
Abstract
Coronavirus Disease 2019 (COVID-19) vaccination has resulted in excellent protection against fatal disease, including in older adults. However, risk factors for post-vaccination fatal COVID-19 are largely unknown. We comprehensively studied three large nursing home outbreaks (20–35% fatal cases among residents) by combining severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) aerosol monitoring, whole-genome phylogenetic analysis and immunovirological profiling of nasal mucosa by digital nCounter transcriptomics. Phylogenetic investigations indicated that each outbreak stemmed from a single introduction event, although with different variants (Delta, Gamma and Mu). SARS-CoV-2 was detected in aerosol samples up to 52 d after the initial infection. Combining demographic, immune and viral parameters, the best predictive models for mortality comprised IFNB1 or age, viral ORF7a and ACE2 receptor transcripts. Comparison with published pre-vaccine fatal COVID-19 transcriptomic and genomic signatures uncovered a unique IRF3 low/IRF7 high immune signature in post-vaccine fatal COVID-19 outbreaks. A multi-layered strategy, including environmental sampling, immunomonitoring and early antiviral therapy, should be considered to prevent post-vaccination COVID-19 mortality in nursing homes.
Similar content being viewed by others


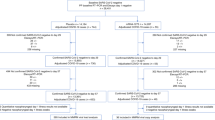
Main
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreaks affecting nursing homes have been a major public health concern since the start of the Coronavirus Disease 2019 (COVID-19) pandemic. During the first epidemic wave, it was estimated that COVID-19 mortality in Belgium was up to 130 times higher inside than outside nursing homes, due to the combined effects of age, sex, frailty and infection risks among residents1. Spatial analyses also indicated an association between the hospitalization incidence and the local density of nursing home residents, thus confirming the important impact of COVID-19 outbreaks in those facilities2. With one of the highest documented COVID-19 mortality rates in the world2, more than half of all COVID-19-related deaths in 2020 in Belgium were linked to nursing homes3. A meta-analysis of the first COVID-19 wave in Spain found that mortality at the facility level was significantly associated with a higher percentage of patients with complex diseases, lower scores on pandemic preparedness measures and higher population incidence of COVID-19 in the surrounding population4.
Nursing home residents are usually characterized by advanced age, a wide arsenal of comorbidities and associated polypharmacy and a decreased function of the immune system, potentially resulting in a higher risk of infections4,5,6,7,8. To protect this highly vulnerable population, the rollout of the vaccination campaign was initially targeted toward older adults and healthcare workers. Vaccination in Belgian nursing homes began in the second half of December 2020, employing mainly the mRNA vaccine BNT162b2. The BNT162b2 vaccine is highly effective at protecting against COVID-19 hospitalization and death, with efficacies of 90–95% reported in phase 3 clinical trials9 and confirmed in large-scale real-life studies10. By March 2021, vaccination coverage (two-dose scheme) among residents of nursing homes had reached 89.4% on a national scale. Starting from September 2021 on, a third or booster dose was administered in nursing homes. Reduction in hospital admissions and mortality among residents of nursing homes on account of vaccination has been reported throughout Europe, such as for a Spanish study that included over 25,000 residents and reported a fatality rate of only 1.6% in the post-vaccination era11. A recent study of 10 European countries, analyzing 240 COVID-19 outbreaks in the post-vaccination era (July–October 2021), identified an average case fatality rate of 5.5% for Belgium, almost half of the European average of 10.2%12. Although the same study identified vaccination status as significantly associated with COVID-19 hospitalization, no association was found with COVID-19 mortality. Strong variability in case fatality ratios has been observed13,14,15, with no major risk factors of fatal post-vaccination COVD-19 identified so far, other than age and comorbidities, mostly due to the limited statistical power in small outbreaks.
Through our nationwide surveillance, we observed only three high fatality rate (>10%) post-vaccination outbreaks in Belgian nursing homes by the end of this study (October 2021). Here we describe a multidisciplinary investigation of these three post-vaccination outbreaks in a collaboration involving the nursing home staff, health inspectors of the respective regional agencies, the national institute for public health (Sciensano), political, academic and governmental stakeholders as well as the National Reference Center of Respiratory Pathogens at the University Hospital and University of Leuven. Thus, we were able to identify demographic and clinical risk factors as well as a unique prognostic gene signature for fatal COVID-19 in vaccinated nursing home residents, revealing actionable public health and precision medicine strategies to mitigate COVID-19 mortality among susceptible older adults in the post-vaccine era.
Results
Epidemiological profile of SARS-CoV-2 nursing home outbreaks
For the largest of the three outbreaks (nursing home A), the first infection was documented in the dementia ward on 17 May 2021, for an 89-year-old woman who developed COVID-19-related symptoms, who was subsequently hospitalized and who died after 2 weeks of hospitalization. A total of 102 cases were documented related to this outbreak between 18 May and 24 June, of which 75 were residents, 25 were staff members and two were family members of staff. All departments of the nursing home were involved, and consecutive screening moments were scheduled. Among 120 residents, 75 were SARS-CoV-2 positive by polymerase chain reaction (PCR) (62.5%; Table 1), whereas only 25 of 146 (17.1%) staff members tested positive (Supplementary Table 1). Timing of diagnosis by a positive PCR result and longitudinal follow-up is illustrated in Fig. 1a, which clearly shows late-onset PCR positivity for a large subset of residents who tested PCR negative at the start of the outbreak. This ‘second wave’ of delayed infections was corroborated by the continuous detection of SARS-CoV-2 by quantitative PCR (qPCR) in aerosol samples taken from the common areas of both staff and residents (Fig. 1b). For 58 of 102 (56.9%) positive cases, whole-genome sequencing (WGS) information was available, identifying the Delta variant (Pangolin lineage B.1.617.2) for all of them. Phylogenetic analysis indicates that all samples from the nursing home cluster were within the same clade, hence suggesting a single introduction event (Fig. 1c). Among the 75 PCR-positive residents, 15 died (case fatality ratio of 20%). Considering all individuals for whom vaccination status was known (Table 1), 96% of residents, but only 66% of staff members, were fully vaccinated. One resident and five staff members were partially vaccinated at the time of the outbreak, whereas one resident and 28.7% of staff members were not vaccinated.

a,b, We report the evolution through time of Ct values measured in both infected residents and staff members (a) and aerosols analyzed in various sections within the nursing home (b). Gray dots refer to negative PCR results. c, Time-scaled phylogenetic analysis involving Delta (B.1.617.2) genomes sampled and sequenced from this outbreak reveals that all 58 full genomes originating from nursing home A are clearly clustered within the overall phylogenetic tree (orange dots), suggesting a single introduction event. The phylogenetic tree is time calibrated, meaning that branch lengths are in units of time (year).
