as biological control agents for both plants and animals due to traits such as spore-forming ability, high efficiency in plant root colonization, and abundant secondary metabolite production [4,5,6,7]. Additionally, B. subtilis has been a major cell and molecular biology model organism for decades [8]. This species has been extensively used for understanding bacteria biofilm formation, industrial production of enzymes and probiotics, and recently, as a proxy demonstrating phage-encoded biosynthetic gene clusters, and a non-photosynthetic bacteria that entrained circadian rhythm [9,10,11].
Bacillus as one of the most extensively studied plant growth promoting rhizobacteria (PGPR), it can competitively colonize plant roots and act as biofungicides, biofertilizers or biopesticides [12]. Despite the fact that members of Bacillus have been used as biological control agents for decades, classification and phylogenetic organization have been elusive for years [13, 14]. A current study demonstrated most Bacillus spp. registered as plant pathogen crop protection products have inconsistent species names in respect to current nomenclature [15]. One factor that caused the extensive polyphyly and misleading classification is the application of loose morphological criteria for assigning distinct species according to their cell shape and the ability to form spores [16]. Furthermore, as a conventional method to inspect the taxonomy of new isolates, members of the Bacillus genus commonly carry multiple copies of the 16S rRNA operon and these not only diverge widely within genomes, but also overlap extensively across different species, making it impossible to use 16S rRNA sequencing for species delineation [17]. Moreover, overall genetic differences are poorly correlated with phenotypic traits, and have even highlighted distinct phenotypes and genetic traits in strains with identical 16S rRNA alleles [18]. Given the wide variety of Bacillus species, specifically considering their role as both pathogens and biocontrol agents, it would appear urgent to develop novel approaches to alleviate the shortcomings of current methods.
To improve precise species-level identification of Bacilli, several alternative loci on the genome have been tested as phylogenetic discriminators of Bacillus species, including genes encoding gyrase subunit A (gyrA), the gyrase subunit B (gyrB), the RNA polymerase beta (rpoB), and elongation factor thermal unstable Tu (tuf) [19,20,21,22,23,24,25,26,27,28]. Caamaño-Antelo et al. isolated 20 foodborne Bacillus strains and analyzed the usefulness of three housekeeping genes, tuf, gyrB, and rpoB in terms of their discriminatory power. The tuf gene exhibited the highest interspecies similarities with sufficient conserved regions for primer matching across species whilst also containing enough variable regions for species differentiation [22]. Another study combined pulse field gel electrophoresis with tuf identification to successfully genotype Bacillus isolates from various environments [27]. Altogether, these studies suggested tuf as a potential phylogenetic marker among Bacillus species, and this gene has moreover been used for similar purposes in other genera [29].
In recent years, microbiologists have shifted their focus from single cultures to more complex microbial communities. This shift has come about partly as a reflection of the natural lifestyle of most bacteria, but more importantly, from observing widely different profiles of multi-cultured bacteria relative to their single-cultured counterparts. Diverse members of Bacillus are involved in ecosystem functions including the degradation of soil organic matter, nitrogen cycle, carbon cycle, phosphorus solubilization, and eco-remediation of pollutants [8]. They also jointly function as plant growth promoters to alleviate abiotic stress or suppress plant pathogens [30,31,32]. In such scenarios, merely identifying individual taxa as Bacillus rather than individual species be insufficient for both basic science and potential microbiome engineering. Therefore, Bacillus community composition needs to be described in these complex settings. However, such analysis relies either on culture-dependent and laborious approaches or resource-extensive metagenomics which is unfeasible for high-throughput analysis. More commonly, amplicon sequencing is used to infer microbial community composition culture-independently and cost-efficiently [33]. Although amplicon analysis of the 16S rRNA gene has been remarkably successful owing to universality in bacteria, this method does not allow confident species identification not only in Bacillus genus but has low resolvability in other medically important genera, e.g., Staphylococcus and Pseudomonas. Thus, alternative molecular markers including cpn60, rpoB, and gyrB have been developed for universal bacterial genotypic identification, and whilst these genes have high discriminatory power, some remain taxa specific [34,35,36]. For instance, gyrB reveals highly similar bacterial community structure within Proteobacteria and Actinobacteria when compared with 16S rRNA, but it has merely 21.5% consistency in Firmicutes [34]. Therefore, amplicon sequence tools have been already developed for few genera that enable species-level resolution, such as rpoD amplicon methods for Pseudomonas and tuf-based methodology for Staphylococcus genus [29, 37].
Since differentiation of Bacillus in mixed communities is highly relevant, but impossible with standard 16S rRNA sequencing, we developed primers for species level differentiation. Specifically, we investigate conserved genes and their corresponding primers for Bacillus species identification using an in-silico amplification approach using Bacillus versus non-Bacillus genomes. Subsequently, tuf gene specific primer pairs were designed and inspected leading to a primer pair with high accuracy and specificity on Bacillus species. Finally, an amplicon sequencing method was tested on an Illumina MiSeq PE300 platform based on the selected primers, along with a customized database for Bacillus taxonomic assignment. The identified tuf2 primers demonstrated almost full coverage of Bacillus species along with discriminatory power approaching whole genome phylogeny. Moreover, the tuf2-based amplicon approach allowed Bacillus profiling in natural communities, which we believe will facilitate the study of potential contributions of Bacilli in relevant ecosystem functions or large-scale exploration of bio-potential Bacillus species.
Material and methods
Primer design
In response to the insufficient availability of differentiating primer pairs for Bacillus genus, an exhaustive search for Bacillus conserved genes was conducted to uncover genes with high phylogenetic discrimination power as candidate targets for primer design. We chose housekeeping genes that were frequently employed in literature and prioritized rpoB, gyrA, and tuf as our candidate genes to design primers. To evaluate the breadth of coverage for these primers, 1149 complete genome sequences of Bacillus were downloaded using ncbi-genome-download with RibDif and added to our Bacillus genome collection (listed in Dataset S1). A few Bacillus genomes were re-identified with TYGS due to the poor annotations of uploaded genomes to improve the phylogenetic inference accuracy. Pan-genome analyses were carried out using roary, showing these three genes (rpoB, gyrA, and tuf) are indeed core genes (presented in >99% of the strains in our Bacillus genome collection) [38, 39]. Meanwhile, previously documented primers targeted on these genes have high amplification rate against the Bacillus genome.
