
Abstract
Here we confirm migration of chlorinated solvents tetrachloroethene and trichloroethene, and co-disposed elemental mercury as dense non-aqueous phase liquid in an aquifer at a scale larger than previously documented in the literature, over 650 m from original surface discharge. This finding enhances the conceptual model explaining extensive contaminated plume persistence by adding structurally controlled dense non-aqueous phase liquid to known mechanisms such as source dissolution and matrix back-diffusion. Following injections of oxidants at a depth greater than 50 m, which effectively destroyed dissolved chlorinated contaminants during a groundwater remediation pilot test in South Carolina, we measured excess chloride higher than attributable to pre-test dissolved concentrations of chlorinated contaminants, and oxidative releases of mercury, which is an opportunistic tracer of non-aqueous phase solvent transport when in its elemental form. The results suggest potentially targeted destruction strategies in disconnected non-aqueous phase liquid accumulation areas may reduce remediation timeframe and support cleanup of sites previously considered technically impracticable.
Similar content being viewed by others

Introduction
Multiphase transport theory clearly allows for non-aqueous phase liquid movement over large distances through porous media as dictated by physical and chemical driving and facilitating parameters such as non-aqueous phase fluid and porous media properties1,2,3,4. However, available literature generally presume that chlorinated solvent dense non-aqueous phase liquid (DNAPL) transport and deposition is limited to a maximum of tens of meters from the release area in most contaminated site scenarios5,6,7,8,9,10,11. The assumption of limited lateral transport is especially true when the contaminant release was discontinued more than a decade before the site was characterized8,12,13. Long and expansive aqueous plumes of contaminated groundwater, sometimes kilometers in length, often found at sites with large releases (>1000 kg) of chlorinated DNAPLs are typically explained primarily by dissolution of the non-aqueous phase solvent into the groundwater near the release area. The dissolved chlorinated contaminant then transports with groundwater to make large aqueous contaminated plumes14,15. Although some researchers15,16 recognized the importance and difficulty of locating and treating small, dispersed DNAPL, they did not recognize the possibility of distant non-aqueous phase transport and in fact warn against identifying a remote DNAPL source from localized high concentration groundwater farther downgradient15. If DNAPL near the release has been eliminated, either by dissolution and transport in groundwater or by direct treatment/extraction, the persistent distal plume is presumed to be sustained primarily by back diffusion of aqueous phase contamination out of the aquifer matrix14,17,18. Indeed, the position that back diffusion is likely more important than residual DNAPL is explicitly stated in guidance documents19. The justification for this assumption is based on analytical and numerical models14,18,20,21, laboratory microcosm and mesocosm experiments22,23,24,25, as well as field data from well-characterized and monitored sites14,17,18,26,27. Unfortunately, the data from these laboratory and field experiments often do not have the resolution to detect theoretically possible small, disperse occurrences (e.g., micro- or even nano-blobs) of non-aqueous phase liquids (NAPL)28,29,30. It is difficult enough to directly detect larger occurrences of NAPL in pools or large ganglia which occur in complex three-dimensional structures (architecture) as a result of spatially and temporally heterogeneous transport in heterogeneous and anisotropic porous media13,29,31,32,33,34,35. The much smaller micro-NAPL occurrences, postulated here, would likely be held within porous media as a residual outside of the predominant groundwater advective pathways and would be more difficult to directly detect or mobilize for collection1,36. Samples distant from the release area are either liquid (likely all aqueous) or solid (e.g., bulk core). Unless these samples have captured a volume with a density of NAPL (e.g., a large number of NAPL micro-blobs) that results in the exceedance of the aqueous solubility of these compounds, NAPL could easily be missed or not easily distinguished from the dissolved component. This potential conceptual model of remote DNAPL is usually not considered in favor of the long-distance transport of a high concentration dissolved phase. More direct methods like in situ optical spectroscopy will also be unlikely to detect NAPL if the optical cross section of the NAPL is too small or if the occurrences of NAPL are three-dimensionally dispersed and complex37,38.
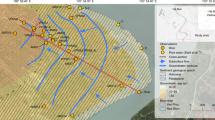
At the United States Department of Energy’s Savannah River Site (SRS) M Area (Aiken, South Carolina) shown in Fig. 1, ~1.7 million kilograms of chlorinated solvents comprised primarily of tetrachloroethene (PCE) and trichloroethene (TCE) were released to the subsurface as DNAPLs in a settling basin and through leaks in a terra cotta sewer line connecting the manufacturing process area to the settling basin from ~1950–198539. The subsurface at SRS is comprised of interbedded layers of sand, silt, and clay consistent with a fluvial, shallow marine, or lagoonal depositional environment typical of the coastal plain sediments in the southeastern United States39,40,41,42. The layering is often discontinuous but there are some locally extensive layers that serve as confining or semi-confining intervals that affect fluid transport42. Groundwater flow in this area is generally to the south towards tributaries leading to the Savannah River41. DNAPL was only collected from one well adjacent to the settling basin but high aqueous concentrations were found in wells along a tortuous pathway to distances more than a kilometer west of the basin (e.g., MSB76: 48 mg l−1 PCE, 32 mg l−1 TCE) and not consistent with the prevailing groundwater flow40. To explain these high concentrations in unexpected areas, a site conceptual model was developed that was based on density driven transport of DNAPL (see Fig. 1c) along fine grain strata below the water table43. A multiple lines of evidence approach was used to construct the site conceptual model that included both conventional and innovative techniques43. High vertical resolution mapping of the subsurface strata using standard drilling, core recovery, and analysis, as well as direct push (primarily cone penetrometer) based tools to differentiate soil type were conducted to identify likely DNAPL flow paths40,43,44. High vertical resolution sampling and analysis of PCE and TCE in groundwater and soil using innovative rapid gas chromatograph headspace methods was able to quantify and differentiate concentrations in samples that sometimes varied by three orders of magnitude yet were separated by <30 centimeters39,41,42,43. Several innovative in situ analytical spectroscopic methods using either direct optical probes advanced by direct push or in situ sampling and analysis were tested at the site and provided information on the heterogeneity of PCE and TCE contamination37,44,45,46. The earliest NAPL FLUTe® deployment was performed at the site and detected discrete occurrences of NAPL within 30 meters of one of the source release areas10,27,39. A partitioning tracer test was also conducted at the site47. Finally, both surface and cross borehole geophysical techniques including electrical conductivity and induction logs, complex resistivity, and seismic were used to identify potential pathways and to try and identify NAPL in the subsurface48,49,50,51. The lines of evidence supported the DNAPL transport conceptual model but could not definitively exclude the possibility of high concentration aqueous transport as the sole cause of the high concentrations downgradient.