The first documented PCR-positive case for nursing home B dates from 20 May 2021, and the presumed index case developed COVID-19 symptoms the day before. Overall, 19 of 29 residents (65.5%) tested positive for SARS-CoV-2, but none of the 17 staff members tested positive on the repetitive screening moments organized between 20 May and 24 June. Despite high cycle threshold (Ct) values for this outbreak (Extended Data Fig. 1), WGS was successful for 19 of 19 (100%) PCR-positive cases, all classified as Gamma variant (Pangolin lineage P.1). Our phylogenetic analysis highlights that all samples clustered together within the more global Gamma phylogeny inferred in our study, again pointing toward the hypothesis of a single introduction event (Extended Data Fig. 1). Overall, seven fatal cases were reported in this outbreak, of which one resident tested negative by PCR. Although this death was classified as COVID-19 related according to World Health Organization (WHO) criteria16, due to severe respiratory symptoms and recent close contact with positive residents, we conservatively used only PCR-positive residents to calculate the case fatality ratio (6/19, 32%). For this nursing home, the vaccination rate was high among residents (86.2%), whereas only 52.9% of the staff members were fully vaccinated at the time of the outbreak. Nevertheless, none of the latter tested positive for SARS-CoV-2.
The post-vaccination outbreak in nursing home C was initially alerted by two cases (related resident and staff) infected with the Delta variant a few days before the large testing initiative for the other residents and staff members (20 July 2021). Twenty-five additional SARS-CoV-2-positive cases were identified during the outbreak. WGS determined the presence of the variant of interest, Mu (Pangolin lineage B.1.621), complemented with the mutation K417N in the spike protein, and, for one isolated staff member without resident contact, an additional Delta infection was identified. The single Delta-infected resident was, therefore, not included for further analysis of the outbreak (Table 1; 27/27 PCR-positive cases (100%) were confirmed by WGS: three Delta and 24 Mu). The Mu variant saw relatively limited circulation in Belgium, resulting in a restricted sampling of related genomic sequences in the local community. Our phylogenetic analysis, however, indicates that PCR-positive cases in this nursing home related to that variant clearly clustered within the overall phylogeny inferred for that variant (Extended Data Fig. 2), again advocating for a single introduction event. Moreover, all 24 PCR-positive cases infected with variant Mu (20 residents and four staff members) were linked to the dementia unit of the nursing home. Overall, seven infected residents died of COVID-19 (7/20, case fatality ratio 35%), and one additional resident died of a COVID-19-unrelated cause. Considering the 229 residents and staff members with known vaccination status, the overall vaccination rate was 98.3%. For the group of PCR-positive residents, 100% were fully vaccinated.
Demographic and clinical profile of SARS-CoV-2 outbreaks
Demographic and clinical risk factors for fatal COVID-19 among residents were identified by multivariable logistic regression models (Table 2), with the best model including age, male sex, non-Delta SARS-CoV-2 variants (Gamma and Mu) and later onset of infection (PCR positivity >7 d after the start of the outbreak). In the sensitivity analysis, only fully vaccinated and PCR-positive residents (n = 107) were included. The results remained statistically significant, with a similar effect size (Supplementary Table 2). The importance of these four factors as predictors of mortality was confirmed by Kaplan–Meier survival estimates (Extended Data Fig. 3) and time-to-event analysis (Cox proportional hazard regression; Supplementary Table 3). Of interest, dementia or peak viral load (nadir cycle quantification (Cq) value) were not predictive of fatal cases in the joint analysis of the three outbreaks (Table 2) but were significant predictors in single nursing homes (Supplementary Table 3). Because nursing home size was found to be a major risk factor for COVID-19 mortality in several countries, including Belgium17,18, we included this as an additional parameter in both logistic and Cox regression models. As shown in Supplementary Table 6, nursing home size was not an independent predictor (in addition to age, sex and late PCR positive) of fatal COVID-19, whereas the preferred model (corrected Akaike information criterion (cAIC)) contained age, sex, late PCR positive and variants/outbreaks as independent predictors.
Digital transcriptomic analysis of SARS-CoV-2 outbreaks
In search of candidate biomarkers for post-vaccine fatal COVID-19, as well as possible therapeutic targets, we opted for nCounter digital transcriptomics for immunovirological profiling of the nasal mucosa, encouraged by previous results19,20,21. For 20 of 28 fatal cases, a sufficient volume of diagnostic nasopharyngeal swabs was available for nCounter analysis, to explore immunological (600 genes representative of the major immune cell types) and virological (SARS-CoV-2 transcripts and ACE2/TMPRSS2 receptors) parameters as possible risk factors for fatal post-vaccine COVID-19. Thus, we carefully matched (age, sex and outbreak) 20 fatal cases (all those with available nasopharyngeal swabs) with 30 PCR-positive non-fatal cases, with similar timing of infection, as well as 10 PCR-negative but SARS-CoV-2-exposed residents. Because these samples were obtained at SARS-CoV-2 diagnosis, before hospitalization or treatment (oxygen and/or dexamethasone), the transcriptomic immune signatures are not modified by immunomodulatory treatment and can be used to predict fatal outcome. In addition, only four of 118 PCR-positive residents had received corticosteroids before their SARS-CoV-2 diagnosis. None of them was a fatal case, and they were not included for nCounter analysis.
As shown in Fig. 2 (volcano plot), a total of 193 human and seven viral gene transcripts were significantly upregulated or downregulated (P < 0.05) when comparing fatal versus non-fatal cases. In addition to the antiviral cytokines IL28A (also known as IFNL2 (interferon-λ2)) and IFNB1 (the gene encoding interferon-beta (IFN-β)), the most upregulated genes were predominantly expressed by innate immune cells: monocytes/macrophages (CX3CR1, TNFSF15, CLEC6A, ITLN1 and LILRB5), natural killer (NK) cells (THY1, CDH5, KIR3DL3, CD160, B3GAT1, NCAM1 and CCL3) and conventional dendritic cells (XCR1). Thus, the predominant immunopathogenic signature of fatal COVID-19 in vaccinated residents represents exacerbated innate immune activation rather than a failed adaptive (B cell and T cell) vaccine response. Likewise, a large subset of B cell genes (CD19, CR2, CD79A, CD79B, PAX5 and CD70), regulatory T cell (Treg) genes (FOXP3 and PTGER4) and cytotoxic CD8 T cell genes (EOMES and PTGER4) were also significantly upregulated in fatal cases, arguing against a curtailed B cell or T cell response or a failure of B cells or T cells to migrate to the nasal mucosa. On the other hand, a generalized downregulation of major histocompatibility complex (MHC) class I-mediated antigen presentation (B2M and HLA-C) was observed across all cell types, in agreement with previous reports demonstrating loss of MHC class I activity at the transcriptomic, epigenomic and functional level22,23,24,25,26,27.

Volcano plot of differentially expressed genes in nasal mucosa of fatal (n = 20) versus age-matched, sex-matched and outbreak-matched non-fatal PCR-positive cases (n = 30), quantified by nCounter digital transcriptomics (uncorrected P values from linear model, negative binomial distribution, dotted line showing P < 0.05, FDR q values provided in Source Data). Selected viral (red circles) and host immune transcripts (blue circles) significantly upregulated or downregulated in fatal versus non-fatal cases are highlighted with gene names. Details on immune genes are given in the Results section. PCR+, PCR positive.
