
Abstract
Many large unit-cell rare-earth transition metal ternary alloys of the type Ra(M1−xM’x)b exhibit non-monotonic ferrimagnetic Curie temperatures (TC) coupled to monotonic composition-controlled magnetization. Its origin remains an important long-standing puzzle in the absence of studies probing their temperature-dependent element-specific magnetism. Here, in order to resolve this issue and identify design principles for new R-M-M’ permanent magnets, we carry out x-ray magnetic circular dichroism (XMCD) for the series Gd6(Mn1−xFex)23, x = 0.0 − 0.75. The results show that the net Mn-moment reduces and switches from parallel to antiparallel for x ≥ 0.2, while the Fe-moment is always antiparallel to the Gd-moment. Kouvel-Fisher analyses of XMCD data reveals distinct sublattice TC’s and 3D Heisenberg criticality. Band structure calculations show magnetic moments and density of states consistent with experiments. The magnetic phase diagram shows three regions characterized by (i) Mn-sublattice bulk-TC > Gd-sublattice TC, (ii) a reduced common-TC for all sublattices, and (iii) Fe-sublattice bulk-TC > Gd-sublattice TC. The Mn-moment switching and gradual increase of Fe-moment combine to cause non-monotonic TC’s with monotonic magnetization. The study indicates the importance of element-specific TC’s for tuning magnetic properties.
Similar content being viewed by others

Introduction
Binary and ternary intermetallics containing rare-earths(R) and transition metals(M) play an important role in the fields of heavy-fermions1,2,3, Non-Fermi-liquids4, magnetic metals2,3,4,5,6, magnetocaloric materials7, etc. In addition to applications as strong permanent magnets, magnetocalorics, thermoelectrics, fracture toughness, ductility, etc., R-M based materials are now also used in phosphors, lasers, energy storage batteries, catalysts, etc. 8
The variety of their applications stem from distinct properties of the R f-electrons and M d-electrons. Typically, f-electrons are localized with large magnetic moments µ and strong spin-orbit coupling (SOC), negligible bandwidths (W) and weak intersite exchange (J’) interactions5,6. In comparison, delocalized M d-electrons in intermetallics show a smaller µ and weaker SOC, but larger W and J’ which results in M d-electrons determining the bulk TC’s. Several intermetallic series Ra(M1−xM’x)b, such as R(M1−xM’x)2, R2(M1−xM’x)17, R6(M1−xM’x)23 with R = Gd-Tm and M, M’ = Mn, Fe, Co show non-monotonic variation of TC’s with a monotonic variation of the total bulk magnetization \({M}_{{Tot}}^{B}\)(x) at low-T, or vice-versa5,6,9,10,11,12,13. In the absence of element specific TC’s, this behavior remains an open question and it is generally considered that three types of exchange interactions are important in R-M-M’ alloys: R-R indirect exchange proceeding via 4f - conduction electrons - 4f states, M-M/M-M’ direct exchange between 3d states, and R-M/M’ indirect exchange via 4f - 5d - 3d states. Early studies based on effective Heisenberg models for large unit-cell systems concluded that M-M exchange > R-M exchange > R-R exchange11,12,13,14,15,16,17. For an isostructural R-M series with R varying from Ce to Yb, the R-M exchange coupling is always ferromagnetic (FM) for light rare-earths and anti-ferromagnetic (AFM) for heavy rare-earths, which constitutes the “FM-AFM rule”. In addition, varying M or M’ can also lead to competition of bulk FM vs. AFM order9,10. Using element-specific T-dependent XMCD to address the interplay of f and d electrons in the series Gd6(Mn1−xFex)23, we resolve these issues by showing that element specific TC’s and changes in the M-M’ exchange compared to R-M and R-R exchange plays a critical role in their unusual magnetism.
The R6Mn23 intermetallics crystallize in the Th6Mn23 (G-phase) type cubic structure and show very interesting magnetic properties9,15,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37. Early work concluded that the R6Mn23 compounds did not apparently fit with the FM-AFM variation of the R-M exchange coupling for the light and heavy rare-earths, respectively18. Polarized neutron diffraction (ND) of isostructural Y6Mn23 at 4.2 K showed that the Mn sublattice consists of up-spin ‘f1, f2’ sites (with magnetic moment µ1 and µ2 respectively), and down-spin ‘b, d’ sites (with magnetic moment µ3 and µ4 respectively). The results show that µ1 = µ2~ +1.8 µB, µ3~ −2.8 µB and µ4~ −2.1 µB, respectively, for the four different sites25. In contrast, Y6Fe23 showed all four Fe sites (b, d, f1, f2) were aligned with an average µFe~ + 1.94µB27,38. Since it is not easy to measure ND of Gd-based structures, it was assumed that Gd6(Mn1−xFex)23 possesses the same ferrimagnetic Mn sublattice structure as Y6Mn23. Also, with Gd-moments interacting more strongly with the ‘b, d’ sites compared to the ‘f1, f2’ sites, it was concluded that the R6Mn23 compounds were not an exception to the FM-AFM rule25,28,33. However, \({M}_{{Tot}}^{B}\) of Y6Fe23 is larger than Gd6Fe23, but that of Y6Mn23 is smaller than Gd6Mn23. Further, upon Fe substitution in Y6Mn23, \({M}_{{Tot}}^{B}\) of Y6(Mn1−xFex)23 increases with increasing x, but in isostructural Gd6(Mn1−xFex)23, \({M}_{{Tot}}^{B}\) reduces on increasing x21,24,27,30,32,36. Thus, magnetism of the composite Fe-Mn sublattice and its relation with the R-sublattice cannot be explained as an additive mixture of the parent compounds and it is necessary to determine the evolution of element-specific µ with x and T.
The non-monotonic behavior of TC’s in Gd6(Mn1−xFex)23 is also seen in Y6(Mn1−xFex)2321,30,32. This suggests that TC’s of Gd6(Mn1−xFex)23 are decoupled from the R-sublattice and originate from the M-M’ sublattices. Magnetocaloric studies of Gd6(Mn1−xFex)23 revealed two maxima in the magnetic entropy changes (∆SM), at T = TC and at a lower T~100 K. It was inferred that the T~100 K maximum could be due to Gd sublattice ordering or due to modifications in the magnetic structure for small x = 0.0 − 0.232. However, element-specific TC’s of the Gd, Mn and Fe sublattices using XAS-XMCD have not been reported to date. In addition, T-dependence of the XMCD signal in terms of Ising, Heisenberg or mean-field type critical behavior would help to model these complex systems.