Next, sequences of candidate genes were then dereplicated with vsearch followed by sequence alignments using MUSCLE v5 [40, 41]. Analysis of multiple sequence alignments was conducted to target conserved regions flanking highly variable regions of 300–600 bp (https://github.com/mikaells/MSA-primers). Potential primers were suggested from these conserved regions. Out of several preliminary primer designed, the primer pair tuf1-F (5’-CACGTTGACCAYGGTAAAACH-3’), tuf1-R (5’-DGCTTTHARDGCAGADCCBTT-3’) and tuf2-F (5’-AVGGHTCTGCHYTDAAAGC-3’), tuf2-R (5’-GTDAYRTCHGWWGTACGGA-3’) targeting a 500 bp sequence of tuf gene showed the top performance characteristic in initial examination and was selected for further evaluation.
In silico evaluation of primers
Initially, RibDif was used to evaluate the usefulness of standard primers for taxonomy, which specifically means primers targeting the V3V4 and V1V9 region of the 16S rRNA gene in bacteria [17]. As this analysis clearly showed the inability of these standard primers in terms of resolving the individual species of Bacillus, we investigated 11 primer sets targeting the genes of 16S rRNA, gyrB, gyrA, and rpoB derived from previous studies along with our two newly designed sets of tuf primers. We used RibDif to download all completed genomes of the Bacillus genus and then evaluated the performance of these primers on this collection through in silico PCR (https://github.com/egonozer/in_silico_pcr).
We followed the standards as described by Lauritsen et al. to benchmark the performance of primers according to two metrics, both of which should encapsulate the functional performance in mixed communities [37]. Specifically, (1) what is the proportion of Bacillus genome amplified and (2) what is the proportion of non-Bacillus genome amplified. Furthermore, the top candidate primers were further evaluated by building phylogenetic trees from the alignments of their resulting amplicons. These phylogenetic trees were inferred using neighbor-joining (NJ) method with the Maximum Composite Likelihood model, and 1000 bootstraps where used to test the strengths of the internal branches of the trees. Trees were visualized in iTol (https://itol.embl.de/). TreeCluster were used to define clusters within the trees and these clusters where then compared to the known taxonomy of the amplicons [42]. Cohens Kappa was calculated with the R package “irr” to infer the degree of agreement between TreeCluster and the known species names [43, 44].
Whole genome sequencing
A short and long read hybrid approach was used to sequence new Bacillus isolates obtained from an ongoing project in our laboratory. Bacterial genomic DNA was extracted using E.Z.N.A.® Bacterial DNA Kit (Omega, Bio-tek, USA, Georgia). The qualities and quantities were evaluated by NanoDrop DS-11+ Spectrophotometer (Saveen Werner, Sweden, Limhamn) and Qubit 2.0 Fluorometer (Thermo Fisher Scientific). The libraries for short-reads sequencing were constructed using the MGI paired-end protocol [45]. Briefly, 300 ng DNA was fragmented to 200–300 bp using segmentase enzyme followed by fragment selection with VAHTS™ DNA Clean Beads (Vazyme; China, Nanjing). Subsequently, end repair, A-tailing reactions and adapter ligation were implemented. After PCR and purification, the libraries were sequenced on the MGISEQ-2000 (MGI Tech Co., Ltd.) platform according to the manufacturer’s instructions to generate 2 × 150 bps paired end reads. For Nanopore sequencing, the rapid barcoding kit (SQK-RBK110.96) was used and these libraries were sequenced with an R9.4.1 flow cell on a MinION device running a 48-h sequencing cycle. The resulting reads were base called and demultiplexed with MinKNOW UI v.4.1.22. For de novo assembly, the NGS short reads were adapter and quality trimmed using fastp v.0.22.0 and the Nanopore reads were adapter trimmed using porechop v.0.2.1 [46, 47]. The trimmed reads from Nanopore were assembled using flye v.2.9.1-b1780, and subsequently the trimmed reads from both platforms and the long read assembly were hybrid assembled with Unicycler v.0.5.0 using the –existing_long_read_assembly option [48, 49]. The completeness and contamination levels of each strain was checked using CheckM v.1.2.2 [50]. The assemblies were then taxonomically assigned and placed in the full-genome, multi locus GTDB-Tk reference tree, using the Classify Workflow of GTDB-Tk v2.1.1 [51]. The tree was subsequently pruned to create a full-genome multi locus tree of the query strains. The chromosomes were annotated using the NCBI Prokaryotic Genome Annotation Pipeline [52].
Comparative identification of Bacillus isolates
Since the primers targeting the tuf gene appeared superior for Bacillus differentiation, we investigated these in detail. Initially, we assessed the accuracy of the tuf primers by comparing the resolving power with other means of Bacillus identification methods, such as 16S rRNA PCR and whole genome sequencing. Complete 16S rRNA genes were PCR-amplified with primer pair 27 F (5’-AGAGTTTGATCCTGGCTCAG-3’) and 1492 R (5’-GGTTACCTTGTTACGACTT-3’) [53]. A 25 µl PCR mixture contains: 2.5 µl 2 mM dNTP, 2.5 µl 10 × DreamTaq Buffer, 0.5 µl of each primer (10 µmol/l), 0.25 µl DreamTaq DNA Polymerase (5 U/µl), 17.5 µl nuclease-free water, and 1.25 µl DNA template. Thermal cycling conditions were 95 °C for 3 min; 30 cycles of 30 s denaturation at 95 °C, 30 s annealing at 55 °C, and 1 min extension at 72 °C; final extension at 72 °C for 10 min. PCR product were purified using NucleoSpin gel and PCR cleanup kit (Macherey-Nagel; Germany, Düren) and sent for Sanger sequencing Eurofins Genomics.