Because the top downregulated genes were most representative of mucosal epithelial cells (PIGR, CD9 and MUC1), the observed exacerbated innate response might represent enhanced migration of innate immune cells but also virus-mediated destruction of the mucosal epithelial cells. In favor of the latter hypothesis, fatal cases were characterized by significantly higher viral transcript levels when measured by nCounter. Transcript levels for spike, envelope, nucleoprotein, ORF1ab, ORF3a and ORF7a genes (Fig. 3a and data not shown, all P < 0.05 with false discovery rate (FDR) correction) were higher in fatal cases compared to non-fatal PCR-positive residents. In addition, antisense SARS-CoV-2 was selectively increased in eight of 20 fatal cases (Fig. 3a) versus PCR-positive cases, indicating heightened intracellular viral replication. Of note, peak viral load (nadir Cq values) or viral load of the first PCR-positive sample, measured by qPCR, was not significantly different between fatal cases and PCR-positive controls (Fig. 3a), underscoring the sensitivity of nCounter digital transcriptomics. Exacerbated viral replication in fatal cases was paralleled by a marked eight-fold increase in viral receptor ACE2 transcript levels (P < 0.001) as well as an unexpected two-fold decrease (P < 0.01) in viral co-receptor TMPRSS2 expression (Fig. 3b).

a–c, Viral transcript levels for spike protein (left: fatal versus PCRpos P = 0.012, fatal versus PCRneg P = 0.000022, PCRneg versus PCRpos P = 0.0089) and ORF1ab antisense RNA (middle), measured by nCounter digital transcriptomics. Right panel shows peak viral load (nadir Cq values) as quantified by qPCR. Viral receptors (ACE2: fatal versus PCRpos P = 0.0009; TMPRSS2: fatal versus PCRpos P = 0.0036, fatal versus PCRneg P = 0.0005, PCRneg versus PCRpos P = 0.0422) (b) and antiviral cytokine IFNB1 (fatal versus PCRpos P = 0.0022, fatal versus PCRneg P = 0.0022) (c) were quantified by nCounter digital transcriptomics. Data are presented as median values ± s.d. d, Left: visualization of best predictive model (multivariate logistic regression, selected by cAIC), including age (not depicted) and ORF7a and ACE2 transcripts. Dashed gray lines indicate the detection limit of SARS-CoV-2 transcripts. Each circle represents a resident, and the size of the circle is proportional to ACE2 normalized expression. Right: comparison of ROC curves of predictive models by univariate (IFNB1) or multivariate (IFNB1/age/sex and age/ORF7a/ACE2) logistic regression. ROC curves showing significant prediction of fatal versus non-fatal COVID-19 according to IFNB1 transcript levels (right), with and without age and sex as additional factors (detailed in the Results section). For a–c, statistical results are from Kruskal–Wallis test with FDR correction for multiple testing (PCRneg n = 10, PCRpos n = 30, fatal n = 20), ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, NS, not significant. PCRpos, PCR positive; PCRneg, PCR negative.
Among all immune genes, IFNB1 transcripts displayed the strongest negative correlation to survival time (starting from the date of PCR-positive diagnosis, Spearman’s ρ = −0.24, P = 0.0024). Corroborating our previous findings in a Belgian cohort of intensive care unit (ICU) patients19, we found that increased IFNB1 transcript levels significantly predicted a fatal outcome (Fig. 3c,d; area under the receiver operating characteristic (AUROC) curve 0.76 (95% confidence interval (CI) 0.63–0.89), P = 0.0013), which was slightly increased by adding age and sex to the model (Fig. 3d; AUROC 0.82 (95% CI 0.71–0.93), P = 0.000064). IFNB1 remained a significant predictor in multivariable logistic regression, independent of age, sex and peak viral load (nadir Cq value), which was also confirmed by time-to-event analysis (Cox proportional hazard models; Table 3).
Lastly, when combining all available demographic, immune and viral parameters, the best predictive model for mortality, according to the cAIC, included age (odds ratio (OR) 1.07, 95% CI 0.98–1.19), increased viral ORF7a (OR 1.67, 95% CI 0.98–3.46) and viral receptor ACE2 (15.43, 95% CI 2.54–165.9) transcript levels, resulting in correct classification of 18 of 20 (90%) fatal cases (AUROC 0.88, 95% CI 078–0.98, P = 0.000002), as visualized in Fig. 3d.
A unique immune signature in post-vaccination fatal COVID-19
To our knowledge, no well-powered study of immune signatures in post-vaccination fatal COVID-19 in the older adult population have been published at present. Thus, no public datasets are currently available for independent validation of our ‘post-vaccine fatal COVID-19’ immune signature in a comparable epidemiological setting. Therefore, we compared published transcriptomic and genomic signatures of pre-vaccination fatal and/or life-threatening COVID-19.
Upon cross-comparison of our ‘post-vaccine fatal COVID-19’ transcriptomic signature with previously described IEI (inborn errors in type I IFN immunity) genes linked to life-threatening COVID-19 (ref. 28), we identified a clear dichotomy between upregulated (IRF7) versus downregulated (IRF3) IEI genes in fatal cases (Fig. 4a). Therefore, we quantified type I IFN signaling score based on nCounter gene expression data (Supplementary Table 5). Of note, type I IFN signaling score was not significantly different between fatal cases and matched controls (Fig. 4b). However, lymphocyte activation, Th17 and Treg differentiation pathways were significantly increased in fatal cases (Fig. 4b), in agreement with our finding of upregulated EOMES, SRC, THY1, RORC, IL6R, FOXP3 and PTGER4 genes (Fig. 2). As shown in Fig. 4c, type I IFN signaling score was highly correlated to IRF7 (ρ = 0.84, P = 7 × 10−14) as well as STAT2 (ref. 29) (ρ = 0.91, P = 6 × 10−20) but not to IRF3 or IFNA2 (both P > 0.05) transcripts. In contrast to IFNA2, but in agreement with our multivariable logistic regression models for mortality, IFNB1 levels were most strongly correlated to IRF7 and TLR7 plasmacytoid dendritic cell (pDC)-specific type I IFN drivers as well as inflammatory targets, such as IL6R (Fig. 4c, lower panel). Similar to IFNB1, we found that IRF3 transcript levels were also able to predict mortality in residents (Fig. 4d; Kaplan–Meier curve, P = 0.0030). In addition, classification of nursing home residents according to IFNB1 levels demonstrated a significant link with lower IRF3 expression as well as higher viral replication and apoptosis, providing a possible molecular and cellular mechanism of action of IFN-β.