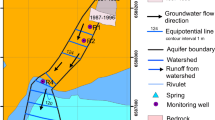
a Location of SRS in South Carolina, United States. b SRS bounded in yellow with M Area location (red circle) highlighted. The Savannah River and surrounding counties including Aiken and Barnwell are marked. c M Area lost lake aquifer zone TCE plume showing groundwater flow direction and the hypothesized DNAPL flow path. Red stars indicate the locations of recovery wells (RWM) used in the groundwater containment and recovery system in the area. Blue and green squares show soil boring locations, while blue, green, and black circles show monitoring well locations. The yellow, blue, green, light pink, and pink shades indicate TCE concentrations from 5–100, >100–1000, >1000–10,000, >10,000–50,000, and >50,000 µg l−1, respectively. The red arrow indicates a possible DNAPL migration path based on density driven transport along fine-grain geologic strata. The location of the ISCO test site is also shown as a purple rectangle. The red triangle shows the location of recovery well RWM 018 that controls groundwater flow in the ISCO test area. Please note that panel c is oriented to Plant North in the SRS coordinate system rather than True North as shown in the legend.
Analysis of the release of NAPL co-contaminants, specifically compounds that preferentially transport in the separate non-aqueous liquid phase, provide an innovative alternative NAPL detection method that supplements direct sampling of parent and breakdown products in solid and liquid bulk phase and the other tools and techniques mentioned above. Example NAPL co-contaminants include hydrophobic organics or inorganics such as polychlorinated biphenyls or elemental mercury (Fig. 2). To be an effective tracer of NAPLs, these compounds must have a low background concentration in the bulk aquifer and must have been co-released with the NAPL as is the case for elemental mercury (Hg) at the Savannah River Site52. Figure 2 is a conceptual model that depicts and describes the processes that result in higher than expected mercury concentrations (up to ~0.003 µM) in groundwater recovered by the SRS M Area pump and treat system. The mercury measured in the groundwater plume has been attributed to a process composed of a series of steps beginning with enhanced solubilization of elemental mercury (Hg0) in solvents within the processing facilities (DNAPL phase mercury concentrations > 5.29 µM)52,53,54. The elemental mercury laden DNAPL was then released as a waste to the subsurface and descended through the vadose zone into the groundwater. The mercury in the DNAPL is slowly oxidized at the NAPL/water interface by the aerobic groundwater. Finally, the oxidized, and now ionic mercury complexes with chloride to generate a relatively mobile form of mercury (with maximum aqueous concentrations about 50 times background). During in situ chemical oxidant applications in the field, the mercury oxidation step is enhanced resulting in the potential to rapidly release significant quantities of mercury over a short time if NAPL is present. The baseline conceptual model of mercury as a co-contaminant in NAPL has thus been extended to support a more definitive assessment of the presence of NAPL in the oxidant injection test area.

Conceptual model explaining why higher than background mercury concentrations are detected in groundwater recovery wells in the M Area of SRS. DNAPL (solid red) comprised of PCE and TCE and co-disposed Hg0 travels downward via density driven flow in a tortuous path through the vadose zone (white), below the water table (blue triangle), then through the aquifer until constrained by fine grain aquifer materials. Hg0 oxidation at the DNAPL:H2O interface occurs under bulk aerobic geochemical conditions and complexes of oxidized ionic mercury (e.g., HgCl20) are formed which are soluble in groundwater. The mobile aqueous phase complexes travel with groundwater and are collected in monitoring and recovery wells. Vermillion, bluish green, and sky blue cross hatched areas indicate qualitative cVOC concentration contours.
The conceptual model depicted in Fig. 2 simplifies DNAPL transport consistent with literature assumptions of interfacial pools and isolated blobs localized to within tens of meters from the release area. However, the conceptual model for mercury laden DNAPL transport can be extended to account for blobs, sheets at interfaces, and micro blobs transported much further away. At this site, it is conceivable that DNAPL was transported along preferential pathways driven by high hydraulic head conditions during the periods of waste solvent release to the subsurface. Ganglia of DNAPL were then stranded by changes in flow conditions after release of the contaminants was stopped55. While there are some general similarities in future projected groundwater concentration trends among the non-exclusive scenarios of high concentration aqueous transport, back/matrix diffusion, and widely distributed NAPL, an improved conceptual understanding of the inter-relationships and spatial pattern for scenarios at a site may provide improved management options. Notably, the potential for rapid remediation in a remote DNAPL source scenario is much higher if the DNAPL is recognized and the approximate location of the DNAPL can be identified and targeted as opposed to assuming only back diffusion is controlling the system, which will only permit access and treatment at the rate of diffusion without disrupting the matrix.
If DNAPL is present in a location, perturbing the subsurface system with oxidant injections that hasten the depletion of the DNAPL would generate two primary results. First, more PCE and TCE would be destroyed than could be accounted for by the aqueous phase mass and second, co-contaminants such as mercury would rise significantly52,53. In addition, if DNAPL were completely eliminated in the zone of capture of a monitoring well, TCE and PCE concentration “rebound” would be greatly and permanently attenuated. In addition to the practical goals of determining the feasibility of a commonly used remedial technique for chlorinated alkene remediation, the oxidant injection pilot test offered the opportunity to more definitively ascertain the remote DNAPL transport conceptual model.
The aim of this work was to provide evidence that DNAPL was transported in the subsurface >650 meters from the original release location and has persisted for more than three decades. The principal innovation of this work was the opportunistic use of co-disposed metallic mercury as a tracer selectively transported in the carrier phase (DNAPL). When DNAPL is intentionally depleted by a localized application of chemical oxidants, the metallic mercury is susceptible to rapid oxidation by the applied chemical amendments. The oxidized mercury, now much more soluble in the aqueous phase than the neutrally valent species, is sensitively detected in groundwater. We performed a field test where oxidant solutions were injected into an aquifer suspected of containing residual DNAPL far from the source/release area. Groundwater flow in this area is controlled by nearby recovery well RWM 018 (Fig. 1). Analytical results from this field pilot and concomitant lab tests show a large depletion of TCE and PCE, and significant increases in mercury concentration in groundwater from wells where increased chloride and oxidant concentrations have been detected after injections. When converted to molar equivalent chlorine and compared to chloride concentration increases after oxidant injection, pre-injection aqueous phase TCE and PCE could not produce the increase in chloride measured in some of the monitoring wells. Although additional time is needed for verification, after more than a year of monitoring following the final oxidant injection event most of the affected monitoring wells have markedly lower concentrations – little to no “rebound”. Despite the lack of direct measurements of a separate phase or concentrations exceeding solubility for the chlorinated solvents, the simplest explanation is that localized DNAPL is present far from the source/release area and is an important contributor to the groundwater plume.