Evaluation of primer performance on amplicon sequencing
We chose 11 distinct Bacillus strains to create a synthetic community to evaluate the in vitro performance of the tuf primers on species resolution through amplicon sequencing. Bacillus thuringiensis 407 cry- (NCBI accession number GCF_000306745.1), Bacillus velezensis SQR9 (CGMCC accession number 5808), Bacillus cereus ATCC 14579 (NCBI accession number GCF_000007825.1), and Bacillus subtilis PS216 (NCBI accession number GCF_000385985.1) were type culture collection strains [54,55,56,57]. The rest seven isolates were obtained from ongoing projects in our laboratory and identified by whole genome sequencing. All bacteria were grown in lysogeny broth (LB; Lennox, Carl Roth, Germany, Limhamn) overnight, and supplemented with 28% glycerol before storing them at −80 °C. DNA extractions of each strain were pooled in equimolar ratio to create a positive control mixture (Bac-DNAmix). To benchmark the performance of tuf2 on amplicon sequencing, primer pairs gyrA3 that were previously applied on Bacillus mock community (gyrA3-F: 5′-GCDGCHGCNATGCGTTAYAC-3′ and gyrA3-R: 5′-ACAAGMTCWGCKATTTTTTC-3′) and universal primers targeting the V3–V4 hypervariable region of the 16S rRNA gene (341 F: 5’-CCTACGGGNGGCWGCAG-3’ and 805 R: 5’-GACTACHVGGGTATCTAATCC-3’) were selected [20, 53]. Short barcodes were attached on all primers for downstream sequence demultiplication as is listed in Table S1.
For tuf2 amplification, a 25 µl PCR mixture contains: 12.5 µl TEMPase Hot Start 2 × Master Mix Blue, 0.8 µl of each primer (10 µmol L−1), 10.6 µl nuclease-free water and 0.3 µl DNA template. The PCR program included initial denaturation for 15 min at 95 °C; 30 cycles of 30 s at 95 °C, 30 s at 47 °C and 1 min at 72 °C; and a final extension for 5 min at 72 °C. For the amplification of 16S rRNA and gyrA, the annealing temperatures were 62 °C and 50 °C, respectively. All PCR products were purified and pooled into equimolar ratios and sequenced on a MiSeq platform using the MiSeq Reagent Kit v3(600-cycle).
Amplicon data analysis
Raw sequence data was processed with the QIIME2 pipeline for all primer sets [58]. Primers and barcodes were removed with cutadapt, and after demultiplexing, amplicons were denoised, merged, and chimera-checked using DADA2 [58, 59]. In Bacillus DNA mixture, all ASVs were assigned to NCBI database for parallel comparison to avoid bias. For natural soil sample, 16S rRNA data were analyzed using standard workflow SILVA database and Naive Bayers classifier [60]. A tuf-specific database was built from all tuf genes of our Bacillus genome collection (available at https://github.com/Xinming9606/BAST). The tuf amplicons of each sample where then taxonomically assigned using BLASTN against the tuf-database, using max_target_seq: 1. was used to assign taxonomy to the representative sequences of amplicon data with the best hits selected as taxonomy names.
Results
Comparative analysis of primer pairs for Bacillus identification
A battery of six genes and primers widely used for Bacillus identification were compared using in silico PCR. Primers were tested against a Bacillus genome collection including 1149 complete genomes downloaded from NCBI in April 2023, along with 41 non-Bacillus genomes corresponding to other well-studied microbes often found in soil (listed in Dataset S1 and Table S2).
Initially, the performance of the normally used primers targeting the 16S rRNA gene was examined to provide a baseline and motivation for our investigation. As they are designed for, the universal primer sets targeting the full (V1V9) and partial (V3V4) parts of the 16S rRNA gene successfully amplified both Bacillus and non-Bacillus strains (Table 1). Using the RibDif2 tool for a more detailed analysis, however, revealed that Bacillus has an exceptionally high allele multiplicity and extensive species overlap, which means that amplicons derived from 16S rRNA gene will rarely be unique for individual species (Table 1 & Fig. 1A) [61]. For example, full-length V1V9 amplicons derived from B. subtilis are indistinguishable from amplicons derived from B. velezensis, B. siamensis and B. amyloliquefaciens. Thus, 16S rRNA gene it is not an ideal molecular marker for the Bacillus genus [18]. The B. subtilis group specific primers Bsub5F and Bsub3R reportedly can identify B. subtilis group exclusively, including species B. subtilis, Bacillus pumilus, Bacillus atrophaeus, Bacillus licheniformis and B. amyloliquefaciens. This primer set had hits on 797 Bacillus genomes out of the 1149 (69.36%), and did not amplify non-Bacillus genomes. Despite a high level of specificity, the lack of broad coverage in these amplified sequences will not provide species-level identification in diverse communities.

A 16S rRNA V3–V4 region Bacillus genome overlap. B tuf2 genome overlap. Nodes represented Bacillus genomes and were connected if genomes have overlapping alleles. Node color described different Bacillus species. Non-connected nodes are excluded. Edge width is proportion to the number of shared alleles.
Next, we investigate primer pairs targeting housekeeping genes. Previous studies reported that the partial sequence of gyrase subunit A sharply separated twelve strains belonging to B. amyloliquefaciens, B. atrophaeus, B. licheniformis, B. mojavensis, B. subtilis and B. vallismortis [62]. Liu and colleagues have also reported their primer pair gyrA3 distinguished six species in the mock community they constructed. During our examination, the gyrA-based primer pairs displayed high specificity for the Bacillus genus with no amplification of species from the other genera. However, the primers targeting the gyrA gene amplified only 39% (450/1149) and 18% (212/1149) of the genomes, using gyrA3-F - gyrA3-R and the gyrA-42f - gyrA-1066r primer combinations, respectively, suggesting lack of discriminatory potential among certain clades within the Bacillus genus.
Similarly, the primer pair targeting a 809-nucleotide region of the rpoB gene displayed limited detection with 26% (297/1149) amplification rate by primers rpoB-fw and rpoB-rev. Surprisingly, some of the published primer sets which claimed to drastically increase resolution and/or discrimination displayed no amplification of Bacillus genomes at all, which was especially evident for gyrB-based primers, such as the BS-F - BS-R and the UP-1S - UP-2Sr sets that were designed for B. cereus group identification. In contrast, the primer pair tufGPF and tufGPR, targeting the tuf gene displayed high amplification rate, although the amplicon generated (791 bp) are longer than the Illumina technology currently supports for overlap, and the resulting sequencing gap complicates analysis and may decrease the resolution of species identification.