In this work, we carry out T -dependent XAS and XMCD studies to determine element-specific magnetic moments and TC’s in Gd6(Mn1−xFex)23, x = 0.0 − 0.75. The results identify the elemental origin of coexisting distinct sublattice TC’s with 3-D Heisenberg-type criticality, and provide an understanding of the non-monotonic TC’s and monotonic magnetization as a function of x in Gd6(Mn1−xFex)23. Moreover, since there are no reported band structure calculations even for the parent compounds, we have carried out band structure calculations of Gd6M23 (M = Mn, Fe) based on the Density Functional Theory with on-site Coulomb energies (DFT + U) for Gd, Mn and Fe. The calculated element specific magnetic moments and total magnetization are in agreement with XMCD results and reported bulk magnetization measurements. In addition, the band structure calculations provide the partial density of states (PDOS) and total DOS which are consistent with experimental valence band spectra measured by Hard x-ray photoemission spectroscopy (HAXPES).
Results and Discussion
XAS and XMCD results
Figure 1a-c shows representative XAS measurements of Gd M-edge and Mn L-edge for x = 0.0 and the Fe L-edge for x = 0.5, respectively. The Gd M-edge, Mn L-edge and Fe L-edge XAS spectra without applied field for all x are shown in Supplementary Fig. 1 and discussed in Supplementary Note 1. The Mn and Fe L-edge XAS spectra show peak positions and shapes matching well with the Mn and Fe metal spectra39,40,41,42,43,44. Figure 1d–f, shows the Gd M-edge, Mn L-edge and Fe L-edge XMCD for in-situ cleaved samples of the Gd6(Mn1−xFex)23 series (x = 0, 0.2, 0.5 and 0.75) at T = 29 K, the lowest sample temperature attained on our spectrometer. The Gd M-edge XAS spectra shown in Fig. 1a give rise to a large XMCD (~ 36%) and are shown for all x in Fig. 1d. Their spectral shape is very similar to earlier XMCD results of Gd intermetallics like GdFe, GdNi, and GdCo42,45,46,47,48,49. The values of spin and orbital magnetic moments (µspin, µorb) of Gd from the measured intensities of XAS and XMCD spectra was determined by a sum rule analysis as detailed in Supplementary Note 2. Based on the Gd3+ configuration, the number of holes for Gd 4f states is taken to be nh(Gd) = 7. The obtained µspin and µorb are shown in Supplementary Table 1 for all x. The µorb of Gd is not exactly zero but very small µorb ≈ 0.13 ± 0.02 µB, which is attributed to a combination of 4f-4f multiplet interactions and the 4f SOC42,50. The total moment of Gd, µGd is very close to 7 µB for all x, corresponding to the localized Gd3+ 4f 7 configuration with S = 7/2.

Representative XAS spectra with an applied field of ±1 T for (a) Gd M-edge, (b) Mn L-edge of the parent compound, and (c) the Fe L-edge of Gd6(Mn0.5Fe0.5)23 measured at T = 29 K, from which we obtained the XMCD spectra, as shown for all x in (d–f). d Gd M-edge, (e) Mn L-edge and (f) Fe L-edge experimental XMCD spectra of the series Gd6(Mn1−xFex)23 (x = 0, 0.2, 0.5 and 0.75) measured at T = 29 K. Arrows in (d–f) show the relative magnitude and orientation of the spins.
For x = 0, although the Mn L-edge XMCD peak signal is small (~4% at hν ~639 eV), it is clear and indicates the net Mn moment µMn is oriented parallel to µGd. Surprisingly, as shown in Fig. 1e for Fe substitution with x = 0.2, the Mn XMCD signal switches to an antiparallel orientation with respect to Gd, and becomes smaller (~2% at hν ~640 eV). This originates from the clear reversal of the L3 and L2 peak intensities of XAS spectra with ± 1 T for x = 0.2 compared to x = 0.0, as shown in Supplementary Fig. 2 and discussed in Supplementary Note 3. The shape and intensity of the Mn XMCD signals are very similar for x = 0.2, 0.5 and 0.75. Thus, our results indicate that in the parent compound Gd6Mn23, the Mn moment associated with 32f1, 32f2 sites are parallel to Gd, while 24b, 4d sites are antiparallel to Gd. This results in a net Mn sublattice magnetization parallel to Gd in Gd6Mn23, as XMCD is proportional to magnetization. Upon Fe substitution, for x ≥ 0.2, the XMCD shows a switching of the net Mn sublattice magnetization from parallel to antiparallel with respect to Gd. In contrast, the Fe XMCD signal indicates that the Fe sublattice magnetization exhibits antiparallel orientation with the Gd moments and increases systematically with x, as shown in Fig. 1f. The Mn and Fe µspin and µorb values were also obtained by sum rule analyses as detailed in Supplementary Note 2. We used nh(Mn) = 4.5 based on values known for Mn alloys44,51,52,53,54. Similarly, we used nh(Fe) = 3.7 for Fe, based on values known for Fe alloys42,43,44. The obtained values of µspin and µorb and total moments (µMn, µFe) are listed in Supplementary Table 2.
Supplementary Table 3 shows \({M}_{{Tot}}^{X}\), the total magnetization using magnetic moments obtained from the XMCD sum rule analysis according to the formula \({M}_{{Tot}}^{X}\) = 6µGd + 23{(1-x) µMn + xµFe}. The obtained values of \({M}_{{Tot}}^{X}\) from XMCD are in good agreement with \({M}_{{Tot}}^{B}\) from magnetization results reported earlier at 1 T21,30,32. Since ref. 30 reported the values at T = 77 K, they are consistently lower by a factor of 1.22 compared to the present results as well as refs. 21,32. measured at lower T. The results show the monotonic reduction in \({M}_{{Tot}}^{B}\) is caused by the systematic increase of µFe aligned antiparallel to µGd, while µMn is very small for Fe substituted cases. It is also noted that even if we assume that all the substituted Fe for x = 0.2 occupies the ‘f1, f2’ sites with µFe aligned antiparallel to µGd, \({M}_{{Tot}}^{X}\) is consistent with \({M}_{{Tot}}^{B}\) because µMn shows a switching, indicating a breakdown of the magnetization as an additive mixture of the parent compounds; this behaviour has not been recognized earlier in the absence of element-specific magnetic moments. Moreover, the results are fairly consistent with the change in magnetic moments µFe and µMn estimated from the 3s core-level HAXPES55, which also showed a monotonic change in total magnetization.
Band structure calculations
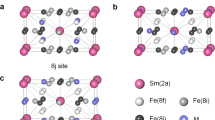
In order to understand the electronic structure responsible for the magnetism, we carried out DFT based spin-polarized generalized-gradient approximation (GGA) band structure calculations with on-site Coulomb energies for Gd6Mn23 and Gd6Fe23. The spin-polarized calculations were carried out using the experimental crystal structures as the starting point (Fig. 2a), i.e. cubic Gd6Mn2356 and Gd6Fe2357, and the details are described in the Methods section. Structural optimizations performed within the GGA-PBE (Perdew-Burke-Ernzerhof) approximation led to cubic cell parameter a in reasonable agreement with the experimental ones: aexp(Gd6Mn23) = 12.54 Å and acal(Gd6Mn23) = 11.78 Å while aexp(Gd6Fe23) = 12.13 Å and acal(Gd6Fe23) = 11.84 Å. Electronic structure calculations were performed for the optimized structures using the simplified (rotationally invariant) approach to the DFT + U, introduced by Dudarev et al. 58. In this approach, the parameters for the Coulomb (U) and Hund’s exchange (J) interactions do not enter separately, and only the difference U-J = UDFT is considered as a parameter.