Results
The mean and standard deviation of analytes that were expected to be indicators of injection influence are provided in Table 1 below. These parameters, mostly compounds comprising the injectates, were expected to change significantly if groundwater in the zone of capture of a particular monitoring well was affected by injection. They are also expected to have a relatively low and uniform distribution in the aquifer.
Manganese and sodium concentrations were used to evaluate the approximate arrival time of potassium permanganate and sodium persulfate, respectively. Arrival times for injection event 1 were determined based on concentrations exceeding the baseline average value plus three standard deviations and are provided in Supplementary Table 1 for each injection event. Since potassium permanganate was injected prior to sodium persulfate in both injection events, it was expected that sodium concentrations would lag manganese concentrations in the monitoring wells. However, this was not always the case since the sample frequency may not have been high enough to differentiate arrival times in some cases and the two injectates may have transported through different paths in others.
PCE and TCE concentrations are not uniformly distributed so baseline conditions are more difficult to establish but clearly decreased in areas that were affected by injections as determined by an increase in the injectate compounds. Figure 3 shows persistent concentrations of chlorinated volatile organic compounds (cVOCs) TCE and PCE exceeding 10 mg L−1 until high concentrations of the oxidants arrive ~4 months after the first injection. In Figs. 3–6, markers represent sample dates and lines between sample dates are included to help identify trends. TCE and PCE are converted to their molar equivalents of chlorine and combined in this figure. A small rebound is evident when permanganate and persulfate concentrations attenuate ~1 year after the first oxidant injection but is extinguished ~1 month after the second injection. Concentration data are provided as Supplementary Data.xlsx and are retained in Open Science Framework repository56: (https://osf.io/azhvy/?view_only=3953a7d698864f279345cd275cee37b5).

Reddish purple and orange lines indicate beginning and end of permanganate and persulfate solution injections, respectively. Vermillion line with vermillion diamonds mark concentrations of cVOCs as chlorine, which is depleted soon after high concentrations of oxidants arrive. Bluish green line with large bluish green triangles mark chloride concentrations, which increases as cVOCs are destroyed by oxidants. Blue line with small blue triangles mark concentration of manganese, which is directly related to permanganate concentration. Yellow line with yellow squares mark concentrations of sodium, which is directly related to persulfate concentration. Only two dates (9/25/18 and 6/9/21) were analyzed in duplicate for chloride and the data ranges are too small to see in this figure (range 4 µM).

Reddish purple and orange lines indicate beginning and end of permanganate and persulfate solution injections, respectively. Vermillion line with vermillion diamonds mark concentrations of cVOCs as chlorine. Bluish green line with large bluish green triangles mark chloride concentrations, which increases as cVOCs are destroyed by oxidants. Sky blue line with sky blue circles mark concentrations of mercury, which significantly increase after oxidant injections. Black line indicates average baseline mercury concentration. Duplicate analyses of chloride were performed on 9/30/20 (range 20 µM) and 6/9/21 (range 1 µM) so conservative error bars of ±10 µM were placed on all chloride data in the figure. A duplicate analysis of mercury (9/30/20) had a data range too small to see in this figure (0.0002 µM).

Reddish purple and orange lines indicate beginning and end of permanganate and persulfate solution injections, respectively. Vermillion line with vermillion diamonds mark concentrations of cVOCs as chlorine, which is depleted after oxidant injection. Bluish green line with large bluish green triangles mark chloride concentrations, which are higher than can be accounted for by cVOC destruction. Sky blue line with sky blue circles mark concentrations of mercury, which significantly increase after the second oxidant injections. Black line indicates average baseline mercury concentration. Duplicate analyses of PCE and TCE were performed on 11/25/19 (range 3 µM combined as chlorine) and 3/16/20 (range 74 µM combined as chlorine) so conservative error bars of ±37 µM were placed on all cVOCs as chlorine data in the figure. Duplicate analyses of mercury were performed on 3/12/19 (range 0.0003 µM), 11/25/19 (range 0.0037 µM), and 3/16/20 (range 0.0005 µM) so conservative error bars of ±0.0019 µM were placed on all mercury data in the figure.

Reddish purple and orange lines indicate beginning and end of permanganate and persulfate solution injections, respectively. Vermillion line with vermillion diamonds mark concentrations of cVOCs as chlorine, which is depleted after oxidant injection. Bluish green line with large bluish green triangles mark chloride concentrations, which are persistently higher than baseline and suggest an additional chloride source apart from aqueous phase cVOC destruction. Sky blue line with sky blue circles mark concentrations of mercury, which significantly increase after the second oxidant injections. Black line indicates average baseline mercury concentration. Only two dates (9/25/18 and 6/9/21) were analyzed in duplicate for chloride and the data ranges are too small to see in this figure (range 4 µM).
An increase of mercury is associated with a depletion of cVOCs and an increase in chloride as illustrated in Fig. 4. In this figure showing analytes measured in WSM002C before, during, and after the second oxidant injection, oxidant concentrations consistently exceeded 5 mM after injection. Initially, mercury concentrations appear to be closely correlated with chloride but the concentration match does not persist. However, it is clear that mercury concentrations are above baseline when chloride concentrations are above baseline. Due to the four order of magnitude difference in concentrations between mercury and chloride, two separate linearly scaled ordinate (y) axes are used in this figure to more clearly show the concentration behavior with time of each. In addition to the significant increase in mercury more than five times above baseline concentration, chloride concentrations exceed the maximum expected values predicted from destruction of cVOCs.
Nearly all of the wells showing an increase in chloride and impacts by oxidants had occurrences of mercury concentrations above the baseline mean plus three standard deviations, and several of the wells show significant increases in average mercury concentrations above baseline under the more stringent statistical requirements of the t-test after injections as seen in Table 2. It can be observed that high or low concentrations of mercury do not necessarily mirror the depletion of high or low concentrations of cVOCs, yet in all cases when chlorinated contaminants are depleted and oxidant arrival is observed, mercury concentrations are measured that are significantly higher than baseline. Figure 5 shows a large increase in mercury concentration in WSM002BB after the first injection but a rapid drop nearly to baseline concentrations despite elevated chloride concentrations and depleted cVOCs as chlorine. Mercury rises and falls with chloride in the latter part of 2019 and the first half of 2020, and rises again after the second injection. The difference in average mercury concentrations after the first injection are not significant under the conservative t-test criteria but there is a statistically significant increase after the second injection. Figure 5 also clearly shows an excess of chloride post injection in the fall of 2019 that cannot be accounted for by the combination of baseline chloride concentrations plus pre-injection chlorine concentrations from aqueous phase PCE and TCE. As in Fig. 4, two linearly scaled ordinate axes are provided to more clearly see the temporal behavior of each of the parameters.