Motivated by the lack of generally applicable and sufficiently differentiating primer pairs for the Bacillus genus, we designed primer pairs targeted on tuf gene. Two sets were investigated, since 4 conserved regions within the tuf gene alignment were available position 58, 517 to 518, and 958 to 1004. Thus, primer pairs where likely to be at 58 to 517 and position 517 to 1004 (Fig. S1B). Profiling the tuf gene for nucleotide diversity showed that these conserved regions flanked variable regions of high nucleotide diversity which may potentially allow species identification (Fig. S1A). In total, 18 tuf-based primer pairs were suggested (Dataset S2). According to the lowest number of degenerate sites and substitutions within the primer sequences, two sets of primers were selected for further evaluation. Both of these primers, now referred to as tuf1 and tuf2, had substantially better performances than the ones hitherto tested: it had the highest coverage at close to 100% amplification rate, along with a notable 0% rate of non-Bacillus amplification from the genomes of the negative controls. Additionally, network visualization of genome overlap provided by RibDif2 revealed that compared to 16Sr RNA and gyrA (Fig. 1A and Fig. S2), tuf2 produces amplicons completely resolving individual members of Bacillus with limited overlap of alleles with other species (Fig. 1B). From these results, both primer sets targeting the tuf gene were selected for further analysis.
Of note, the predicted primer pairs for rpoB and gyrA loci had high number of degenerate sites and the most conserved fragments in the alignments remained highly diverse. For example, only two primer pairs between position 1549 and position 1927 were suggested for rpoB alignment, with more than 10 sequences incorporating three degenerate nucleotides (Fig. S3). Thus, primer design is challenging on such non-conserved fragments.
Phylogenetic analysis of Bacillus species based on different sequence approaches
To evaluate the discrimination resolution of the selected tuf primers, phylogenetic trees of their in silico PCR amplicons alignments were created for both along with the amplicons generated with 16S rRNA V3–V4 primers as comparison (Fig. 2). Two approaches were applied to analyze the phylotaxonomic distribution obtained using the tuf1, tuf2 and 16S rRNA gene amplicons: neighbor-joining trees were used to show the phylogenetic distribution of the amplicons and the TreeCluster program was then used to cluster the amplicons on the basis of their position in these trees. The reasoning for this was to compare the phylogenetic positioning with known taxonomy, which consequently reveals how well these amplicons can group their parent genome correctly. The phylogenetic tree of tuf2 amplicons grouped the amplicons in correspondence with the published species names of their parent genomes. Divergent clades were interspersed by different species with the main division of subtilis clade and cereus clade. Few branches displayed inadequate separation of nodes, although this was mainly due to poor annotation or species misnaming, e.g., certain B. velezensis isolates were originally proposed as B. amyloliquefaciens. The inter-rater reliability analysis using Cohen’s kappa showed a substantial agreement (kappa = 0.721, z = 35.6, p < 0.001) between known species names and tuf2 amplicon clusters. The tree of the tuf1 amplicons performed substantially worse in this regard, having much less systematic clustering of each taxa (kappa = 0.326, z = 21.9, p < 0.001). Thus, we do not recommend tuf1 primers for Bacillus identification. As expected, the tree based on 16S rRNA genes exhibited worse intraspecific phylogenetic resolution than tuf2, such as failing to delineate the distinct groups within B. cereus, B. anthracis, and B. thuringiensis. Moreover, TreeCluster annotation revealed distinct species having identical 16S rRNA V3–V4 sequences as well as multiple instances of one genome having 16S rRNA alleles in several clades of the tree (kappa = 0.63, z = 26.9, p < 0.001). Noteworthy, 60% of Bacillus genomes exhibit multiple alleles and V3V4 sequences were extensively dereplicated for tree construction.

A 16 S rRNA, (B) tuf1, (C) tuf2. Species names were annotated by whole genome sequencing. The outer ring was colored based on published species names and circles on the tips denoted these species have identical amplicons. Alkalihalobacillus clausii was used as the outgroup to root the tree.
To validate the accuracy of the tuf2 primer pairs in vitro, we compared the phylogenetic affiliation between amplicons of 16S rRNA, tuf2, and complete genomes of twenty soil-derived isolates (Fig. 3). The same genomic DNAs were used for Sanger sequencing and whole genome sequencing. The tuf2-based tree clearly delineated seven distinct clusters with high bootstrap values and in good agreement with the tree structure depicted based on the whole genomes. For instance, tuf2-tree grouped environmental strains AQ13, D8_B_37, G1S1 etc. in the Peribacillus cluster as expected since the sequence identity with Peribacillus simplex was >98% shown on NCBI-blast (Table S3). Similarly, isolates were accurately grouped that belong to B. subtilis, B. licheniformis, B. velezensis, although it demonstrated lower resolution on the identification of Bacillus altitudinis, B. pumilus and B. safensis. The 16S rRNA gene-based tree lacked concordance with full genome-based phylotaxonomics and led to an unreliable phylogenetic signal due to the prevalence of multiple copies of 16S rRNA in Bacilli in addition to the high genetic similarity of 16S rRNA genes between these species. It is worth mentioning that in vitro assay tuf2 primers did not exhibit any unspecific amplification of negative control Pseudomonas aeruginosa, Streptomyces iranensis, and Clavibacter michiganensis in line with expectations from in silico tests (Fig. S4).

A Phylogenetic tree constructed by partial tuf gene, (B) complete genome, and (C) 16S rRNA gene. Bacillus spp. were recently isolated or strains with publicly available genomes in NCBI GenBank was obtained using the Neighbor-Joining method. Numbers depicted on the branches indicate bootstrap values.
In summary, the tuf2 primers developed in this study is an effective tool to identify the species-level taxonomy within the Bacillus genus with high phylogenetic discrimination power comparable to the methods based on complete genome.