a, Venn diagram shows overlap between gene transcripts upregulated (‘up Fatal’) or downregulated (‘down Fatal) in fatal cases versus PCR-positive controls (quantified by nCounter digital transcriptomics) and the gene mutations (IEI) identified in life-threatening COVID-19 (ref. 28) (pre-vaccine era). The five IEI genes not differentially expressed between cases and controls are TICAM1, TBK1, UNC93B1, IFNAR1 and TLR3. b, Pathway scores (calculated by nSolver from gene expression profiling by nCounter) for lymphocyte activation (P = 0.043), Th17 (P = 0.028) and Treg differentiation (P = 0.022) were increased in fatal cases versus PCR-positive controls, whereas type I IFN signaling was not (t-test with Welch’s correction). No pathways were significant after stringent Bonferroni correction for multiple testing. Data are presented as median values ± s.d. Red circles: fatal cases; green circles: PCR-positive controls. c, Spearmanʼs correlation of type I signaling score (upper panel) and IFNB1 expression (lower panel) with drivers of IFN signaling (STAT2, IRF7, IRF3, IFNA2 and TLR7) and inflammation (IL6R), across all 50 residents (20 fatal cases and 30 PCR-positive controls). d, Kaplan–Meier curve demonstrating significantly lower (log-rank test) survival in nursing home residents with ‘IRF3 low’ status (nCounter normalized expression below the median). e, Classification of nursing home residents into ‘IFNB1 high’ versus ‘IFNB1 low’ (below or above 100 normalized counts) reveals a significant link with IRF3 expression (Mann–Whitney test P = 0.000011), intracellular viral replication (measured as SARS-CoV-2 antisense RNA, Mann–Whitney test P = 0.000072) and apoptosis score (calculated by nSolver, Mann–Whitney test P = 0.044) in upper airway mucosa. Data are presented as median values ± s.d. ****P < 0.0001, **P < 0.01, *P < 0.05, NS, not significant. PCR+, PCR positive.
In addition to the genetic link to type I IFN signaling, anti-type I IFN neutralizing antibodies have been shown by several groups to be an additional risk factor for life-threatening COVID-19 (refs. 30,31,32,33). Because no serum samples were available from the fatal cases, we cross-examined our fatal COVID-19 immune gene signature with the LAIR1 biomarker recently described by van der Wijst et al.33 as strongly correlated to anti-IFN auto-antibodies. Confirming its antagonistic role in IFN/antiviral signaling, LAIR1 level was positively correlated with peak viral load (nadir Cq value, P = 7.0 × 10−5, ρ = −0.49, n = 50; Extended Data Fig. 4). Similar to peak viral load (Fig. 2 and Tables 2 and 3), LAIR1 transcript levels were not able to predict survival in this cohort (data not shown). However, we found that LAIR1 transcript level was significantly and negatively correlated with IRF3 (the major upstream driver of type I IFN production in epithelial cells) in fatal cases (ρ = −0.59, P = 0.0067, n = 20), whereas no significant correlation was observed for matched PCR-positive controls (ρ = −0.037, P = 0.84, n = 30; Extended Data Fig. 4). This demonstrated a major difference in the type I IFN pathway regulation between fatal cases and controls, probably more pronounced in the epithelial cells of the upper airway mucosa, in agreement with Zhang et al.34.
Taken together, cross-examination of published transcriptomic and genomic pre-vaccine fatal COVID-19 signatures highlights the unique innate and adaptive immune signature observed in post-vaccination fatal COVID-19 in nursing home residents.
Discussion
We comprehensively studied three large outbreaks in Belgian nursing homes with high fatality ratios (20–35%), which resulted in several epidemiologically and clinically relevant insights into the ongoing ‘arms race’ between vaccines and SARS-CoV-2 variants of concern. First, whole-genome phylogenetic analyses indicated that each outbreak stemmed from a single introduction event, although with different variants (Delta, Gamma and Mu). Second, our study confirms previous reports of the independent relationship of older age and male sex with fatal COVID-19, yet is the first to associate Gamma and Mu variants and late onset of PCR positivity with fatal post-vaccination COVID-19 among older adults. Our findings evoke that even non-dominant variants of concern (Gamma) or variants of interest (Mu) can result in high mortality, similar to the dominant variant of concern (Delta at the time of this study, May–August 2021) in specific high-risk settings. Third, environmental sampling revealed that SARS-CoV-2 could be detected in aerosol samples of common spaces (used by either residents or staff) up to 52 d after the initial infection. Fourth, gene expression profiling of nasopharyngeal swabs identified candidate immunological (IFNB1 and IRF3) and virological (ORF7A and ACE2) biomarkers for early monitoring of post-vaccine breakthrough cases in high-risk older adults, which might not be limited to nursing homes.
Indeed, increased IFNB1 transcript levels are highlighted as a significant independent predictor of fatal post-vaccination COVID-19, extending our previous findings in critical COVID-19 (ref. 19). Although IFN-β therapy was beneficial in small phase 2 clinical trials35,36, subsequent larger trials identified no benefit37 or even an association with a longer ICU stay38, thus underscoring our previous findings on endogenous IFN-β expression in ICU patients19. As previously proposed39,40, these apparently conflicting effects of type I IFN can be explained by a two-phase model, in which early IFN results in antiviral protection41, whereas late IFN exerts a deleterious pro-inflammatory effect. In support of this hypothesis, type I IFN scores were strongly correlated to STAT2 levels (Fig. 4c, ρ = 0.91, P = 6.8 × 10−20), for which our group previously demonstrated a simultaneous antiviral and pathogenic in vivo role in a COVID-19 hamster model29. In addition, we found that IFNB1 transcripts were strongly correlated (ρ = 0.84, P = 6.8 × 10−17) to IL-6 receptor (the target of tocilizumab) expression. Thus, our study suggests IL6/IL6R signaling as a plausible ‘downstream’ therapeutic target in IFNB1-overexpressing patients with COVID-19, which should be investigated in future clinical trials.
Regarding the clinical use of transcriptomic biomarkers in COVID-19, only nCounter technology was able to reliably detect IFNB1 as well as other low-abundance transcripts (MASP2 and THY1) when compared to single-cell RNA sequencing (RNA-seq) analysis of both nasal mucosa23 and blood24 (data not shown). Moreover, 10 cytokine transcripts found to be overexpressed in fatal cases by nCounter (IFNA1, IFNA2, IFNB1, IL2, IL3, IL17B, IL17F, IL20, IL21 and IL26) were undetectable or extremely low in several single-cell RNA-seq datasets22,23,24. In addition, only a small subset of these cytokines has been reproducibly detected at the protein level as biomarkers of COVID-19 disease severity and mortality, as evidenced by a recent meta-analysis42. In addition, this study also found that nCounter technology outperformed conventional qPCR (Fig. 2a) for virological monitoring of nasopharyngeal swabs to instruct COVID-19 clinical management.