Another example of an excess of chloride post-injection is provided in Fig. 6. In this figure the chloride concentration does not exceed the pre-injection combination of equivalent PCE and TCE chlorine plus baseline chloride concentration. However, high chloride persists in excess of the expected contributions of chlorine from aqueous phase cVOCs in the pore volume plus slow matrix diffusion. This post-injection persistently high chloride while TCE and PCE in groundwater are non-detect is observed in several of the wells and suggests an alternative, readily available source of chloride (e.g., DNAPL). Similar to Fig. 5, despite several measurements much higher after oxidant injections than pre-test measurements, the statistical difference in average mercury concentrations after the first injection is not significant under the conservative t-test criteria. However, there is a statistically significant increase in mercury after the second injection. As in Figs. 4, 5, due to the four order of magnitude difference in concentrations between mercury and chloride, two separate linearly scaled ordinate (y) axes are used in this figure to more clearly show the concentration behavior with time of each.
To determine if oxidant contact will extract mercury from sediments under controlled conditions, lab tests were performed. Since no core was collected and retained during the monitoring and injection well drilling performed for this pilot test, core collected from borings in the general vicinity of the pilot test but >10 years prior to this test were obtained from the SRS core storage facility. The plastic wrapped core is stored in dedicated, polymer coated fiberboard boxes in a climate-controlled storage area with limited access at SRS, which is a secure United States Department of Energy facility. Background mercury contained within the sediment would likely be in an oxidized state (solid oxides or sulfides) and not subject to a significant amount of volatilization loss. Therefore, experiments using the two oxidants to try and extract mercury for comparison to the field results are useful to demonstrate that the observed increase in mercury seen in the field is due to transported mercury and not due to extraction of ambient mercury within the solid matrix. Core was selected from eight different borings and subsamples were collected from specific elevations of the core to provide measurements from sediments that may have been exposed to DNAPL or high concentration aqueous cVOC liquids, and sediments that were likely not exposed. Measured concentrations of mercury under oxidant leaching conditions ranged from 7.0E-04–3.6E-03 µM, which is not statistically different from the measurements of mercury used to derive the field baseline value. Control samples of the oxidant solutions used for the injections showed mercury content of <4.0E-04 µM as indicated by the manufacturer’s certificates of analysis. The complete report on the laboratory analyses is provided as Supplementary Note 1.
Discussion
As part of metal fabrication and other processes, chlorinated solvents as DNAPLs were released through a process sewer line to the unlined M Area settling basin. DNAPLs were released to the vadose zone and ultimately into aquifers from both the sewer line and the basin. Process history indicates elemental mercury was released along with the chlorinated solvents and other process wastes. DNAPL has been collected from a well adjacent to the basin (source/release area) and analyzed for mercury content. Analyses indicate elemental mercury concentrations of 5.29 µM in the DNAPL. To test the SRS conceptual model of remote DNAPL transport as an important contributor to persistent, high concentrations of cVOCs in the aqueous phase plume, three lines of evidence were pursued in response to the injections of oxidant far from the source/release area and can be described by the following questions: (1) Were chloride concentrations higher post-injection than can be explained by a combination of baseline concentrations and conversion of PCE and TCE to chloride by oxidation?; (2) Did PCE and TCE concentrations recover to near pre-test levels after oxidants have diminished?; and (3) Were mercury concentrations significantly higher than pre-injection concentrations? The most important and least equivocal line of evidence is the last one.
Total mercury concentration measurements in the relevant groundwater monitoring wells prior to oxidant injections averaged ~0.002 µM but concentrations in several of these wells post oxidant injection increased by more than an order of magnitude to levels of 0.02–0.05 µM. In all groundwater samples from wells affected by the oxidant injections and showing a decrease in cVOCs and an increase in chloride, mercury concentrations were measured that were significantly higher than baseline concentrations. In several of the wells, the high measurements of mercury were sustained, likely as a result of sustained depletion of DNAPL that was in the monitoring well’s zone of capture. In other wells, discrete measurements of high mercury concentrations were interspersed with low measurements, possibly as a result of depletion of available DNAPL in the zone of capture pathways to the well.
The oxidant injectate solutions contained no mercury (verified by certificates of analysis and Savannah River National Laboratory measurements of starting oxidant solution at 0.0004 µM discussed in Supplementary Note 1). In controlled laboratory experiments, measurements of mercury leaching from analogous, but uncontaminated M Area sediments when exposed to oxidants at similar concentrations and durations to the field experiment show low concentrations (0.0007–0.0036 µM) not statistically different from baseline measurements used in the field test. Measurements of total mercury in groundwater from the site pump and treat recovery wells addressing the contaminant plume prior to oxidant injections have shown similar low, but above uncontaminated background concentrations53. The release of DNAPL transported co-contaminants is strong evidence in support of a conceptual model of the transport and persistence of DNAPL far from the location of their release and that distal DNAPL is an important source of persistent and high concentrations of PCE and TCE in the extended groundwater plume.
Although, it is too early to determine if cVOC rebound has been eliminated or significantly diminished because all of the monitoring wells have oxidant concentrations that are still above baseline, it is possible to evaluate excess chloride produced. It is clear that both the concentration and persistence of chloride beyond what can reasonably be explained by the sole conversion of an aqueous cVOC source has been observed. It is possible, assuming a large matrix diffusive area and reservoir, yet it does not seem likely that back diffusion could provide enough TCE and PCE to sustain the high chloride concentrations observed over the observed timeframe of this test57,58,59. The excess chloride data provide support to the mercury data strongly implicating remote transport of DNAPL.
During their release, DNAPL from the M Area settling basin and groundwater would have been subjected to much higher head forces (>30 m vadose zone) which likely resulted in different flow pathways for both groundwater and DNAPL than the current groundwater now follows. As a result, DNAPL could have been deposited and stranded (by snapoff and other processes) in locations which are not in the prevailing advective flow paths as determined by background groundwater gradients and the current aquifer recovery network55. The slow dissolution of and diffusion from residual DNAPL could be a significant source of continued groundwater contamination in addition to matrix diffusion from fine grain sediments59.