Amplicon sequencing of Bacillus synthetic community
Next, we investigated whether the tuf2 primer pairs could be applied for high-throughput amplicon sequencing. To evaluate the specificity and efficiency of tuf2, we compared these with the frequently employed 16S rRNA primers (V3–V4) and a newly published primer set (gyrA3) that was suggested to have the potential for Illumina sequencing of complex Bacillus community [20, 53]. A defined DNA mixture containing 11 Bacillus species was assembled and sequenced by the three sets of primers.
As expected, 16S rRNA V3V4 amplicon sequencing performed poorly, only identifying 4 out of 11 species and instead overestimating species in the B. subtilis group or the B. cereus group, resulting in B. cereus abundance being highly overinflated. Within the subtilis clade, B. velezensis was three-fold larger than expected. Apart from the 16S rRNA gene-based identification only provided correct detection of four species, this method instead inaccurately reported Priestia aryabhattai within the sample composition which was not added to the DNA mixture (Fig. 4). The approach with the primer pairs of gyrA3 locus resolved 8 species, including B. altitudinis, B. licheniformis, B. velezensis that were previously validated during development of these primer pair, but largely overestimated the proportion of B. pumilus and B. safensis (Table 2). In comparison, the tuf2 amplicon method was able to identify nine strains but missed B. amyloliquefaciens and B. cereus, other species have closer correspondence to expected abundance and lower deviation. These data suggest not only that tuf specific primers can reveal molecular variation at species level, but also complements and potentially outperforms 16S rRNA amplicon sequencing in complex Bacillus community studies. Noteworthy, all amplicons were assigned to NCBI database for parallel comparison which includes partial sequences and incorrect information, thus, mapping to our customized tuf database would highly improve the accurateness of results.

The first bar represents the theoretical abundances in the Bacillus DNA mixture followed by abundances detected using the V3–V4 16S rRNA, gyrA, and tuf 2 sequencing approaches, respectively.
Profiling Bacillus species in natural soil sample
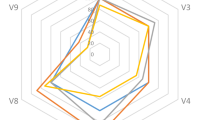
To elucidate the feasibility of the newly developed primer pairs to dissect the composition of the Bacillus genus in natural soil samples, we used the tuf2 primers, since they performed best in our analysis, and compared it with V3V4 sequencing. Using V3–V4 16S rRNA gene-based amplification, 53,517 reads were mapped for classification per sample on average, from which 99% of the reads were unidentifiable on the species level. A total of 741 families were found, with the dominant families belonging to Xanthobacteraceae (9%), Chthoniobacteraceae (3.2%), Isosphaeraceae (3.8%), Bacillaceae (6.1%). Out of 70 Bacillus ASVs, only 2 were annotated as Bacillus sp. and B. simplex in the environmental samples. It is conceivable to improve the species designation by assigning the sequence reads a defined Bacillus database, however the issue of heterogeneity and multiplicity of the 16S rRNA gene still remain. For tuf2 sequencing, after filtering, denoising and chimera removing, an average of 28,231 reads per sample were available. Rarefaction curve showed saturated sequencing depth for all samples with 601 ASVs assigned (Fig. S5). With these primers, species level composition could be achieved, showing the predominant species in this soil sample to be B. badius, Bacillus dafuensis, Bacillus infantis, and Bacillus weihaiensis, which presumably can be considered the correct composition of the fraction classified as Bacillus in the 16S rRNA analysis (Fig. 5). B. velezensis, B. mycoides, and B. amyloliquefaciens were also detected in natural soil with lower abundance. Our results demonstrated the ability of tuf2 as a complementary analysis when employing 16S rRNA analysis for specifically profiling Bacillus species in natural soil. As an example, one would infer that since ~10% of Bacillus is B. velezensis and ~5% of overall bacteria is Bacillus, the total abundance of B. velezensis is ~0.5%.

A Relative abundance of the most abundant bacterial families, and (B) relative abundance of the most abundant Bacillus species in natural soil sample collected from Dyrehaven (55.791000, 12.566300).
Discussion
The Bacillus genus is one of the most predominant bacterial genera in soil and exerts fundamental roles in soil ecology (i.e., the cycling of soil organic matter) and in plant growth promotion (e.g., nitrogen fixation, nutrient acquisition) [8, 63, 64]. Nevertheless, in nature, rather than existing as a solitary microbiome comprising a single species, Bacillus spp. exist as a part of complex microbial communities and therefore may execute ecosystem functions by diverse species. Mandic-Mulec et al. highlighted the urgent need to develop primer sets to estimate the contributions of Bacillaceae members to specific relevant ecosystem functions [8]. Meanwhile, sustainable agriculture has boosted the demand for employing biological agents rather than chemical ones, and members of the Bacillus genus has come into focus as a joint PGPR group. Altogether, methods that can accurately identify Bacillus spp. and systematically profile Bacillus communities in a natural soil has now become of vital importance.
Here, we designed and scrutinized primer pairs that can dissect the Bacillus genus on species level. Moreover, a corresponding bioinformatic pipeline has been employed that allows simple analysis of Illumina Miseq300 platform-based data on QIIME2 enabling rapid identification and selection of soils with specific Bacillus communities for further analysis and culturing.
Previous studies have designed and used Bacillus-specific primer sets targeting non-universal regions of the 16S rRNA gene for rapid taxonomic identification, and alternative biomarkers rpoB, gyrB, and gyrA have been proposed to resolve the limited intra-specificity as well. However, use of these genes as universal markers in Bacillus performed poorly, as evident by gyrB and rpoB primer sets tested in our study had no amplification against the Bacillus genome collection [21, 23, 24, 65]. The lack of in silico amplification might potentially be caused by the lack of adjustment in annealing temperature and internal walking primers were not introduced in our test. Primer sets gyrA-42f and gyrA-1066r limited within B. subtilis group amplification but would still provide accurate classification and works for single isolate identification. In case of pathogenic Bacillus species, it is recommended to conduct polyphasic analyses that go beyond solely genome sequencing. Methods such as microscopy, biochemical tests, or phage-based approaches can swiftly aid the identification process, facilitating clinical diagnosis regarding their potential pathogenicity to humans [66].