We would like to highlight that, due to the challenging circumstances of the outbreaks (sudden high mortality, extremely high work burden on staff with emergency measures and quarantine, closing of the nursing homes for all visitors and family members not allowed to visit terminally ill residents), all our research analyses (qPCR, SARS-CoV-2 WGS, phylogenetics and immune gene expression profiling) were limited to the diagnostic samples (nasal swabs). Additional (blood) samples for antibody or genetic testing in the fatal cases were logistically and ethically not possible. However, a cross-comparison with published (pre-vaccine) transcriptomic22 and genomic28 signatures for fatal and life-threatening COVID-19 revealed a surprising dichotomy between IRF3-mediated IFN/antiviral signaling and IR7-mediated IFN/antiviral signaling, which our data suggest as ‘protective’ versus ‘deleterious’, respectively (Fig. 4). Of interest, this dichotomy also provides a possible molecular and cellular mechanism of action for IFN-β, linking IRF7/TLR7 overactivation in pDCs to IL-6R-mediated inflammation, triggering the destruction of IRF3-expressing epithelial cells through apoptosis (Fig. 4), similar to previous findings43,44,45. Notably, a recent study, published during the reviewing process of our study, confirmed the significant link between IFN-β-induced transcriptomic changes and severe COVID-19 in the aging brain46. Moreover, the contrasting antiviral and/or pro-apoptotic effects of IFN-β versus IFN-α were shown previously in other pathologies44,45,46,47,48. Importantly, anti-IFN-β neutralizing antibodies are infrequent (≤1% of critical COVID-19), in contrast to anti-IFN-α or anti-IFN-ω antibodies, which occur in up to 20% of older patients and fatal COVID-19 (ref. 49). Taken together, our findings reveal a need to refine the ‘generic’ type I IFN response (easily quantified by nCounter digital transcriptomics, this study and refs. 50,51 or qPCR arrays41,52), according to subtypes (IFN-α, IFN-β and IFN-ω), cellular context (epithelial cells and pDCs) and upstream signaling (IRF3 versus IRF7) to accurately predict ‘protective’ versus ‘deleterious’ clinical outcomes.
Finally, our finding of increased viral receptor ACE2, enhanced intracellular viral replication and later onset of PCR positivity in fatal cases hints at a therapeutic window for early antiviral therapy at the start of an outbreak, supported by the recent availability of effective oral antivirals53,54,55,56,57,58. The significantly higher mortality with late onset of infection (PCR positivity >7 d) was observed in each of the three outbreaks (Fig.1 and Extended Data Figs. 1 and 2). We hypothesize that this increased mortality might be due to a higher infectious dose, linked to the exposure to multiple concomitant viral shedders, as compared to early infections. This hypothesis is supported by our demonstration of prolonged detection of SARS-CoV-2 by aerosol PCR in several common rooms of each nursing home. Thus, our study indicates that biomarker-guided clinical trials evaluating the role of early antiviral therapy during post-vaccination nursing home outbreaks, and conceivably also among susceptible community-dwelling older adults, are warranted.
Limitations of this study include missing demographic (8.4% of 657), clinical (2.4% of 620) and vaccination (26.5% of 574) data, although no data were missing for fatal cases. Due to the unpredictable and sudden onset of these large-scale COVID-19 outbreaks in nursing homes, no baseline serum samples were available before the three outbreaks, nor from fatal cases, to compare the levels of vaccine-elicited SARS-CoV-2 neutralizing antibodies or anti-IFN type I auto-antibodies. Moreover, the observational nature of the study and the heterogeneity among three outbreaks (three different variants in nursing homes with different characteristics) might result in residual confounding factors, although the vaccination rates were highly similar among the nursing homes (Supplementary Table 1), and nursing home size did not predict COVID-19 mortality in our study (Supplementary Table 6). Lastly, we did not have specific data on staff pandemic preparedness and population incidence of COVID-19 in the surrounding population, which Suñer et al.4 identified as major predictors of (pre-vaccine) COVID-19 mortality in a large retrospective study of Spanish nursing homes. A major strength of this study is the simultaneous vaccination of residents in each nursing home (prioritized in the national vaccination campaign) and the defined onset (outbreaks) of SARS-CoV-2 infections, thus eliminating any possible bias in waning vaccine efficacy between fatal and non-fatal cases. Because this study was performed before vaccination booster doses were offered to older adults in Belgium (starting in September 2021), the risk factors identified herein might not be directly applicable in (recently) boosted older adult populations but remain highly relevant in the global context, in which currently only 63% of people have received an initially full vaccination protocol (two doses), and only 33% have received a booster dose59, as exemplified by recent high Omicron COVID-19 mortality among unvaccinated older adults in Hong Kong60.
In conclusion, high case fatality ratios in susceptible older adults can be observed with various SARS-CoV-2 variants—that is, Delta, Gamma and Mu. Broad immunovirological profiling of nasal mucosa by nCounter transcriptomics allowed prediction of fatal COVID-19 in diagnostic samples, whereas standard qPCR viral load quantification did not. The best predictive models for mortality comprised IFNB1 or age, viral ORF7a and ACE2 receptor transcripts, whereas comparison with pre-vaccine fatal COVID-19 signatures uncovered a unique IRF3 low/IRF7 high immune signature in post-vaccine fatal COVID-19 outbreaks. A multi-layered strategy including environmental sampling, immunomonitoring and early antiviral therapy should be considered to prevent post-vaccination COVID-19 mortality in nursing homes.
Methods
Data collection
Demographic and clinical characteristics, including comorbidities, were compiled from health records provided by the individual nursing homes. The primary outcome was COVID-19-related death, as defined by WHO criteria16. All residents, as well as the large majority of staff members, received the BNT162b2 (Comirnaty (Pfizer)) vaccine. This work was framed within the role of the National Reference Centre for Respiratory Pathogens UZ/KU Leuven (as defined by the Royal Decree of 9/2/2011), as approved by the UZ/KU Leuven ethical committee for research (S66037). No written informed consent was obtained for the use of human data and samples; all individuals involved were orally informed of the setup, context and objectives of the study, and all individuals provided oral consent.
Quantification of viral loads
Consecutive screening events were organized in all three nursing homes, first testing symptomatic individuals, followed by collective and repeated testing after the identification of a positive case. Next to nasopharyngeal swabs of residents and staff, aerosol samples were collected using the AerosolSense instrument (Thermo Fisher Scientific). After RNA extraction, samples were tested by the TaqPath COVID-19 CE-IVD RT–PCR kit (Thermo Fisher Scientific). More details can be found in the Supplementary Methods.
WGS and phylogenetic analyses
Samples with a sufficiently high viral load (>1,000 copies per milliliter) were subjected to WGS using the ARTIC Network protocol version 3.17 (ref. 61) or as described by Freed et al.62 and sequenced with Oxford Nanopore Technologies ARTIC library preparation. Complete sequences were recovered using the ARTIC analysis pipeline and typed using Pangolin and NextClade. Specifically, and to investigate if those outbreaks could have been induced by multiple introduction events in the nursing home, we aimed to contextualize the position of those infectious cases in a more global phylogenetic tree built from the analysis of an alignment made of (1) the viral genomes collected in the considered nursing home and sequenced in the context of the present study as well as (2) the genomic sequences of the same variant available for Belgium at the time of the outbreak and (3) a subtree of the European Nextstrain build containing all the genomic sequences of that variant at the time of the outbreak. A time-calibrated maximum likelihood phylogenetic tree was constructed using IQ-TREE version 2.0.3.19 (ref. 63) (GTR model)64 and TreeTime version 0.8.4.22 (ref. 65). Extended protocols are available in the Supplementary Methods.