DNAPL can be transported by advection, capillary, density, or other forces that can overcome pore entry pressure limits. It can even be transported by condensation from the gas phase at a steam/pressure front. Chlorinated solvent DNAPL cannot be deposited in natural porous media by diffusion through the aqueous phase and subsequent agglomeration. Therefore, we can assume that DNAPL in the subsurface can be accessed by advective (e.g., amendment injections) or other forces and not limited by slow liquid diffusion. This would eliminate one impediment to technical practicability as a result of time to remediate because of access limited by the rate of diffusion.
Conclusions
As a result of a pilot scale oxidant injection test to remediate persistent high concentrations of PCE and TCE in the western sector plume of the Savannah River Site, we have identified the presence of DNAPL >650 m from the release. Significant increases in concentrations of total mercury above baseline, some more than an order of magnitude higher, were observed in all of the monitoring wells where oxidant constituent compounds were measured above baseline. In addition, chloride concentrations in excess of what would be expected from oxidative conversion of PCE and TCE were observed. The large increase in mercury concentrations indicates both the presence and destruction of local residual DNAPL in the volume of influence of the oxidant injections since elemental mercury was used in the manufacturing process, and dissolves in and can be transported in DNAPL but essentially not in aqueous phase liquids. The excess chloride data support the more definitive mercury data. The determination of DNAPL far from the source area suggests a conceptual model that requires consideration of alternatives to contaminant persistence in this area solely as a result of dissolved phase transport from the release area and local matrix diffusion. It also may suggest different remediation strategies that could reduce cleanup time since DNAPL has >1000 times the contamination potential of a similar mass or volume of aqueous phase contaminant and the reduction of that potent source could have a great impact on the persistence and magnitude of aqueous contaminant concentrations. The identification of a remote NAPL rather than a large-scale matrix diffusion contaminant source scenario may allow the conversion of a technically impracticable scenario to one where the site can be effectively remediated.
Methods
Monitoring and injection wells
The pilot test injection and monitoring well locations were selected to be in an area with persistent high concentrations of chlorinated solvents and consistent with the dominant groundwater flow gradient created by recovery well RWM 018. Recovery well RWM 018 was installed with a 15.2 m screen (elevations between 39.4 and 54.6 mamsl) across the upper and lower lost lake aquifer zones and is located ~180 m downgradient from the pilot test injection wells. Monitoring wells and the first set of injections wells were installed in the lost lake aquifer zone to depths >50 m below ground surface using rotasonic drilling in the spring of 2018 while the second set of injection wells were installed in the spring of 2020. The location of the proximal monitoring wells and both sets of injection wells are shown in Fig. 7.

Reddish purple circles mark the locations of the injection wells used in the first test. Blue triangles mark the location of the injection wells used in the second test. Vermillion squares mark the locations of the proximal monitoring wells. The black line indicates the direction of groundwater flow from A to A’ and is used to mark the cross-sectional view in Fig. 8. The sky blue arrow in the direction from A to A’ indicates that recovery well RWM 018 which controls groundwater flow in the test area is ~180 m from the injection wells.
An approximate cross section with stratigraphy is shown in Fig. 8. As described in the introduction, the subsurface at SRS is heterogeneous and anisotropic but it has some locally extensive layering. The stratigraphic section in Fig. 8 highlights some of the commonly identified, fine grain layers that appear to control fluid flow and separate aquifer zones.

The orange shaded area indicates heterogeneous sand, clay, and silt vadose zone materials. The black dotted lines indicate discontinuous strata with higher concentrations of clay. The blue line with blue triangle indicates the water table. The blue, sky blue, and light sky blue shaded areas indicate the unconfined, upper lost lake, and lower lost lake aquifer zones, respectively. The bluish green line indicates the relatively continuous green clay confining stratum. The dark red line indicates the relatively continuous upper clay confining stratum.
The first set of eight 5-cm diameter PVC injection wells (WSI001B, C; WSI002B, C; WSI003B, C; and WSI004B, C) shown in Fig. 3, were installed with 4.6 m screens in either the upper lost lake aquifer zone (C wells), or the lower lost lake aquifer zone (B wells). Descriptive details for the first set of injection wells are provided as Supplementary Table 2.
The second set of four 5-cm diameter PVC injection wells (WSI005B, C; and WSI006B, C) were installed with 1.5 m screens based on the results of the first injection which didn’t sufficiently address the shallower of the two target zones. Descriptive details for the second set of injection wells are provided as Supplementary Table 3.
Eight 5-cm diameter PVC monitoring wells (WSM001BB,B,CC,C; and WSM002BB,B,CC,C) were installed with 1.5 m screens allowing greater vertical resolution in the two lost lake aquifer zones (see Fig. 8). An additional four 5-cm diameter PVC monitoring wells (WSM003BB,B,CC,C) were installed ~70 m downgradient of the injection wells. Monitoring well details are provided as Supplementary Table 4. The radial distance between the first set of injection wells (WSI001,WSI002,WSI003,WSI004) and the proximal monitoring wells (WSM001,WSM002) with overlapping screen elevations ranges between 9.1 and 19.4 m. The average radial distance between the second injection wells (WSI005,WSI006) and proximal monitoring wells with overlapping screen elevations ranges between 3.7 and 11.7 m. The radial distance between injection wells and distal monitoring wells with overlapping screen elevations range between 71.4 and 79.8 m. Groundwater elevations in all monitoring wells fluctuate between ~61 and 66 mamsl. Nomenclature for the relevant wells and soil borings addressing lost lake aquifer zone contamination that are shown in Fig. 1 include MSB and MW indicating M area characterization or monitoring, and RWM indicating M area recovery wells.
Injections
For the first phase of the field test, injections of 18,927 liters of 267.5 mM potassium permanganate solution (Carus RemOx S, Peru, IL) in each of 8 injection wells were performed in 11 working days between 8/30/18 and 9/14/18. Injections of 18,927 liters of 296.0 mM sodium persulfate (Peroxychem Klozur SP, Philadelphia, PA) and 16.9 mM sodium hydroxide (Brenntag 25% NSF, North Charleston, SC) solution in each of the same 8 wells were performed in 8 working days between 9/18/18 and 10/1/18 directly after the permanganate injections. Both the potassium permanganate and sodium persulfate were received as dry granules and were mixed with potable water from the site to make the oxidant solutions. Details of the phase 1 injection are provided as Supplementary Tables 5, 6.
For the second phase, injections of 19,987 liters of 253.5 mM potassium permanganate solution in each of four injection wells were performed in four working days between 8/27/20 and 9/1/20 and injections of 19,230 liters of 291.4 mM sodium persulfate and 16.9 mM sodium hydroxide aqueous solution in each of the same four wells were performed in three working days between 9/3/20 and 9/8/20 directly after permanganate injections. Details of the phase 2 injection are provided as Supplementary Tables 7, 8.