a The crystal structure of Gd6Mn23 showing the magnetic moments from band structure calculations of the Gd-site, Mn ‘b, d’ sites and Mn ‘f1, f2’ sites. b The valence band spin-integrated PDOS and total DOS of Gd6Mn23 obtained from the DFT + U calculations with \({U}_{{Mn}}^{{DFT}}\) = 0.75 eV and \({U}_{{Gd}}^{{DFT}}\) = 6.5 eV, together with the bulk-sensitive valence band spectrum measured by HAXPES. The calculations identify the contributions of the Gd 4f, Gd 5d, Mn 4s and Mn 3d PDOS contributions to the experimental spectrum. c The valence band spin-integrated PDOS and total DOS of Gd6Fe23 obtained from the DFT + U calculations. The calculations identify the contributions of the Gd 4f, Gd 5d, Fe 4s, and Fe 3d PDOS contributions to the experimental spectrum. d The valence band spin-integrated total DOS of Gd6(Mn0.25Fe0.75)23 is approximated by an additive mixture of 25% Gd6Mn23 and 75% Gd6Fe23 calculations, plotted together with the bulk-sensitive valence band spectrum measured by HAXPES55.
It is known that even for Gd metal, the XAS-XMCD behaves like a typical atomic Gd3+ 4f 7 configuration with S = 7/2. Hence, the ground state can be described by a single Slater determinant and the one electron DFT is valid. In fact, it was shown early on that the two main parameters, the average energies of the Coulomb Uff and exchange Jff interactions could be calculated using the local spin-density approximation59,60. The resulting values of Uff = 6.7 eV and Jff = 0.7 eV gave DOS consistent with the experimental N − 1 and N + 1 final states for one electron-removal (PES) and electron-addition (inverse-PES) spectra61, with a \({U}_{{Gd}}^{{eff}}\)= Uff + 6Jff ≈ 11 eV. In our case, using the Dudarev approach, we varied \({U}_{{Gd}}^{{DFT}}\) (= Uff − Jff) from 6.0 eV to 12 eV and confirmed that \({U}_{{Gd}}^{{DFT}}\) = 6.5 eV gave a suitable match with the Gd 4f PDOS in valence band HAXPES spectrum (Fig. 2b). Further, the average magnetic moment of Gd is calculated to be μGd = +7.18 μB, in good agreement with the localized Gd3+ 4f 7 configuration and the sum rule analysis discussed above. Assuming Jff = 0.7 eV, we obtain \({U}_{{Gd}}^{{eff}}\) = Uff + 6Jff = \({U}_{{Gd}}^{{DFT}}\) + 7Jff = 11.4 eV. Similarly, in order to obtain calculated Mn magnetic moments close to the experimental values, \({U}_{{Mn}}^{{DFT}}\) was varied from 0.0 to 2.5 eV. It was found that\(\,{U}_{{Mn}}^{{eff}}\) = \({U}_{{Mn}}^{{DFT}}\) = 0.75 eV gave magnetic moments close to the experimental values, and hence, Udd = 1.65 eV if we consider Jdd = 0.9 eV60.
Next, we discuss the obtained values of magnetic moments from the band structure calculations in comparison with the experimental values. The net magnetic moments per unit cell and per atom from the band structure calculations and the sum rule analysis are listed in Table 1 in comparison with the ND data and the Van Vleck analysis. The net magnetic moments were obtained using the average atomic magnetic moments from the band structure calculations as detailed in Supplementary Note 4. The average calculated magnetic moments for the ‘f1, f2’ sites (with μ1~ +1.87 μB and μ2~ +1.76 μB (Fig. 2a) shows good agreement with values known from ND studies. However, the average magnetic moment of ‘b, d’ sites (with μ3~ −2.95 μB and μ4~ −2.57 μB (Fig. 2a) is slightly higher compared to ND results. The magnetic moments are also consistent with values estimated from a Van Vleck analysis of 3s core-level photoemission spectra55. The average magnetic moment of Gd is calculated to be μGd = +7.18 μB, also in good agreement with the localized Gd3+ 4f 7 configuration as well as the result from sum rule analysis discussed above. The calculated moments with antiferromagnetic coupling between the Mn ‘b, d’ sites with Mn ‘f1, f2’ sites leads to a net Mn moment parallel to the Gd moment, and gives a total magnetization MTot = 53.8 μB. This value is consistent with bulk magnetization M = 54.7 μB measured at 5 T32. It is slightly higher compared to the \({M}_{{Tot}}^{X}\) from sum rule analysis measured at 1 T, as expected, because magnetic field dependent studies indicated a weak metamagnetic behavior with an increase around 4.5 T32.
The valence band total DOS and spin-integrated PDOS of Gd6Mn23 obtained from the DFT + U calculations with said optimal parameters is shown in Fig. 2b, together with the bulk-sensitive valence band spectrum measured by HAXPES. The spin-resolved PDOS are discussed in Supplementary Note 4 and shown in Supplementary Fig. 3. The calculated spectra were obtained by applying the known photoionization cross-sections (PICS) at 10 keV62, and convoluted by a Gaussian function (1.0 eV FWHM for Mn 3d, Mn 3s and Gd 5d PDOS; and 1.5 eV FWHM for Gd 4f PDOS). As can be seen in Fig. 2b, the Gd 4f PDOS is positioned at ~7 eV binding energy (BE) below EF, quite like Gd metal60, while Mn 3d and Gd 5d PDOS are spread from EF to nearly 6 eV BE. The Mn 4s states are spread from EF to nearly 8 eV and show sizable intensity due to higher cross-sections for the incident hard x-ray energies62, leading to a bump feature at about 4.5 eV BE. On the other hand, at and near EF, the total DOS is dominated by Gd 5d PDOS with a weaker contribution from Mn 3d states at EF, and nearly similar contributions from both for the feature at ~3 eV BE. As discussed in Supplementary Note 4, the spin resolved PDOS shows that the localized Gd3+ configuration leads to 4f 7 up-spin states well-separated from the 4f 7 down-spin states due to the large \({U}_{{Gd}}^{{eff}}\) = 11.4 eV. The Mn 3d up-spin and down-spin states show a relatively large bandwidth, but nonetheless, the \({U}_{{Mn}}^{{eff}}\) = \({U}_{{Mn}}^{{DFT}}\) = 0.75 eV leads to weak splitting in up and down spin states, with a net Mn magnetic moment parallel to the Gd moments.