Short tuf gene sequences have been reported as a reliable molecular marker for investigating the evolutionary distances between Lactobacillus and Bifidobacterium, and employed for the identification of Staphylococcus [29, 67, 68]. Our results demonstrated that tuf gene had superior performance on the specificity and range, evident by the 100% amplification rate of 1149 Bacillus genomes and no unspecific amplification of negative controls. When using our tuf primers for the identification of soil isolates, Sanger sequencing aligned exactly with the results we obtained from the complete genome demonstrating the versatile use of tuf primers.
An important reason that 16S rRNA gene gained widespread use is the universality in bacteria combined with highly conserved regions that facilitate universal primer targets flanked by variable regions that are suited for metataxonomics on next-generation sequencing platforms. However, based on previous analysis conducted by RibDif, it has been found out that most genomes of Bacillus have multiple alleles of V3V4 region, and 39 out of 50 species have V3V4 alleles that are not unique to particular species [17]. As per RibDif analysis, a community containing B. subtilis analyzed with V3V4 metaxonomics will also incorrectly suggest several unique ASVs due to the multiple alleles of B. subtilis and hence overestimate the richness the sample. In a sample containing B. thurigiensis, one may even incorrectly infer the presence of no less than 14 other species, as all these have V3V4 alleles shared between one another. As a result, amplicons of the V3V4 region of the 16S rRNA gene cannot be used to differentiate species of Bacillus. Therefore, we aimed to generate primers which can separate species of Bacillus phylogenetically. When designing primers that targeted the tuf gene, consensus sequences were identified at the beginning (58–59 bp) and the middle (517–518 bp) of tuf gene that comprise highly variable regions among those regions that allow species differentiation. The tuf1 and tuf2 amplicons were adapted to Illumina Miseq300 platform that allows straightforward analysis of amplicon sequencing results. Our primers incorporate traits that make them applicable universally in the Bacillus genus where highly variable regions allow for species identification and sequence in high-throughput contexts. While all amplicons were assigned to NCBI database for parallel comparison, a customized tuf gene database could potentially improve the resolution of species identification [37]. For instance, retrieving gene sequences encoding members exclusively belonging to the protein family TIGR00485 that translate elongation factor Tu (EF-Tu) gene and use corresponding nucleotide sequences as our database [34]. With the rapid development of long-read sequencing and the study of Bacillus evolutionary history, the tuf database can be expanded, and a more accurate phylogenomics of Bacillus can be established, which could potentially lead to strain-level differentiation in the future.
The here described tuf amplicon sequencing approach demonstrates species-specific detection with highly similar results across biological replicates. Given the natural variability of Bacillus community structure in different environments, certain species within Bacilli could be more prevalent than others. For future experiments, examining various mixture combinations, rather than equal ratios, could be used to validate the accuracy of the tuf amplicon sequencing approach within a synthetic context. However, additional experiments that incorporate metagenome sequencing on same samples to evaluate the performance could be more efficient. Importantly, variation in total bacterial load between samples restricts the ability to reflect absolute concentrations of individual Bacillus species and bias might be introduced by exclusion of rare species or over-representation of certain species [69]. Quantitative methods that complement the tuf amplicon sequencing approach could improve the resolution of Bacillus community profiling. Meanwhile, applications of the tuf primer pairs on different samples from diverse environments, such as rhizosphere, sediments will further examine the sensitivity of tuf amplicon methodology.
The genus Bacillus has complex ecological behaviors and participates in various ecosystem functions. To gain a comprehensive understanding of the lifestyle of Bacillus in their natural habitats, such as soil and plant rhizosphere, the development of Bacillus amplicon sequencing tool (BAST) becomes indispensable. For example, what influence do environmental factors have on the diversity and community structure of Bacillus? Do Bacillus members actively compete and affect other members of the soil and rhizosphere community? Additionally, do members of Bacillus exhibit varying contributions to distinct ecosystem functions? In agricultural settings, BAST will facilitate accurate identification of Bacillus and aid plant-microbiome interactions study. Furthermore, in studies involving isolation of Bacillus genus from harsh environments characterized by high salinity and drought, our amplicon sequencing tool would foster the identification of bio-potential isolates that could aid plants in alleviating abiotic stress. In addition, evaluating the performance of PGPRs in terms of coexistence, anti-interference, and stabilization is crucial where BAST provides a way to track and identify the species in field.
In summary, we designed novel primers and compared with previously documented primers for identification of Bacilli at species level. We have exploited our tuf gene-targeting primers to accurately classify Bacillus on the species level and applied for high-throughput sequencing as a complementary tool in addition to standard 16S rRNA amplicon sequencing. The Bacillus amplicon sequencing tool (BAST) could be potential applied on tracking bio-inoculant activeness in field, guiding exploration of bio-potential strains in field and understanding ecological roles of Bacillus species in natural habitats,
Data availability
The raw sequencing data has been deposited to NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA960711 and PRJNA976106. All code is available at https://github.com/Xinming9606/BAST.
References
Parte AC, Sardà Carbasse J, Meier-Kolthoff JP, Reimer LC, Göker M. List of Prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int J Syst Evol Microbiol. 2020;70:5607–12.
Saxena AK, Kumar M, Chakdar H, Anuroopa N, Bagyaraj DJ. Bacillus species in soil as a natural resource for plant health and nutrition. J Appl Microbiol. 2020;128:1583–94.
Ehling-Schulz M, Lereclus D, Koehler TM. The Bacillus cereus group: Bacillus species with pathogenic potential. Microbiol Spectr. 2019;7:GPP3-0032-2018.
Mahapatra S, Yadav R, Ramakrishna W. Bacillus subtilis impact on plant growth, soil health and environment: Dr. Jekyll and Mr. Hyde. J Appl Microbiol. 2022;132:3543–62.
Rabbee MF, Ali MS, Choi J, Hwang BS, Jeong SC, Baek K. Bacillus velezensis: a valuable member of bioactive molecules within plant microbiomes. Molecules. 2019;24:1046.
Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, et al. Comparative analysis of the complete genome sequence of the plant growth–promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol. 2005;25:1007–14.
Jers C, Strube ML, Cantor MD, Nielsen BKK, Sørensen OB, Boye M, et al. Selection of Bacillus species for targeted in situ release of prebiotic galacto-rhamnogalacturonan from potato pulp in piglets. Appl Microbiol Biotechnol. 2017;101:3605–15.
Mandic-Mulec I, Stefanic P, van Elsas JD. Ecology of Bacillaceae. Microbiol Spectr. 2015;3:1–24.
Eelderink-Chen Z, Bosman J, Sartor F, Dodd AN, Kovács ÁT, Merrow M. A circadian clock in a nonphotosynthetic prokaryote. Sci Adv. 2021;7:eabe2086.
Arnaouteli S, Bamford NC, Stanley-Wall NR, Kovács ÁT. Bacillus subtilis biofilm formation and social interactions. Nat Rev Microbiol. 2021;19:600–14.
Dragoš A, Andersen AJC, Lozano-Andrade CN, Kempen PJ, Kovács ÁT, Strube ML. Phages carry interbacterial weapons encoded by biosynthetic gene clusters. Curr Biol. 2021;31:3479–89.
Pérez-García A, Romero D, de Vicente A. Plant protection and growth stimulation by microorganisms: biotechnological applications of Bacilli in agriculture. Curr Opin Biotechnol. 2011;22:187–93.
Gupta RS, Patel S, Saini N, Chen S. Robust demarcation of 17 distinct Bacillus species clades, proposed as novel Bacillaceae genera, by phylogenomics and comparative genomic analyses: description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus Bacillus limiting it only to the members of the Subtilis and Cereus clades of species. Int J Syst Evol Microbiol. 2020;70:5753–98.
Bhandari V, Ahmod NZ, Shah HN, Gupta RS. Molecular signatures for Bacillus species: demarcation of the Bacillus subtilis and Bacillus cereus clades in molecular terms and proposal to limit the placement of new species into the genus Bacillus. Int J Syst Evol Microbiol. 2013;63:2712–26.
Dunlap CA. Taxonomy of registered Bacillus spp. strains used as plant pathogen antagonists. Biol Control. 2019;134:82–86.
Fritze D. Taxonomy of the genus Bacillus and related genera: the aerobic endospore-forming bacteria. Phytopathology. 2004;94:1245–8.
Strube ML. RibDif: can individual species be differentiated by 16S sequencing? Bioinform Adv. 2021;1:vbab020.
Maughan H, Van der Auwera G. Bacillus taxonomy in the genomic era finds phenotypes to be essential though often misleading. Infect Genet Evol. 2011;11:789–97.
Chun J, Bae KS. Phylogenetic analysis of Bacillus subtilis PB6 based on 16S rRNA and partial gyrA nucleotide sequences. Antonie Leeuwenhoek. 2000;78:123–7.
Liu Y, Štefanič P, Miao Y, Xue Y, Xun W, Zhang N, et al. Housekeeping gene gyrA, a potential molecular marker for Bacillus ecology study. AMB Express. 2022;12:133.
Wang LT, Lee FL, Tai CJ, Kasai H. Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA-DNA hybridization in the Bacillus subtilis group. Int J Syst Evol Microbiol. 2007;57:1846–50.
Caamaño-Antelo S, Fernández-No IC, Böhme K, Ezzat-Alnakip M, Quintela-Baluja M, Barros-Velázquez J, et al. Genetic discrimination of foodborne pathogenic and spoilage Bacillus spp. based on three housekeeping genes. Food Microbiol. 2015;46:288–98.
Yamada S, Ohashi E, Agata N, Venkateswaran K. Cloning and nucleotide sequence analysis of gyrB of Bacillus cereus, B. thuringiensis, B. mycoides, and B. anthracis and their application to the detection of B. cereus in rice. Appl Environ Microbiol. 1999;65:1483–90.
La Duc MT, Satomi M, Agata N, Venkateswaran K. gyrB as a phylogenetic discriminator for members of the Bacillus anthracis–cereus–thuringiensis group. J Microbiol Methods. 2004;56:383–94.
Ki J-S, Zhang W, Qian PY. Discovery of marine Bacillus species by 16S rRNA and rpoB comparisons and their usefulness for species identification. J Microbiol Methods. 2009;77:48–57.
Mohkam M, Nezafat N, Berenjian A, Mobasher MA, Ghasemi Y. Identification of Bacillus probiotics isolated from soil rhizosphere using 16S rRNA, recA, rpoB gene sequencing and RAPD-PCR. Probiotics Antimicro Prot. 2016;8:8–18.
Draganić V, Lozo J, Biočanin M, Dimkić I, Garalejić E, Fira D, et al. Genotyping of Bacillus spp. isolate collection from natural samples. Genetika. 2017;49:445–56.
Blackwood KS, Turenne CY, Harmsen D, Kabani AM. Reassessment of sequence-based targets for identification of Bacillus species. J Clin Microbiol. 2004;42:1626–30.
Strube ML, Hansen JE, Rasmussen S, Pedersen K. A detailed investigation of the porcine skin and nose microbiome using universal and Staphylococcus specific primers. Sci Rep. 2018;8:12751.
Zakavi M, Askari H, Shahrooei M. Maize growth response to different Bacillus strains isolated from a salt-marshland area under salinity stress. BMC Plant Biol. 2022;22:367.
Wei Z, Gu Y, Friman V-P, Kowalchuk GA, Xu Y, Shen Q, et al. Initial soil microbiome composition and functioning predetermine future plant health. Sci Adv. 2015;5:eaaw0759.
Zahra ST, Tariq M, Abdullah M, Azeem F, Ashraf MA. Dominance of Bacillus species in the wheat (Triticum aestivum L.) rhizosphere and their plant growth promoting potential under salt stress conditions. PeerJ. 2023;11:e14621.
Armougom F, Raoult D. Exploring microbial diversity using 16S rRNA high-throughput methods. J Comput Sci Syst Biol. 2009;2:74–92.