Sampling and analysis
All wells were sampled and analyzed for cVOCs, anions, mercury, metals, and radionuclides. The proximal monitoring wells were sampled twice before the first injection, then weekly for 2 months during and immediately after the injections, then monthly for 4 months, and quarterly 6 months post-injection. The distal monitoring wells were sampled twice before the first injection, then quarterly. Other monitoring wells on site were sampled semi-annually. Only a small percentage of groundwater samples were analyzed as replicates with the majority reported as single samples. All relevant groundwater analytical data including replicate data are provided in the Supplementary Data file and are available in the OSF data repository (https://osf.io/azhvy/?view_only=3953a7d698864f279345cd275cee37b5). Standard analytical methods were used for all parameters and are listed in Supplementary Table 9.
Measured mass concentrations of parameters were converted to molar concentrations for several reasons including facilitating comparison between injection constituents to determine oxidant arrival time, and allowing mass balance calculations on cVOC destruction. It was assumed that the injectate solutions would oxidize TCE and PCE to carbon dioxide and chloride. By converting the pre-test concentrations of TCE and PCE to chlorine molar equivalents, it was possible to combine the two. This normalization allows a direct comparison to determine if the measured increase in chloride is consistent with pre-injection dissolved cVOC concentrations or if there is an excess of chloride indicating the potential for DNAPL.
Mercury background and oxidant leaching tests
Additional total mercury analyses of analogous soil cores and leachate from laboratory oxidation tests were completed in the fall of 2021 to determine if mercury released during oxidation could result from non-DNAPL related leaching from aquifer solids. Since core from the monitoring and injection wells used for the oxidant injection tests was not recovered and retained, previously collected and stored core from M Area borings in the vicinity of the test were used to determine background concentrations of total mercury in aquifer solids and measure total mercury extracted by oxidants contacting those sediments. The report on these analyses is provided as Supplementary Note 1. All soil samples were homogenized and the solids were placed in quartz sample boats and directly analyzed for total mercury. For the oxidant leaching experiments, a control solution (oxidant with no sediments) and the sediment samples were equilibrated with oxidant solution (solid solution ration 1.2:1) for 48 h. The supernate was collected using a syringe and filtered (0.45micron). A 0.3 mL subsample was placed into a quartz boat for direct analysis of total mercury. All samples, solids and leachate liquids, were analyzed for total mercury using a Milestone Direct Mercury Analyzer (DMA-80) by sequential pyrolysis, catalysis, amalgamation, thermal desorption, and atomic absorption spectroscopy (EPA Method 7473). Solids were analyzed in triplicate and leachate liquids were analyzed in duplicate.
Baseline calculations
To determine average baseline values for the analytes used to evaluate the effects of oxidant injection (apart from PCE and TCE), pre-injection values from WSM001 and WSM002 (4/25/18, 7/17/18) were averaged with all data from the WSM003 wells (3/22/18 to 9/15/21), MSB101 wells (1/23/09 to 8/26/21), and MSB107 wells (9/24/14 to 9/15/21) which are all near the test site and appear to have been unaffected by the injections. Initially, if an analyte value exceeded the median plus three standard deviations after the injections, it was assumed that this value was significantly different from baseline as a result of the injections. Because the principal monitoring wells (WSM001, WSM002) were only sampled twice prior to injections, values from other wells (MSB101, MSB107, and WSM003) in the area that were not affected by the injections were used to help establish baseline values for the analytes of interest. The pre-injection averages of the principal monitoring wells all were within one standard deviation of the ensemble baseline averages. Subsequent analyses used standard t-test evaluations (two-tailed, unequal variance) to determine whether the null hypothesis was statistically disproved under the assumption of a 95% confidence interval.
The MSB101 wells are upgradient of the injection area and the rest of the wells are downgradient of the injections. All of the wells are in the capture zone of recovery well RWM018 but have different screen elevation intervals that are distributed across both the lower and upper lost lake aquifer zones. When averaged, the analytical measurements from all of these wells result in a standard deviation that is larger than one that would be derived from measurements of wells at the same elevation. This has the effect of making a departure from the null hypothesis more difficult to achieve with statistical significance, resulting in a conservative approach to determine change as a result of injection.
In all monitoring wells but one, the initial TCE concentration was at least an order of magnitude higher than the PCE concentration. The cVOC compounds were converted to moles of chlorine and compared to separately measured chloride for several figures shown in the results.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The datasets generated during the current study are available in the Open Science Framework repository by link to the following: https://osf.io/azhvy/?view_only=3953a7d698864f279345cd275cee37b5
References
Schwille, F. Dense Chlorinated Solvents in Porous and Fractured Media—Model Experiments (Lewis Publishers, 1988).
Cohen, R. M. & Mercer, J. W. DNAPL Site Evaluation (C.K. Smoley, 1993).
Pankow, J. F. & Cherry, J. A. Dense Chlorinated Solvents and Other DNAPLs in Groundwater: History, Behavior, and Remediation (Waterloo Press, 1996).
Illangasekare, T. H., Ramsey, J. L., Jensen, K. H. & Butts, M. B. Experimental study of movement and distribution of dense organic contaminants in heterogeneous aquifers. J. Contam. Hydrol. 20, 1–25 (1995).
Mackay, D. M. & Cherry, J. A. Groundwater contamination: pump-and-treat remediation. Environ. Sci. Technol. 23, 630–636 (1989).
Anderson, M. R., Johnson, R. L. & Pankow, J. F. Dissolution of dense chlorinated solvents into groundwater. 3. Modeling contaminant plumes from fingers and pools of solvent. Environ. Sci. Technol. 26, 901–908 (1992).
Einarson, M. D. & Mackay, D. M. Peer reviewed: predicting impacts of groundwater contamination. Environ. Sci. Technol. 35, 66A–73A (2001).
Rivett, M. O., Dearden, R. A. & Wealthall, G. P. Architecture, persistence and dissolution of a 20 to 45 year old trichloroethene DNAPL source zone. J. Contam. Hydrol. 170, 95–115 (2014).
Heron, G., Bierschenk, J., Swift, R., Watson, R. & Kominek, M. Thermal DNAPL source zone treatment impact on a CVOC plume. Groundw. Monit. Remediat. 36, 26–37 (2016).
Fjordbøge, A. S. et al. Integrity of clay till Aquitards to DNAPL migration: assessment using current and emerging characterization tools. Groundw. Monit. Remediat. 37, 45–61 (2017).