Similarly, DFT + U calculations were carried out for Gd6Fe23, and the results gave the average magnetic moment of Fe consistent with the experimental value, µFe = −2.39 µB for \({U}_{{Fe}}^{{eff}}={U}_{{Fe}}^{{DFT}}\)=\(\,0.75\) eV i.e. with Udd = 1.65 eV and Jdd = 0.9 eV. For Gd in Gd6Fe23, a value of µGd = +7.55 µB was obtained for \({U}_{{Gd}}^{{eff}}=\) 11.4 eV. Since a full calculation for substituted compounds requires an extremely large unit cell, we have approximated the calculated valence band total and spin-integrated PDOS of Gd6(Mn0.25Fe0.75)23 as an additive mixture of 25% Gd6Mn23 and 75% Gd6Fe23. The same analysis for the total DOS of Gd6(Mn1−xFex)23, x = 0.0 - 0.75 is shown in Supplementary Fig. 4. The calculated valence band total and spin-integrated PDOS of Gd6(Mn0.25Fe0.75)23 obtained from the DFT + U calculations is shown in Fig. 2d, together with the bulk-sensitive valence band spectrum measured by HAXPES. The spin-resolved PDOS are discussed in Supplementary Note 4. The calculated spectra were obtained by applying the known PICS as for Gd6Mn23. A fairly good match is obtained between the calculated and experimental spectrum. In particular, it is seen that contribution from Fe 3d and 4s PDOS (Fig. 2c) shows higher relative intensities compared to Mn 3d and 4s PDOS (Fig. 2b), leading to a small shape change between 3 and 5 eV BEs in Gd6(Mn0.25Fe0.75)23 compared to Gd6Mn23. More importantly, the Gd 5d and Fe 3d PDOS at EF and within 2 eV BE are enhanced and broadened leading to a rounding of the sharp feature at EF seen in Gd6Mn23. The results show that DFT + U calculations help to identify the Gd 4f, Gd 5d, Mn 4s and Mn 3d PDOS contributions to the experimental spectra.
Kouvel-Fisher analyses to characterize element-specific TC’s
Early studies15,21,30,32 used \({M}_{{Tot}}^{B}(T)\) to determine the Curie temperature TC derived from the magnetic ordering of the M sublattice, while the Gd sublattice TC for the series Gd6(Mn1−xFex)23 has not been reported. As the magnetization of Gd is proportional to the Gd M-edge XMCD intensity I(T), it provides a direct measure of the sublattice ordering in Gd6(Mn1−xFex)23. We have thus measured the Gd sublattice TC for the series Gd6(Mn1−xFex)23 using T-dependent Gd M5-edge XMCD intensity I(T) and the results are summarized in Fig. 3a. As seen from Fig. 3a, the normalized XMCD intensity IX(T) of Gd for the series reduces systematically on increasing T. The Gd sublattice TC’s could be directly obtained by a power law least-squares fit to the equation IX(T) = I0(1 − T/TC)β (where I0 is the intensity at T = 0 K, β is the critical exponent, and IX(T) = I(T)/I(T = 29 K), indicating the critical behavior of the XMCD signal. The fit matches with IX(T) over a limited T-range below TC, as shown in Fig. 3a. The XMCD results show that the Gd sublattice exhibit values of TC = 273.5 K, 172 K, 135 K and 273.5 K (with an error bar of ±5 K) for x = 0.0, 0.2, 0.5 and 0.75, respectively (Supplementary Table 3). In comparison, \({M}_{{Tot}}^{B}(T)\) shows TC = 489 K, 176 K, 120 K and 309 K for x = 0.0, 0.2, 0.5 and 0.75 (error bars shown in Supplementary Table 3), respectively30,32. The TC for x = 0.0 and x = 0.75 was determined from \({M}_{{Tot}}^{B}\)(T) measured by a Physical Property Measurement System (PPMS) as shown in Supplementary Fig. 5 and Supplementary Fig. 6, respectively, and discussed in Supplementary Note 530,32. The Gd sublattice TC’s are lower than the bulk magnetization TC’s only when the bulk TC > 273.5 K i.e. for x = 0.0 and 0.75. On the other hand, when the bulk TC < 273.5 K (i.e., for x = 0.2 and 0.5), Gd TC also gets reduced together with the bulk TC. This clearly shows that the Gd sublattice TC equals the transition metal M sublattice only for intermediate x and it is lower for x = 0.0 and 0.75 compared to \({M}_{{Tot}}^{B}\hskip-1pt(T)\) results. More importantly, by plotting the normalized XMCD intensity as a function of T/TC − 1, we can scale the critical behavior for the entire Gd6(Mn1−xFex)23 series. Figure 3b shows that IX(T) below TC for all x falls on a single curve near TC with β = 0.38 ± 0.01, indicating validity of the three-dimensional (3-D) Heisenberg model63. For x = 0.0, a small deviation at T ~100 K was also observed in \({M}_{{Tot}}^{B}\)(T) results measured with 1 T applied magnetic field15.

a T-dependence of the Gd M5-edge XMCD intensity of Gd6(Mn1−xFex)23 fitted by a power law with β = 0.38 ± 0.01 (dashed black line). b Critical scaling behavior for the Gd6(Mn1−xFex)23 series. The inset shows temperature dependence of Gd M-edge XMCD intensity of Gd6(Mn0.25Fe0.75)23 at a few selected temperatures, and a full figure showing the spectra for more temperature values is shown in Supplementary Fig. 7 and described in Supplementary Note 6. c T-dependence of the Gd M5-edge XMCD intensity and \({M}_{{Tot}}^{B}(T)\) of Gd6(Mn0.8Fe0.2)23 fitted by a power law with β = 0.38 ± 0.01 (black line). d, e Show the power-law and Kouvel-Fisher analyses fits for \({M}_{{Tot}}^{B}(T)\) (black empty circles), respectively. The Gd M5-edge XMCD intensity IX(T) plotted together (red empty circles) also follows the Kouvel-Fisher analysis of \({M}_{{Tot}}^{B}(T)\) near TC. f T-dependence of the Gd M5-edge (blue squares) and Fe L3-edge (red squares) XMCD intensity of Gd6(Mn0.5Fe0.5)23 fitted to a power law with β = 0.37 ± 0.01 (dashed blue-red line). g, h Show the power-law and Kouvel-Fisher analyses, respectively, for both Gd M5-edge and Fe L3-edge XMCD intensities plotted together. i T-dependence of the Gd M5-edge (blue squares) and Fe L3-edge (red squares) XMCD intensity of Gd6(Mn0.25Fe0.75)23 fitted by a power law with β = 0.38 ± 0.01 (blue and red lines, respectively). j, k Show the power-law and Kouvel-Fisher analyses for Gd M5-edge XMCD intensities and (l, m) show the power-law and Kouvel-Fisher analyses for Fe L3-edge XMCD intensities, respectively. The red and blue lines in (d, e, g, h, j–m) are the fits below and above TC, respectively. All XMCD were obtained with an applied field of ±1 T. Error bars represent standard error (SE).