Immunovirological profiling by digital transcriptomics (nCounter)
To identify immune and viral risk factors, 600-plex target profiling was performed by digital nCounter transcriptomics (NanoString) in a subset of residents (n = 60). RNA was extracted from nasopharyngeal swabs as described above and used for hybridization to pre-specified Human Immunology V2 and customized SARS-CoV-2 panels, as described previously19,20,21. Pathway score analyses and cell type deconvolution were performed using nSolver software (NanoString). Details on the gene lists and pathways are provided in Supplementary Table 5.
Analysis of publicly available RNA-seq data
Bulk RNA-seq data from both nasopharyngeal and blood samples, as well as corresponding gene signatures of fatal versus non-fatal COVID-19 in hospitalized patients, were obtained from Lee et al.22. Single-cell RNA-seq data23 of nasopharyngeal samples of 19 patients with COVID-19 (eight moderate and 11 critical, according to WHO classification) and five healthy controls were obtained from https://doi.org/10.6084/m9.figshare.12436517; single-cell RNA-seq data from peripheral blood mononuclear cell (PBMCs) of patients with COVID-19 were obtained from http://www.covidcellatlas.com/ (ref. 24).
Statistics and reproducibility
Owing to the nature of the study (nationwide comprehensive mapping of high-fatality SARS-CoV-2 outbreaks in nursing homes), no statistical methods were used to pre-determine sample sizes, and data collection and analysis were not performed blinded to the clinical outcome (fatal COVID-19). Demographic and clinical data (COVID symptoms, detailed pre-existing comorbidities, clinical outcome from all residents and pre-COVID pharmacological data; the level of detail differed per nursing home) were collected from electronic health records provided by the nursing homes and hospitals. Missing data were not imputed, and only individuals with all available parameters respective to the specific model were included. Stepwise logistic regression was used to identify risk factors for fatal COVID-19, and the best model was selected using cAIC. Kaplan–Meier estimates of survival were calculated up to 60 d after the first SARS-CoV-2 PCR-positive case in each nursing home outbreak. Selected predictors were confirmed by Cox proportional hazard regression, defining survival in days since PCR diagnosis. In sensitivity analyses, only fully vaccinated (defined as two BNT162b2 doses received at least 14 d before the start of the outbreak) and PCR-positive residents were included. Because transcriptomic data did not follow a normal distribution (determined by Kolmogorov–Smirnov test), non-parametric Kruskal–Wallis test (with FDR correction for multiple testing) was used to compare three groups (PCR-negative, PCR-positive and fatal cases) and Mann–Whitney test for two groups (fatal versus non-fatal). Pathway scores were normally distributed and analyzed using a t-test (with Welch’s correction when needed, fatal versus non-fatal groups). All statistical tests were two-sided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are included in the manuscript, Source Data files and supplementary files. Demographic, clinical and nCounter data are available from the authors upon reasonable request, due to privacy protection. All Source Data and code related to phylogenetic analysis (Fig. 1 and Extended Data Figs. 2 and 3) are available for download at https://www.zidu.be/SI_data.zip. The following GISAID IDs corresponding to SARS-CoV-2 genomes were generated as part of this study: EPI_ISL_2289002, EPI_ISL_2301430, EPI_ISL_2304141, EPI_ISL_2304143, EPI_ISL_2348574-78, EPI_ISL_2348580-86, EPI_ISL_2348587-92, EPI_ISL_2626083-96, EPI_ISL_2864473-74, EPI_ISL_2864478, EPI_ISL_2864483, EPI_ISL_2864485, EPI_ISL_2864489, EPI_ISL_2864573-76, EPI_ISL_2864707-10, EPI_ISL_2864714-15, EPI_ISL_2864717-21, EPI_ISL_2886237, EPI_ISL_3118412-26, EPI_ISL_4007338, EPI_ISL_4008034, EPI_ISL_4008052, EPI_ISL_4348705, EPI_ISL_4348711, EPI_ISL_4348959, EPI_ISL_4354278, EPI_ISL_4358318, EPI_ISL_4571448-51 and EPI_ISL_5349110.
Code availability
All code necessary to reproduce the findings of this study is provided in the Supplementary Information: https://www.zidu.be/SI_data.zip.
References
Hardy, J. O. et al. A world apart: levels and determinants of excess mortality due to COVID-19 in care homes: the case of the Belgian region of Wallonia during the spring 2020 wave. Demogr. Res. 45, 1011–1040 (2021).
Dellicour, S. et al. Investigating the drivers of the spatio-temporal heterogeneity in COVID-19 hospital incidence—Belgium as a study case. Int. J. Health Geogr. 20, 29 (2021).
Gillain, S., Belche, J.-L. & Moreau, J.-F. COVID-19 epidemic in the nursing homes in Belgium. J. Nursing Home Res. 6, 40–42 (2020).
Suñer, C. et al. A retrospective cohort study of risk factors for mortality among nursing homes exposed to COVID-19 in Spain. Nat. Aging 1, 579–584 (2021).
Collier, D. A. et al. Age-related immune response heterogeneity to SARS-CoV-2 vaccine BNT162b2. Nature 596, 417–422 (2021).
Grange, Z. et al. Characteristics and risk of COVID-19-related death in fully vaccinated people in Scotland. Lancet 398, 1799–1800 (2021).
Juthani, P. V. et al. Hospitalisation among vaccine breakthrough COVID-19 infections. Lancet Infect. Dis. 21, 1485–1486 (2021).
Thompson, D.-C. et al. The impact of COVID-19 pandemic on long-term care facilities worldwide: an overview on international issues. Biomed. Res. Int. 4, 8870249 (2020).
Thomas, S. J. et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine through 6 months. N. Engl. J. Med. 385, 1761–1773 (2021).
Haas, E. J. et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalizations, and deaths following a nationwide vaccination campaign in Israel: an observational study using national surveillance data. Lancet 397, 1819–1829 (2021).
Cabezas, C. et al. Associations of BNT162b2 vaccination with SARS-CoV-2 infection and hospital admission and death with covid-19 in nursing homes and healthcare workers in Catalonia: prospective cohort study. BMJ 374, n1868 (2021).
Suetens, C. et al. Increasing risk of breakthrough COVID-19 in outbreaks with high attack rates in European long-term care facilities, July to October 2021. Euro Surveill. 26, 2101070 (2021).
Rivasi, G. et al. Course and lethality of SARS-CoV-2 epidemic in nursing homes after vaccination in Florence, Italy. Vaccines (Basel) 9, 1174 (2021).
Faggiano, F. et al. An outbreak of COVID-19 among mRNA-vaccinated nursing home residents. Vaccines (Basel) 9, 859 (2021).
McMichael, T. et al. Epidemiology of COVID-19 in a long-term care facility in King County, Washington. N. Engl. J. Med. 382, 2005–2011 (2020).
World Health Organization. WHO criteria for COVID-19-related death. https://www.who.int/news-room/commentaries/detail/estimating-mortality-from-covid-19 (2020).
Aalto, U. L. et al. COVID-19 pandemic and mortality in nursing homes across USA and Europe up to October 2021. Eur. Geriatr. Med. 13, 705–709 (2022).