Yang, L., Wang, X., Mendoza-Sanchez, I. & Abriola, L. M. Modeling the influence of coupled mass transfer processes on mass flux downgradient of heterogeneous DNAPL source zones. J. Contam. Hydrol. 211, 1–14 (2018).
Stroo, H. F. et al. Chlorinated ethene source remediation: lessons learned. Environ. Sci. Technol. 46, 6438–6447 (2012).
Steelman, C. M. et al. The importance of transects for characterizing aged organic contaminant plumes in groundwater. J. Contam. Hydrol. 235, 103728 (2020).
Parker, B. L., Chapman, S. W. & Guilbeault, M. A. Plume persistence caused by back diffusion from thin clay layers in a sand aquifer following TCE source-zone hydraulic isolation. J. Contam. Hydrol. 102, 86–104 (2008).
Guilbeault, M. A., Parker, B. L. & Cherry, J. A. Mass and flux distributions from DNAPL zones in sandy aquifers. Ground Water 43, 70–86 (2005).
Anderson, M. R., Johnson, R. L. & Pankow, J. F. Dissolution of dense chlorinated solvents into ground water: 1. dissolution from a well-defined residual source. Ground Water 30, 250–256 (1992).
Chapman, S. W. & Parker, B. L. Plume persistence due to aquitard back diffusion following dense nonaqueous phase liquid source removal or isolation. Water Resour. Res. https://doi.org/10.1029/2005WR004224 (2005).
Liu, C. & Ball, W. P. Back diffusion of chlorinated solvent contaminants from a natural aquitard to a remediated aquifer under well-controlled field conditions: predictions and measurements. Ground Water 40, 175–184 (2002).
Sale, T., Parker, B. L., Newell, C. J. & Devlin, J. F. Management of Contaminants Stored in Low Permeability Zones—A State of the Science Review. 348.0 (Defense Technical Information Center, 2013).
Farhat, S. K. et al. Vertical discretization impact in numerical modeling of matrix diffusion in contaminated groundwater. Groundw. Monit. Remediat. 40, 52–64 (2020).
Borden, R. C. & Cha, K. Y. Evaluating the impact of back diffusion on groundwater cleanup time. J. Contam. Hydrol. 243, 103889 (2021).
Chapman, S. W., Parker, B. L., Sale, T. C. & Doner, L. A. Testing high resolution numerical models for analysis of contaminant storage and release from low permeability zones. J. Contam. Hydrol. 136–137, 106–116 (2012).
Newman, M. A., Hatfield, K., Hayworth, J., Rao, P. S. C. & Stauffer, T. Inverse characterization of NAPL source zones. Environ. Sci. Technol. 40, 6044–6050 (2006).
Sale, T. C., Zimbron, J. A. & Dandy, D. S. Effects of reduced contaminant loading on downgradient water quality in an idealized two-layer granular porous media. J. Contam. Hydrol. 102, 72–85 (2008).
Yang, M., Annable, M. D. & Jawitz, J. W. Back diffusion from thin low permeability zones. Environ. Sci. Technol. 49, 415–422 (2015).
Ball, W. P., Liu, C., Xia, G. & Young, D. F. A diffusion-based interpretation of tetrachloroethene and trichloroethene concentration profiles in a groundwater aquitard. Water Resour. Res. 33, 2741–2757 (1997).
Broholm, K., Feenstra, S. & Cherry, J. A. Solvent release into a sandy aquifer. 1. Overview of source distribution and dissolution behavior. Environ. Sci. Technol. 33, 681–690 (1999).
Broholm, M. M. et al. Characterization of chlorinated solvent contamination in limestone using innovative FLUTe® technologies in combination with other methods in a line of evidence approach. J. Contam. Hydrol. 189, 68–85 (2016).
Kang, X. et al. Improved characterization of DNAPL source zones via sequential hydrogeophysical inversion of hydraulic‐head, self‐potential and partitioning tracer data. Water Resour. Res. https://doi.org/10.1029/2020WR027627 (2020).
Maldaner, C. H. et al. Quantifying groundwater flow variability in a poorly cemented fractured sandstone aquifer to inform in situ remediation. J. Contam. Hydrol. 241, 103838 (2021).
Ferrari, A., Jimenez-Martinez, J., Borgne, T. L., Méheust, Y. & Lunati, I. Challenges in modeling unstable two-phase flow experiments in porous micromodels. Water Resour. Res. 51, 1381–1400 (2015).
McMillan, L. A., Rivett, M. O., Wealthall, G. P., Zeeb, P. & Dumble, P. Monitoring well utility in a heterogeneous DNAPL source zone area: Insights from proximal multilevel sampler wells and sampling capture-zone modelling. J. Contam. Hydrol. 210, 15–30 (2018).
Arshadi, M., De Paolis Kaluza, M. C., Miller, E. L. & Abriola, L. M. Subsurface source zone characterization and uncertainty quantification using discriminative random fields. Water Resour. Res. https://doi.org/10.1029/2019WR026481 (2020).
Yuan, Q., Ma, Z., Wang, J. & Zhou, X. Influences of dead‐end pores in porous media on viscous fingering instabilities and cleanup of NAPLs in miscible displacements. Water Resour. Res. https://doi.org/10.1029/2021WR030594 (2021).
Stewart, L. D., Chambon, J. C., Widdowson, M. A. & Kavanaugh, M. C. Upscaled modeling of complex DNAPL dissolution. J. Contam. Hydrol. 244, 103920 (2022).
Wilson, J.L., Conrad, S.H., Mason, W.R., Peplinski, W., and Hagan, E. Laboratory Investigation of Residual Liquid Organics from Spills, Leaks, and the Disposal of Hazardous Wastes in Groundwater. 267 (US Department of Energy, 1990).
Rossabi, J. et al. Field tests of a DNAPL characterization system using cone penetrometer-based raman spectroscopy. Groundw. Monit. Remediat. 20, 72–81 (2000).
Einarson, M., Fure, A., St. Germain, R., Chapman, S. & Parker, B. DyeLIFTM: A new direct-push laser-induced fluorescence sensor system for chlorinated solvent DNAPL and other non-naturally fluorescing NAPLs. Groundw. Monit. Remediat. 38, 28–42 (2018).
Jackson, D. G. Characterization Activities to Determine the Extent of DNAPL in the Vadose Zone at the A-014 Outfall of A/M Area (US Department of Energy, 2000).
Jackson, D. G., Payne, T. H., Looney, B. B. & Rossabi, J. Estimating the Extent and Thickness of DNAPL within the A/M Area of the Savannah River Site (IAEA, 1996).