We have carried out a power-law analysis for the Gd6(Mn0.8Fe0.2)23 bulk magnetization \({M}_{{Tot}}^{B}(T)\) from a PPMS measurement using the same power-law with IX(T) replaced by the intensity of \({M}_{{Tot}}^{B}(T)\) below TC and compared it with the T-dependent Gd XMCD data to test the validity of the method. As shown in Fig. 3c, the power law analysis of the normalized \({M}_{{Tot}}^{B}(T)\) was then used to determine TC independently and gave values of TC = 175 K with β = 0.38. These values of TC and β are consistent with values obtained from T-dependence of the Gd M5-edge XMCD intensity for Gd6(Mn0.8Fe0.2)23 from Fig. 3b. It is noted that we could not obtain the T- dependent XMCD intensity of transition metal Mn or Fe for x = 0.2 due to very low XMCD intensities. As a further check of the ordering behavior, we also carried out a power law analysis for the \({M}_{{Tot}}^{B}(T)\) data above TC to the equation χX(T)−1 = \({\chi }_{0}^{-1}\)(T/TC − 1)γ. Here, χX(T) is replaced by the intensity of \({M}_{{Tot}}^{B}(T)\) above TC, χ0 is critical amplitude and γ is the critical exponent. The above-TC (χX(T)−1) power law analysis gives a TC value of 174.5 K (with γ = 1.28), and this TC value is comparable to the value obtained from the below-TC (IX(T)) analysis (with β = 0.38), as shown in Fig. 3d and listed in Supplementary Table 4.
In order to accurately determine the critical behavior, we then carried out a Kouvel-Fisher analysis of the \({M}_{{Tot}}^{B}(T)\) signal as shown in Fig. 3e. Accordingly, the critical exponents could be determined from the equations IX(T)/(dIX(T)/dT)=(T − TC)/β and χX(T)−1/(dχX(T)−1/dT)=(T − TC)/γ64 with IX(T), χX(T) replaced by intensity of \({M}_{{Tot}}^{B}(T)\) below and above TC, respectively. From a linear fit to the experimental bulk magnetization \({M}_{{Tot}}^{B}(T)\) data, we have obtained values of TC = 175 K, β = 0.38 and γ = 1.30, and these values are very consistent with the power law analyses. It is noted that the values of β confirm that \({M}_{{Tot}}^{B}(T)\) of Gd6(Mn0.8Fe0.2)23 follows the 3-D Heisenberg-type critical behavior. We have then plotted the Gd M-edge XMCD intensity IX(T) on the same scale in Fig. 3d, e, for Gd6(Mn0.8Fe0.2)23. The good match between the bulk magnetization \({M}_{{Tot}}^{B}(T)\) (black empty symbols) and the Gd XMCD intensity IX(T) (red empty symbols) indicates that a Kouvel-Fisher analysis can be reliably used for determining the TC and critical exponents from XMCD measurements.
Thus, we have similarly carried out a power-law and Kouvel-Fisher analysis for Gd M-edge and Fe L-edge XMCD intensity of Gd6(Mn0.5Fe0.5)23. Similar to the Gd M-edge XMCD intensity, the magnetization of Fe sublattice is proportional to the Fe L-edge XMCD intensity I(T). As shown in Fig. 3f–h, the T-dependence of Fe and Gd XMCD intensities IX(T) for Gd6(Mn0.5Fe0.5)23 exhibits a very similar T-dependence. To confirm the Fe sublattice TC we carried out a power law analysis for the data and obtained a TC = 135 K for Fe with β = 0.37 ± 0.01. The TC is consistent with \({M}_{{Tot}}^{B}\)(T)21,30 within experimental error. Further, the TC and β values for Fe XMCD are also consistent with the TC and β values of the Gd XMCD (Fig. 3b), as discussed earlier. Similarly, we also carried out a power law analysis for the XMCD signal above TC. The above-TC (χX(T)−1) power law analysis also showed very similar TC values compared to the values obtained from the below-TC (IX(T)) analysis, with γ = 1.35 ± 0.02 as listed in Supplementary Table 4. From the Kouvel-Fisher results, the analyses showed values of TC = 134.4 K, β = 0.36 ± 0.01 and γ = 1.33 ± 0.02.
To study the inter-relation between sublattices, we compared the T-dependent XMCD of Gd and Fe moments for x = 0.75 which showed a Gd sublattice TC ~ 273.5 K, while \({M}_{{Tot}}^{B}(T)\)-studies showed a TC = 309 K32. The normalized IX(T) of Gd and Fe decrease systematically on increasing T but deviate from each other and become nearly zero at different T as shown in Fig. 3i–m. We confirm this point by fitting the T-dependence of Fe XMCD intensity IX(T) to the power law used above and find TC(Fe) = 306 K and β = 0.37 ± 0.01. The TC(Fe) is consistent with \({M}_{{Tot}}^{B}(T)\)32, while β is consistent with the Gd XMCD which showed a TC(Gd) = 273.5 K, as discussed in Fig. 3b. We also carried out a power law analysis for the XMCD signal above TC.
Before doing the power law and Kouvel-Fisher analysis for the XMCD data above TC, we ensured that the x = 0.75 sample is actually in the paramagnetic phase. We have measured the XMCD for the full spectral range (not shown) for 22 different temperatures, and the extracted XMCD intensities at the Fe L3 peak are plotted in Fig. 3i. Supplementary Fig. 8 shows the Fe L-edge XMCD measured for Gd6(Mn0.25Fe0.75)23 from 29 K to 323 K over the full spectral energy range for a subset of temperatures, and is discussed in Supplementary Note 7. In particular, it is clearly seen in Supplementary Fig. 8 that the XMCD signal at T = 316 K and 323 K is reversed compared to all the temperatures below TC = 306 K. In the ferrimagnetic phase with T < TC, the XMCD of Gd and Fe showed opposite signs. However, above TC, the sign of the XMCD signal did not change for the Gd XMCD but the sign of the Fe XMCD switched and showed the same sign as the Gd signal. Thus, the XMCD signal corresponds to the paramagnetic phase. The XMCD signal observed above TC is attributed to the disordered local moments aligned by the applied magnetic field65.
The above-TC (χX(T)−1) power law analysis shows very similar TC values compared to the values obtained from the below-TC (IX(T)) analysis, as listed in Supplementary Table 4. Further, β = 0.37 ± 0.01 and γ = 1.35 ± 0.02 for Fe and Gd are very similar to each other (Supplementary Table 4). As shown in Fig. 3k, m, we also carried out a Kouvel-Fisher analysis of the Gd and Fe XMCD signals above and below TC. From the linear fits to the data, we have obtained values of TC, β and γ values for the Gd and Fe sublattice ordering, very consistent with the power law analyses. The results again show that a Kouvel-Fisher analysis can be reliably used for determining the TC and critical exponents from XMCD measurements.