Mbalayen, F. et al. The COVID-19 pandemic and responses in nursing homes: a cross-sectional study in four European countries. Int. J. Environ. Res. Public Health 19, 15290 (2022).
Menezes, S. M., Braz, M., Llorens-Rico, V., Wauters, J. & Van Weyenbergh, J. Endogenous IFNβ expression predicts outcome in critical patients with COVID-19. Lancet Microbe 2, e235–e236 (2021).
Llorens-Rico, V. et al. Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat. Commun. 12, 6243 (2021).
Fukutani, K. F. et al. In situ immune signatures and microbial load at the nasopharyngeal interface in children with acute respiratory infection. Front. Microbiol. 9, 2475 (2018).
Lee, G. C. et al. Immunologic resilience and COVID-19 survival advantage. J. Allergy Clin. Immunol. 148, 1176–1191 (2021).
Chua, R. L. et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020).
Unterman, A. et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat. Commun. 13, 440 (2022).
Yoo, J. S. et al. SARS-CoV-2 inhibits induction of the MHC class I pathway by targeting the STAT1-IRF1-NLRC5 axis. Nat. Commun. 12, 6602 (2021).
Vigón, L. et al. Impaired cytotoxic response in PBMCs from patients with COVID-19 admitted to the ICU: biomarkers to predict disease severity. Front. Immunol. 12, 665329 (2021).
Castro de Moura, M. et al. Epigenome-wide association study of COVID-19 severity with respiratory failure. EBioMedicine 66, 103339 (2021).
Zhang, Q. et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 370, eabd4570 (2020).
Boudewijns, R. et al. STAT2 signaling restricts viral dissemination but drives severe pneumonia in SARS-CoV-2 infected hamsters. Nat. Commun. 11, 5838 (2020).
Goncalves, D. et al. Antibodies against type I interferon: detection and association with severe clinical outcome in COVID-19 patients. Clin. Transl. Immunol. 10, e1327 (2021).
Bastard, P. et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med. 218, e20210554 (2021).
Bastard, P. et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, eabd4585 (2020).
van der Wijst, M. G. P. et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 13, eabh2624 (2021).
Zhang, Q., Bastard, P., COVID Human Genetic Effort, Cobat, A. & Casanova, J.-L.Human genetic and immunological determinants of critical COVID-19. Nature 603, 587–598 (2022).
Monk, P. D., Marsden, R. J. & Tear, V. J. Safety and efficacy of inhaled nebulised interferon beta-1a (SNG001) for treatment of SARS-CoV-2 infection: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Respir. Med. 9, 196–206 (2021).
Alavi Darazam, I. et al. Role of interferon therapy in severe COVID-19: the COVIFERON randomized controlled trial. Sci. Rep. 11, 8059 (2021).
Kalil, A. C. et al. Efficacy of interferon beta-1a plus remdesivir compared with remdesivir alone in hospitalised adults with COVID-19: a double-bind, randomised, placebo-controlled, phase 3 trial. Lancet Respir. Med. 9, 1365–1376 (2021).
Ader, F. et al. An open-label randomized controlled trial of the effect of lopinavir/ritonavir, lopinavir/ritonavir plus IFN-β-1a and hydroxychloroquine in hospitalized patients with COVID-19. Clin. Microbiol. Infect. 27, 1826–1837 (2021).
Vinh, D. C. et al. Harnessing type I IFN immunity against SARS-CoV-2 with early administration of IFN-β. J. Clin. Immunol. 41, 1425–1442 (2021).
Park, A. & Iwasaki, A. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27, 870–878 (2020).
Lopez, J. et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J. Exp. Med. 218, e20211211 (2021).
Hu, H. et al. Increased circulating cytokines have a role in COVID-19 severity and death with a more pronounced effect in males: a systematic review and meta-analysis. Front. Pharmacol. 13, 802228 (2022).
Van Weyenbergh, J., Wietzerbin, J., Rouillard, D., Barral-Netto, M. & Liblau, R. Treatment of multiple sclerosis patients with interferon-beta primes monocyte-derived macrophages for apoptotic cell death. J. Leukoc. Biol. 70, 745–748 (2001).
Sancéau, J., Hiscott, J., Delattre, O. & Wietzerbin, J. IFN-β induces serine phosphorylation of Stat-1 in Ewing’s sarcoma cells and mediates apoptosis via induction of IRF-1 and activation of caspase-7. Oncogene 19, 3372–3383 (2000).
Dierckx, T. et al. IFN-β induces greater antiproliferative and proapoptotic effects and increased p53 signaling compared with IFN-α in PBMCs of adult T-cell leukemia/lymphoma patients. Blood Cancer J. 7, e519 (2017).
Mavrikaki, M. et al. Severe COVID-19 is associated with molecular signatures of aging in the human brain. Nat. Aging 2, 1130–1137 (2022).
Leal, F. E. et al. Comprehensive antiretroviral restriction factor profiling reveals the evolutionary imprint of the ex vivo and in vivo IFN-β response in HTLV-1-associated neuroinflammation. Front. Microbiol. 9, 985 (2018).
Menezes, S. M. et al. CD80+ and CD86+ B cells as biomarkers and possible therapeutic targets in HTLV-1 associated myelopathy/tropical spastic paraparesis and multiple sclerosis. J. Neuroinflammation 11, 18 (2014).
Bastard, P., Zhang, Q., Zhang, S. Y., Jouanguy, E. & Casanova, J. L. Type I interferons and SARS-CoV-2: from cells to organisms. Curr. Opin. Immunol. 74, 172–182 (2022).
Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020).
Gómez-Carballa, A. et al. A multi-tissue study of immune gene expression profiling highlights the key role of the nasal epithelium in COVID-19 severity. Environ. Res. 210, 112890 (2022).
Pescarmona, R. et al. Comparison of RT–qPCR and NanoString in the measurement of blood interferon response for the diagnosis of type I interferonopathies. Cytokine 113, 446–452 (2019).
Khoo, S. H. et al. Optimal dose and safety of molnupiravir in patients with early SARS-CoV-2: a phase I, open-label, dose-escalating, randomized controlled study. J. Antimicrob. Chemother. 76, 3286–3295 (2021).
Fischer, W. A. 2nd et al. A phase 2a clinical trial of molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci. Transl. Med. 14, eabl7430 (2022).
Abdelnabi, R. et al. Molnupiravir inhibits replication of the emerging SARS-CoV-2 variants of concern in a hamster infection model. J. Infect. Dis. 224, 749–753 (2021).
Sun, F., Lin, Y., Wang, X., Gao, Y. & Ye, S. Paxlovid in patients who are immunocompromised and hospitalised with SARS-CoV-2 infection. Lancet Infect. Dis. 22, 1279 (2022).
Najjar-Debbiny, R. et al. Effectiveness of paxlovid in reducing severe COVID-19 and mortality in high risk patients. Clin. Infect. Dis. 76, e342–e349 (2023).
Saravolatz, L. D., Depcinski, S. & Sharma, M. Molnupiravir and nirmatrelvir–ritonavir: oral COVID antiviral drugs. Clin. Infect. Dis. 76, 165–171 (2023).