Eddy, C. A., Looney, B. B., Dougherty, J. M., Hazen, T. C. & Kaback, D. S. Characterization of the Geology, Geochemistry, Hydrology and Microbiology of the In-Situ Air Stripping Demonstration Site at the Savannah River Site (US Department of Energy, 1991).
Jackson, D. G. Evaluating DNAPL Source and Migration Zones: M-Area Settling Basin and the Western Sector of A/M Area, Savannah River Site (US Department of Energy, 2001).
Looney, B. B. Assessing DNAPL contamination, A/M-Area, Savannah River site: phase 1 results. Environ. Sci. https://doi.org/10.2172/10161455 (1992).
Rossabi, J. Recent advances in characterization of vadose zone Dense non-Aqueous Phase Liquids (DNAPL) in heterogeneous media. Environ. Eng. Geosci. 9, 25–36 (2003).
Rossabi, J. & Nave, S. E. Characterization of DNAPL Using Fluorescence Techniques (US Department of Energy, 1998).
Milanovich, F. P., Brown, S. B., Colston, B. W. Jr., Daley, P. F. & Langry, K. C. A fiber-optic sensor system for monitoring chlorinated hydrocarbon pollutants. Talanta 41, 2189–2194 (1994).
Looney, B. B. et al. Test Plan for Single Well Injection/Extraction Characterization of DNAPL (SciTech Connect., 1995).
Guo, Q. et al. Integrating hydraulic tomography, electrical resistivity tomography, and partitioning interwell tracer test datasets to improve identification of pool-dominated DNAPL source zone architecture. J. Contam. Hydrol. 241, 103809 (2021).
Grimm, R. E., Olhoeft, G. R., McKinley, K., Rossabi, J. & Riha, B. Nonlinear complex-resistivity survey for DNAPL at the Savannah river site A-014 outfall. J. Environ. Eng. Geophys. 10, 351–364 (2005).
Ellefsen, K. J., Nelson, P. H., Horton, R. J. & Wright, D. L. Summary of geophysical investigations for DNAPL remediation at Savannah River Site, South CArolina (U.S. Geological Survey, 1997).
Almpanis, A., Gerhard, J. & Power, C. Mapping and monitoring of DNAPL source zones with combined direct current resistivity and induced polarization: a field‐scale numerical investigation. Water Resour. Res. https://doi.org/10.1029/2021WR031366 (2021).
Jackson, D. G., Denham, M. E. & Looney, B. B. A Framework for the Transport and Release of Mercury from DNAPL. In Proc. 5th International Conference on Remediation of Chlorinated and Recalcitrant Compounds (Battelle Press, 2006).
Looney, B. B., Denham, M. E., Vangelas, K. M. & Bloom, N. S. Removal of mercury from low-concentration aqueous streams using chemical reduction and air stripping. J. Environ. Eng. 129, 819–825 (2003).
Mroczek, E. K. The solubility of elemental mercury in water between 30 and 210 C. In Proc. 19th Workshop on Geothermal Reservoir Engineering 223–229 (Stanford University, 1994).
Foster, A., Trautz, A. C., Bolster, D., Illangasekare, T. & Singha, K. Effects of large-scale heterogeneity and temporally varying hydrologic processes on estimating immobile pore space: a mesoscale-laboratory experimental and numerical modeling investigation. J. Contam. Hydrol. 241, 103811 (2021).
Rossabi, J. Supplementary Data. https://osf.io/azhvy/?view_only=3953a7d698864f279345cd275cee37b5 (2022).
Ayral-Cinar, D. & Demond, A. H. Effective diffusion coefficients of DNAPL waste components in saturated low permeability soil materials. J. Contam. Hydrol. 207, 1–7 (2017).
Ayral-Çınar, D. & Demond, A. H. Accumulation of DNAPL waste in subsurface clayey lenses and layers. J. Contam. Hydrol. 229, 103579 (2020).
Schaefer, C. E., White, E. B., Lavorgna, G. M. & Annable, M. D. Dense nonaqueous-phase liquid architecture in fractured bedrock: implications for treatment and plume longevity. Environ. Sci. Technol. 50, 207–213 (2016).
Looney, B. B. & Jackson, D. G. Impacts of In-Situ Chemical Oxidation (ISCO) on the Stannous Chloride System’s Ability to Remove Mercury at the M-1 Air Stripper, SRNS-STI-2021-00122 (EPA, 2021).
Acknowledgements
This work was supported by Savannah River Nuclear Solutions and the US Department of Energy Environmental Management under Contract Number DE-AC09-08SR2. The Savannah River National Laboratory is operated by Battelle Savannah River Alliance for the U.S. Department of Energy under Contract No. 89303321CEM000080. Publisher acknowledges the U.S. Government license to provide public access under the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan). We thank Joao Cardoso-Neto, Jeff Ross, Branden Kramer, Bob Van Pelt, John Bradley and Chris Bergren from Savannah River Site Area Completions Project, and Seth Dray, Matthias Malin, Gregory Kinsman, and Allison Walker from Northwinds Group for endorsing this project, permitting access, logistical, and administrative support. We thank Zack Poole, Blair Mitchell, Geoff Ives, Robert Sullivan, Greg Powers, and John Haselow from Redox Tech, LLC, and Brian Riha, Austin Coleman and Richard Walker from Savannah River National Laboratory for field and technical support.
Author information
Authors and Affiliations
Contributions
J.R. co-designed and managed the field experiments and provided the majority of the analysis, interpretation, and writing. D.G.J. co-designed the field experiments and contributed to the analysis of the data and review of the manuscript. H.H.V. performed the laboratory mercury leachability experiments and analysis and contributed to the analysis of the field data. B.B.L. contributed to the writing, interpretation, and review of the manuscript and initiated the conceptual model.
Corresponding author
Ethics declarations
Competing interests
J.R. has the following competing interest. He is the minority partner in a company that performs in situ soil and groundwater remediation. It is not clear whether the findings in this article will result in financial gain or loss to this company since the new conceptual model suggested could promote either more or less opportunities for the author’s company depending on the specific site scenario. Most likely, the publication of this article will have no effect on the first author’s company. The other authors have no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Reza Shams and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Edmond Sanganyado and Clare Davis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rossabi, J., Jackson, D.G., Vermeulen, H.H. et al. Dense non-aqueous phase liquid chlorinated contaminant detected far from the source release area in an aquifer. Commun Earth Environ 3, 223 (2022). https://doi.org/10.1038/s43247-022-00556-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-022-00556-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