For the parent compound with x = 0.0, i.e. Gd6Mn23, it is known from \({M}_{{Tot}}^{B}\)(T) that the sample TC = 489 K32. Since the Mn XMCD signal is very small at T = 29 K, we could not measure the Mn sublattice T-dependent XMCD as a function of T. However, using the \({M}_{{Tot}}^{B}\)(T) data reported earlier, we have confirmed that it also follows a power law with β = 0.38 ± 0.01 and TC = 489 K, as shown in Supplementary Fig. 5. Similarly, we have confirmed for x = 0.75 that \({M}_{{Tot}}^{B}\)(T) follows a power law with β = 0.36 ± 0.01 and TC = 309 K as shown in Supplementary Fig. 6. Considering the origin of the maxima observed in magnetic entropy measurements, it is clear that the high temperature maximum matches the bulk TC, while the low temperature maximum for x = 0.0 at T~100 K is not due to the Gd sublattice TC, which is measured to be 273.5 K. Instead, the entropy maxima at T~100 K is related to the XMCD step observed at T~100 K, consistent with T-dependent magnetization with 1 T magnetic field reported earlier15 (Supplementary Fig. 5). It is noted that in the fit for x = 0.0, the Gd XMCD with TC = 273.5 K (Fig. 3b) matched with the Gd XMCD for x = 0.75 (Fig. 3j, k). Accordingly, for all x, element specific TC’s, critical exponents β as well as γ, obtained from a power-law and Kouvel-Fisher analyses of T-dependent XMCD results, are listed in Supplementary Table 4. It is seen from Supplementary Table 4 that for all x, β = 0.37 ± 0.01, and is close to the theoretical estimate of β = 0.36563, indicating a robust 3D Heisenberg criticality in Gd6(Mn1−xFex)23.The exponent γ = 1.35 ± 0.02 is also close to the theoretically expected γ = 1.386 and only for x = 0.2, the value of γ (= 1.29 ± 0.02) deviates a little from for the 3D Heisenberg model63.
In comparison, we would like to clarify that element specific XMCD in combination with magnetic measurements was used to study a variety of magnetic phenomena in rare-earth materials66,67,68. For example, T-dependent study of Co K-edge and Pr L-edge XMCD on the compound La0.75Pr0.5Co2P2, the Co sublattice showed a FM TC1 = 167 K and the Pr sublattice showed a TC2 = 66 K66. In a study of the origin of perpendicular magnetic anisotropy in amorphous NdxCo1−x thin films, XMCD at the Co L2,3- and Nd M4,5-edges was used to show a decoupling of Nd and Co moments67. Regarding single element critical behavior, using T-dependent Eu L-edge and M-edge XMCD, it was shown that ferrimagnetic Eu0.95Fe4Sb12 exhibits a mean-field power-law exponent (β = 0.52 ± 0.05), while a reference FM clathrate material Eu8Ga16Ge30 showed a 3D Heisenberg-type exponent (β = 0.356 ± 0.03)68. However, multi-element critical behavior using T-dependent XMCD with Kouvel-Fisher analyses showing distinct TC’s has not been reported earlier and our study shows it provides valuable insights to understand multi-element magnetic systems.
In Fig. 4a, we summarize the element specific TCs, \({M}_{{Tot}}^{X}\) and \({M}_{{Tot}}^{B}\), while Fig. 4b shows the Mn and Fe magnetic moments (µMn, µFe) as a function of x, obtained from experimental results and analyses. The TC plot can be divided into three regions: (i) For 0.0 < x ≤ 0.15, the bulk TC is determined by Mn sublattice, and Gd moments exhibit a lower TC = 273.5 K compared to Mn moments. (ii) In the intermediate composition range 0.15 < x ≤ 0.72, the Gd and Fe sublattices show the same TC. (iii) For 0.72 < x ≤ 1.0, the Fe moments determine the bulk TC, which is larger than the Gd sublattice TC = 273.5 K. The results thus show coexistence of the 3-D Heisenberg-type critical behavior for the Gd and Mn/Fe sublattice spins even when the sublattices have different TC’s.

a Summary of element specific TCs, \({M}_{{Tot}}^{X}\) and \({M}_{{Tot}}^{B}\) as a function of x. The plots of TC(Gd) (empty red circle  ) and TC(Fe) (red cross circle
) and TC(Fe) (red cross circle  ) can be divided into three regions: (i) For 0.0 < x ≤ 0.15, bulk TC is determined by Mn sublattice, and TC(Gd) = 273.5 K < bulk TC. (ii) For 0.15 < x ≤ 0.72, the Gd and Fe sublattices show same TC = bulk TC. (iii) For 0.72 < x ≤ 1.0, the Fe moments determine the bulk TC, and TC(Gd) = 273.5 K < bulk TC. b Summary of Mn and Fe magnetic moments (µMn, µFe) as a function of x, showing switching of Mn moments for x ≥ 0.2 plotted with magnetic moments from 3s HAXPES analysis55. Error bars represent standard error (SE).
) can be divided into three regions: (i) For 0.0 < x ≤ 0.15, bulk TC is determined by Mn sublattice, and TC(Gd) = 273.5 K < bulk TC. (ii) For 0.15 < x ≤ 0.72, the Gd and Fe sublattices show same TC = bulk TC. (iii) For 0.72 < x ≤ 1.0, the Fe moments determine the bulk TC, and TC(Gd) = 273.5 K < bulk TC. b Summary of Mn and Fe magnetic moments (µMn, µFe) as a function of x, showing switching of Mn moments for x ≥ 0.2 plotted with magnetic moments from 3s HAXPES analysis55. Error bars represent standard error (SE).
Figure 4b shows a relatively abrupt reduction of the Mn moment upon Fe substitution, with a switching of the net Mn moment µMn from parallel (x = 0.0) to antiparallel (x = 0.2) orientation with respect to Gd moments µGd. The Fe moments show a nearly linear gradual increase with x up to x = 0.75. The intermediate x region exhibits a nearly flat minimum of TC for x = 0.3-0.5, with the Fe sublattice moment effectively overcoming the Mn sublattice and in region (iii), the Fe sublattice is dominantly responsible for determining the Curie temperature. Thus, the Mn-moment switching and gradual increase in Fe-moment cause the non-monotonic TC’s and monotonic decrease in magnetization with increasing x. It is clear that region (i) and (iii) are dominated by M-M and M’-M’ exchange, respectively, and implies a weaker R-R exchange in these regions. In region (ii), M-M’ exchange gets reduced below the R-R exchange of regions (i) and (iii). However, TC of the Gd, Mn and Fe sublattices get reduced together, and indicates that R-(M, M’) exchange is active and coupled to M-M’ exchange in region (ii), and results in lowest TC. As discussed in the introduction, earlier studies did not recognize the possible role of R-(M,M’) exchange but from the present results of the Gd sublattice TC with respect to bulk TC, we can conclude that (R-M,M’) exchange is active in region (ii). This evolution of exchange interactions has not been recognized in earlier studies and indicates the importance of element-specific TC’s for tuning magnetic properties. The study shows that power-law and Kouvel-Fisher analyses of T-dependent XMCD provides a reliable method to precisely investigate the role of element-sensitive magnetism in any general Ra(M1−xM’x)b series of alloys.