Barret M, Briand M, Bonneau S, Préveaux A, Valière S, Bouchez O, et al. Emergence shapes the structure of the seed microbiota. Appl Environ Microbiol. 2015;81:1257–66.
Vos M, Quince C, Pijl AS, Hollander M, de, Kowalchuk GA. A comparison of rpoB and 16S rRNA as markers in pyrosequencing studies of bacterial diversity. PLoS One. 2012;7:e30600.
Ren Q, Hill JE. Rapid and accurate taxonomic classification of cpn60 amplicon sequence variants. ISME Commun. 2023;3:77.
Lauritsen JG, Hansen ML, Bech PK, Jelsbak L, Gram L, Strube ML. Identification and differentiation of Pseudomonas species in field samples using an rpoD amplicon sequencing methodology. mSystems. 2021;6:e00704–21.
Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3.
Meier-Kolthoff JP, Göker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019;10:2182.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
Edgar RC. MUSCLE5: High-accuracy alignment ensembles enable unbiased assessments of sequence homology and phylogeny. Nat Commun. 2022;3:6968.
Balaban M, Moshiri N, Mai U, Jia X, Mirarab S. TreeCluster: clustering biological sequences using phylogenetic trees. PLoS One. 2019;14:e0221068.
Cohen J. A coefficient of agreement for nominal scales. Educ Psychol Meas. 1960;20:37–46.
Gamer M, Lemon J, Gamer MM, Robinson A, Kendall’s W Package ‘irr’. Various coefficients of interrater reliability and agreement 2012; https://cran.r-project.org/web/packages/irr/irr.pdf.
Li Q, Zhao X, Zhang W, Wang L, Wang J, Xu D, et al. Reliable multiplex sequencing with rare index mis-assignment on DNB-based NGS platform. BMC Genom. 2019;20:215.
Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90.
Wick RR, Judd LM, Gorrie CL, Holt KE. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb Genom. 2017;3:e000132.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595.
Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–6.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55.
Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2019;36:btz848.
Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016;44:6614–24.
Frank JA, Reich CI, Sharma S, Weisbaum JS, Wilson BA, Olsen GJ. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl Environ Microbiol. 2008;74:2461–70.
Sun X, Xu Z, Xie J, Hesselberg-Thomsen V, Tan T, Zheng D, et al. Bacillus velezensis stimulates resident rhizosphere Pseudomonas stutzeri for plant health through metabolic interactions. ISME J. 2021;16:774–87.
Lereclus D, Arantès O, Chaufaux J, Lecadet MM. Transformation and expression of a cloned δ-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol Lett. 1989;60:211–7.
Periago PM, van Schaik W, Abee T, Wouters JA. Identification of proteins involved in the heat stress response of Bacillus cereus ATCC 14579. Appl Environ Microbiol. 2002;68:3486–95.
Stefanic P, Mandic-Mulec I. Social interactions and distribution of Bacillus subtilis pherotypes at microscale. J Bacteriol. 2009;191:1756–64.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–D596.
Murphy R, Strube ML. RibDif2: expanding amplicon analysis to full genomes. Bioinform Adv. 2023;3:vbad111.
Chun J, Bae KS. Phylogenetic analysis of Bacillus subtilis and related taxa based on partial gyrA gene sequences. Antonie Van Leeuwenhoek. 2000;78:123–7.
Blake C, Christensen MN, Kovács ÁT. Molecular aspects of plant growth promotion and protection by Bacillus subtilis. Mol Plant-Microbe Interact. 2021;34:15–25.
Kloepper JW, Ryu C-M, Zhang S. Induced systemic resistance and promotion of plant growth by Bacillus spp. Phytopathology. 2004;94:1259–66.
Yamamoto S, Harayama S. PCR amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of Pseudomonas putida strains. Appl Environ Microbiol. 1995;61:1104–9.
Irenge LM, Gala J-L. Rapid detection methods for Bacillus anthracis in environmental samples: a review. Appl Microbiol Biotechnol. 2012;93:1411–22.
Hwang SM, Kim MS, Park KU, Song J, Kim E-C. tuf gene sequence analysis has greater discriminatory power than 16S rRNA sequence analysis in identification of clinical isolates of coagulase-negative Staphylococci. J Clin Microbiol. 2011;49:4142–9.
Ventura M, Canchaya C, Meylan V, Klaenhammer TR, Zink R. Analysis, characterization, and loci of the tuf genes in Lactobacillus and Bifidobacterium species and their direct application for species identification. Appl Environ Microbiol. 2003;69:6908–22.
Porath-Krause A, Strauss AT, Henning JA, Seabloom EW, Borer ET. Pitfalls and pointers: an accessible guide to marker gene amplicon sequencing in ecological applications. Methods Ecol Evol. 2022;13:266–77.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Chen K, Xie S, Iglesia E, Bell AT. A PCR test to identify Bacillus subtilis and closely related species and its application to the monitoring of wastewater biotreatment. Appl Microbiol Biotechnol. 2001;56:816–9.
Robertes MS, Nakamura LK, Cohan FMY. Bacillus mojavensis sp. nov., Distinguishable from Bacillus subtilis by sexual isolation, divergence in DNA sequence, and differences in fatty acid composition. Int J Syst Evol Microbiol. 1994;44:256–64.
Acknowledgements
XX was supported by a Chinese Scholarship Council fellowship. GM was supported by the Lendület-Programme of the Hungarian Academy of Sciences (LP2020-5/2020). This project was supported by the Danish National Research Foundation (DNRF137) for the Center for Microbial Secondary Metabolites and the Novo Nordisk Foundation within the INTERACT project of the Collaborative Crop Resiliency Program (NNF19SA0059360).
Author information
Authors and Affiliations
Contributions
XX and ÁTK conceived and designed the study. LJDN and LS provided genome sequence data. GM performed MiSeq. MLS provided analysis tools and supervised the bioinformatic aspects. XX and ÁTK wrote the manuscript; all authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Nielsen, L.J.D., Song, L. et al. Enhanced specificity of Bacillus metataxonomics using a tuf-targeted amplicon sequencing approach. ISME COMMUN. 3, 126 (2023). https://doi.org/10.1038/s43705-023-00330-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43705-023-00330-9