Our World in Data. Coronavirus (COVID-19) Vaccinations. https://ourworldindata.org/covid-vaccinations
Smith, D. J. et al. COVID-19 mortality and vaccine coverage—Hong Kong Special Administrative Region, China, January 6, 2022–March 21, 2022. MMWR Morb. Mortal. Wkly Rep. 71, 545–548 (2022).
ARTIC Network. SARS-CoV-2. https://artic.network/ncov-2019
Freed, N. E., Vlkova, M., Faisal, M. B. & Silander, O. K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc/ 5, bpaa014 (2020).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures Math. Life Sci. 17, 57–86 (1986).
Sagulenko, P., Puller, V. & Neher, R. A. TreeTime: maximum-likelihood phylodynamic analysis. Virus Evol. 4, vex042 (2018).
Acknowledgements
We would like to acknowledge the numerous diagnostics laboratories involved in performing the consecutive screenings in the three nursing homes and for sharing detailed RT–qPCR results. Special thanks go to the representatives of the three nursing homes for providing additional and detailed information to make this collaboration fruitful: the (nursing) staff of Nos Tayons in Nivelles (with specific support of P. Gilbert), of Les Cytises nursing home in Braives and of Ter Burg in Zaventem (with special thanks to N. Deblaere, R. Verschueren and K. Boydens). UZ Leuven, as national reference center for respiratory pathogens, is supported by Sciensano, which is gratefully acknowledged. J.M.C. was supported by the HONOURs Marie-Sklodowska-Curie training network (grant 721367). This work is also supported by ‘Interne Fondsen KU Leuven/Internal Funds KU Leuven’ project 3M170314 and C3/20/105 awarded to P.M. The sequencing capacity of this work was supported, in part, by a COVID-19 research grant of ‘Fonds Wetenschappelijk Onderzoek’/Research Foundation Flanders (grant G0H4420N) awarded to P.M., G.B. and E.A. S.D. is supported by the ‘Fonds National de la Recherche Scientifique’ (FNRS, Belgium) and by the European Union Horizon 2020 project MOOD (grant agreement no. 874850). G.B. acknowledges support from the Internal Funds KU Leuven (grant C14/18/094) and from the Research Foundation–Flanders (‘Fonds voor Wetenschappelijk Onderzoek–Vlaanderen’, G0E1420N and G098321N), the latter grant also for S.D. J.V.W. is supported by the Research Foundation–Flanders (G0A0621N and G065421N). P.V. is a senior clinical investigator of the Research Foundation–Flanders. K.K.A. is supported by Research Foundation–Flanders (FWO G0G4220N). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
Conceptualization: L.C., E.K., G.B., P.M., E.A., S.D. and J.V.W.; methodology and experiments: S.M.M., M.S., T.W.B., J.M.C., B.V., B.V.H., M.B., E.W., J.V.E., M.W., P.V., J.M.D., F.D., K.D., J.R., R.D.M., J.N., K.L. and Consortium; formal analysis and investigation: L.C., S.L.H., S.D. and J.V.W.; data resources: J.M.D., F.D., K.D., J.R., R.D.M., C.B., A.D., C.B., B.C. and Consortium; data curation: L.C., E.K. and J.V.W.; writing—original draft preparation: L.C., E.K., G.B., P.M., E.A., S.D. and J.V.W.; writing—review and editing: L.C., E.K., G.B., P.M., E.A., S.D., J.V.E., J.M.C., F.D., B.C., K.L., Consortium and J.V.W.; visualization: S.L.H., S.D. and J.V.W.; project administration: S.G. and M.V.R.; funding acquisition: E.A., P.M., G.B., S.D. and J.V.W. All authors read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
K.L. received consultancy fees from MRM Health and Merck Sharp & Dohme, speaker fees from Pfizer and Gilead and service fees from Thermo Fisher Scientific and TECOmedical, all outside the reported work. The other authors report no competing interests.
Peer review
Peer review information
Nature Aging thanks Giuseppe Novelli, Emma Thomson and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overview of the outbreak in nursing home B (Gamma/P.1).
We report the evolution through time of Ct values measured in both infected residents (a) and aerosols analyzed in the lounge of the nursing home (b). Grey dots refer to negative PCR results. In addition, we also report the time-scaled phylogenetic analysis involving Gamma (P.1) genomes sampled and sequenced from this outbreak (c), showing one phylogenetic cluster (zoomed in the red circle, scale bar corresponds to the full tree) among 6 full genomes (orange dots), likely corresponding to a single introduction event into nursing home B. The phylogenetic tree is time-calibrated, meaning that branch lengths are in units of time (year).
Extended Data Fig. 2 Overview of the outbreak in nursing home C (Mu/B.1.621).
We report the evolution through time of Ct values measured in both infected resident/staff members (a) and aerosols analyzed in various sections within the nursing home (b). Grey dots refer to negative PCR results. In addition, we also report the time-scaled phylogenetic analysis involving Mu (B.1.621) genomes sampled and sequenced from this outbreak (c), showing one phylogenetic cluster (with short branch lengths) among 24 full genomes (orange dots) likely corresponding to a single introduction event. The phylogenetic tree is time-calibrated, meaning that branch lengths are in units of time (year).
Extended Data Fig. 3 Univariate and multivariate Kaplan-Meier survival curves.
Kaplan-Meier survival curves comparing (a) SARS-CoV-2 variants Delta vs. non-Delta (Gamma/Mu, Log-rank test, p = 0.28), (b) Age above or below the median (86 years, Log-rank test, p = 0.078); (c) Onset of SARS-CoV-2 diagnosis (PCR+): early (0–7 days) vs. late (>7 days), with regard to the start of the respective outbreaks, Log-rank test, p = 0.40; (d) Combined probability of age, sex, variant, and late onset of diagnosis (Log-rank test, p = 1.1 × 10−7).
Extended Data Fig. 4 LAIR1 expression (a surrogate marker for anti-type I IFN neutralizing antibodies) correlates with IRF3 expression in fatal cases only.
(a) LAIR1 expression (nasal mucosa, quantified by nCounter digital transcriptomics) correlates positively with peak SARS-CoV-2 viral load (that is negatively with nadir Cq value) across all residents. (b) LAIR1 expression does not correlate with IRF3 expression (nasal mucosa) in PCR-positive residents. (c) LAIR1 expression correlates negatively with IRF3 expression (nasal mucosa) in fatal COVID-19 cases. All correlations Spearman.
Supplementary information
Supplementary Information
Supplementary Information and Supplementary Methods
Supplementary Tables
Supplementary Tables 1–6
Source data
Source Data
Statistical source data for Figs. 2–4
Source Data
Source data for Extended Data Figs. 3 and 4
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuypers, L., Keyaerts, E., Hong, S.L. et al. Immunovirological and environmental screening reveals actionable risk factors for fatal COVID-19 during post-vaccination nursing home outbreaks. Nat Aging 3, 722–733 (2023). https://doi.org/10.1038/s43587-023-00421-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-023-00421-1