Methods
Sample preparation and characterization
The Gd6(Mn1−xFex)23 were synthesized using stoichiometric amounts of high-purity metals (Gd 99.9 wt.% from Rhodia, Mn 99.99 wt.%, and Fe 99.8 wt.% from Alfa Aesar) by melting them in a high frequency induction furnace (CELES) under pure argon atmosphere. The crystal structure was verified to be cubic by powder X-ray diffraction, using a Philips X-Pert Pro Diffractometer, Cu Kα)32,35,36, and it confirmed the absence of impurity phases. The chemical purity and composition of each sample was checked by microprobe analysis (CamecaSX 100) on mirror polished powder samples dispersed in a cold resin. The purity was evaluated from backscattered electron (BSE) micrographs on different particles and the chemical composition was confirmed to be the nominal composition from an average of six randomly chosen pinpoints on the sample surface.
Spectroscopy experiments
XAS and XMCD measurements were performed at the Dragon Beamline (BL 11A) of the Taiwan Light Source. The samples were cleaved in-situ in an ultrahigh vacuum (UHV) chamber of 8 × 10−10 mbar at T = 29 K. The total electron yield (TEY) method was used to measure XAS and XMCD across the Gd M4,5-edges (3d − 4f), Mn L2,3-edges (2p − 3d) and Fe L2,3-edges (2p − 3d) with circularly polarized light. An external magnetic field of ±1 T was applied along the surface with a circular polarization degree of 80% and was 30o with respect to circularly polarized light direction. The total energy resolution at the Fe L-edge was 0.2 eV for the XAS-XMCD spectra. The incident photon energy has an accuracy of ±10 meV at Fe L-edge and ±20 meV at Gd M-edge. The photon energy was calibrated using a reference Mn metal sample, Fe metal sample, MnO sample, Fe2O3 sample, and Dy metal sample. The sample was cooled using a liquid-He flow-type cryostat, and the measurements were carried out from T = 29 K to T = 340 K. The net orbital and spin moments of Gd, Mn as well as Fe were derived using the well-known sum rules for x-ray magnetic circular dichroism69,70.
Calculation methods
All calculations were performed with Density Functional Theory (DFT) using the Vienna ab initio simulation package (VASP)71,72,73. Spin-polarized calculations were performed with plane-wave basis set and projector-augmented wave (PAW) method73. The following electrons were treated explicitly: 3s2 3p6 3d6 4s1 (Mn), 3s2 3p6 3d7 4s1 (Fe), 5s2 6s2 5p6 5d1 4f 7 (Gd). The strong on-site Coulomb interaction of localized electrons was treated through the DFT + U approach61. Moreover, the on-site Coulomb energy for Mn and Fe, \({U}_{{Mn}}^{{DFT}}\) and \({U}_{{Fe}}^{{DFT}}\) were varied from 0.0 to 2.5 eV and for Gd, \({U}_{{Gd}}^{{DFT}}\) was varied from 6.0 to 12 eV to obtain magnetic moments close to the experimental values. The optimal magnetic moments (s, p, d, f) for a unit cell containing 116 atoms corresponding to 4 formula units of Gd6M23 (M = Mn, Fe) obtained from the calculations are listed in Supplementary Table 5 for Gd6Mn23, and for Gd6Fe23 in Supplementary Table 6. The one-electron Kohn–Sham orbitals were expanded in a plane-wave basis set with a kinetic energy cutoff of 360 eV. Total energies were minimized until the energy differences were less than 10−4 eV between two electronic cycles. The reciprocal space integration was approximated with a Monkhorst–Pack k-point grid of 9 × 9 × 9.
Data availability
The data sets generated/analyzed during the current study are available from the corresponding author on request.
References
Stewart, G. R. Heavy-fermion systems. Rev. Mod. Phys. 56, 755–787 (1984).
Mazet, T. et al. Nonpareil Yb Behavior in YbMn6Ge6−xSnx. Phys. Rev. Lett. 111, 096402 (2013).
Eichenberger, L. et al. Possible room-temperature signatures of unconventional 4f -electron quantum criticality in YbMn6Ge6−xSnx. Phys. Rev. B 101, 020408 (2020).
Stewart, G. R. Non-Fermi-liquid behavior in d- and f-electron metals. Rev. Mod. Phys. 73, 797–855 (2001).
Taylor, K. Intermetallic rare-earth compounds. Adv. Phys. 20, 603 (1971).
Wallace, W. E. Rare earth intermetallics, Academic press, New York London, 1973).
Roy, S. B. In Handbook of Magnetic Materials (ed Buschow, K.) 203–316 (Elsevier, Amsterdam, 2014).
Balaram, V. Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact. Geosci. Front. 10, 1285–1303 (2019).
Campbell, I. A. Indirect exchange for rare earths in metals. J. Phys. F: Metal Phys. 2, L47–L50 (1972).
Brooks, M. S. S. et al. 3d-5d band magnetism in rare earth-transition metal intermetallics: total and partial magnetic moments of the RFe2 and (R=Gd-Yb) Laves phase compounds. J. Appl. Phys. 70, 5972–5976 (1991).
Hilscher, G. & Rais, H. Magnetic properties and molecular field coefficients of Er6(Fe1−xMnx)23. J. Phys. F: Metal Phys. 8, 511 (1978).
Duc, N. H. An evaluation of the R-T spin coupling parameter in the rare earth–transition metal intermetallics. Physica status solidi (b) 164, 545–552 (1991).
Duc, N. H., Hien, T. D., Givord, D., Franse, J. J. M. & de Boer, F. R. Exchange interactions in rare earth-transition metal compounds. J. Magn. Magn. Mater. 124, 305–311 (1993).
Franse, J. J. M. & Radwánski, R. J. Chapter 5 Magnetic properties of binary rare-earth 3d-transition-metal intermetallic compounds 307–501 (Elsevier, 1993).
Buschow, K. & Sherwood, R. Magnetic properties and hydrogen absorption in rare-earth intermetallics of the type RMn2 and R6Mn23. J. Appl. Phys. 48, 4643–4648 (1977).
Crowder, C. & James, W. A review of the magnetic structures and properties of the R6M23 compounds and their hydrides. J. Less Common Metals 95, 1–15 (1983).
Bessais, L. Structure and Magnetic Properties of Intermetallic Rare-Earth-Transition-Metal Compounds: A Review. Materials 15, 1996–1944 (2022).
DeSavage, B. F., Bozorth, R. M., Wang, F. E. & Callen, E. R. Magnetization of the Rare-Earth Manganese Compounds R6Mn23. J. Appl. Phys. 36, 992–993 (1965).
Kirchmayr, H. Magnetic Properties of Rare Earth Manganese Compounds. IEEE Trans. Magn. 2, 493–499 (1966).
Kirchmayr, H. Magnetic Properties of the Compound Series Y(MnxFe1−x)2 and Y6(MnxFe1−x)23. J. Appl. Phys. 39, 1088–1089 (1968).
Kirchmayr, H. & Steiner, W. Magnetic order of the compound series RE6(Fe1−xMnx)23 (RE = Y and Gd). J. Phys. Colloq. 32, C1-665–C1-667 (1971).
Malik, S., Takeshita, T. & Wallace, W. Hydrogen Induced Magnetic. Ordering in Th6Mn23. Solid State Commun. 23, 599–602 (1977).
Hardman, K., James, W., Déportes, J., Lemaire, R. & de la Bathie, R. P. Magnetic properties of R6Mn23 compounds. J. Phys. Colloq. 40, C5–C204–C5–C205 (1979).
James, W., Hardman, K., Yelon, W. & Kebe, B. Structural and magnetic properties of Y6(Fe1−xMnx)23. J. Phys. Colloq. 40, C5-206–C5-208 (1979).
Delapalme, A., Déportes, J., Lemaire, R., Hardman, K. & James, W. J. Magnetic interactions in R6Mn23 rare earth intermetallics. J. Appl. Phys. 50, 1987–1989 (1979).
Buschow, K. Magnetic properties of the ternary hydrides of Nd6Mn23 and Sm6Mn23. Solid State Commun. 40, 207–210 (1981).
Hardman, K., Rhyne, J. J. & James, W. J. Magnetic structures of Y6(Fe1−xMnx)23 compounds. J. Appl. Phys. 52, 2049–2051 (1981).
Parker, F. T. & Oesterreiche, H. Analysis of Magnetic Interactions and Structure in R6Mn23. Appl. Phys. A 27, 65–69 (1982).
Buschow, K., Gubbens, P., Ras, W. & der Kraan, A. V. Magnetization and Mössbauer effect study of Dy6Mn23 and Tm6Mn23 and their ternary hydrides. J. Appl. Phys. 53, 8329–8331 (1982).
Nagai, H., Oyama, N., Ikami, Y., Yoshie, H. & Tsujimura, A. The magnetic properties of psuedo-binary compounds and Gd(MnxFe1−x)2 and Gd6(MnxFe1−x)23. J. Phys. Soc. Japan 55, 177–183 (1986).
Nagai, H. et al. The anomalous behaviour of the electrical resistivities of Gd(Fe and Mn)2 and Gd6(Fe and Mn)23. J. Magn. Magn. Mater. 1131, 177–181 (1998).
Lemoine, P., Ban, V., Vernière, A., Mazet, T. & Malaman, B. Magnetocaloric properties of Gd6(Mn1−xFex)23 alloys (x ≤ 0.2). Solid State Commun. 150, 1556–1559 (2010).
Lemoine, P., Vernière, A., Mazet, T. & Malaman, B. Magnetic and magnetocaloric properties of R6Mn23 (R=Y and Nd and Sm and Gd-Tm and Lu) compounds. J. Magn. Magn. Mater. 323, 2690–2695 (2011).
Lemoine, P., Vernière, A., Mazet, T. & Malaman, B. Magnetic and magnetocaloric properties of R6Mn23 (R = Y and Sm and Tb and Dy and Ho and Er) compounds. J. Alloys Compd. 578, 413–418 (2013).
Lemoine, P., Vernière, A., Mazet, T. & Malaman, B. Magnetic and magnetocaloric properties of Gd6(Mn1−xCox)23 compounds (x ≤ 0.3). J. Alloys Compd. 680, 612–616 (2016).
Lemoine, P. Contribution à l’étude des propriétés structurales et magnétiques de composés intermétalliques isotypes de CeScSi et Th6Mn23 PhD thesis (Université Henri Poincaré and Nancy, 2011).
Dong, P. L. et al. Structural and Magnetic and Magnetocaloric Effect of Gd6(Mn1−xFex)23 Compounds. J. Low Temp. Phys. 195, 221–229 (2019).
Coehoorn, R. Calculated electronic structure and magnetic properties of Y-Fe compounds. Phys. Rev. B 39, 13072–13085 (1989).
Ouardi, S. et al. Magnetic dichroism study on Mn1.8Co1.2Ga thin film using a combination of x-ray absorption and photoemission spectroscopy. J. Phys. D: Appl. Phys. 48, 164007 (2015).
Winterlik, J. et al. Electronic, magnetic and structural properties of the ferrimagnet Mn2CoSn. Phys. Rev. B 83, 174448 (2011).
Sangaletti, L. et al. Electronic properties of the ordered metallic Mn:Ge(111) interface. Phys. Rev. B 72, 035434 (2005).
Elmers, H. J. et al. Exchange coupling in the correlated electronic states of amorphous GdFe films. Phys. Rev. B 88, 174407 (2013).
Mangin, S. et al. Magnetization reversal in exchange-coupled GdFe/TbFe studied by x-ray magnetic circular dichroism. Phys. Rev. B 70, 014401 (2004).
Kallmayer, M. et al. Magnetic properties of Co2Mn1xFexSi Heusler alloys. J. Phys. D: Appl. Phys. 39, 786 (2006).
Thole, B. T. et al. 3d x-ray-absorption lines and the 3d94f n+1 multiplets of the lanthanides. Phys. Rev. B: Condens. Matter 32, 5107–5118 (1985).
Svitova, A. L. et al. Magnetic moments and exchange coupling in nitride cluster fullerenes GdxSc3-xN@C80 (x = 1–3). Dalton Trans. 43, 7387–7390 (2014).
Okane, T. et al. X-ray Magnetic Circular Dichroism Study of Ce0.5Gd0.5Ni. JPS Conf. Proc. 3, 011028(1)–011028(6) (2014).
Chuang, C. W. et al. Electronic structure investigation of GdNi using x-ray absorption and magnetic circular dichroism and hard x-ray photoemission spectroscopy. Phys. Rev. B 101, 115137 (2020).
Streubel, R. et al. Experimental Evidence of Chiral Ferrimagnetism in Amorphous GdCo Films. Adv. Mater. 30, 1800199 (2018).
Wybourne, B. G. Energy Levels of Trivalent Gadolinium and Ionic Contributions to the Ground-State Splitting. Phys. Rev. 148, 317–327 (1966).
Kimura, A. et al. Magnetic circular dichroism in the soft-x-ray absorption spectra of Mn- based magnetic intermetallic compounds. Phys. Rev. B 56, 6021–6030 (1997).
Klaer, P. et al. Localized magnetic moments in the Heusler alloy Rh2MnGe. J. Phys. D: Appl. Phys. 42, 084001 (2009).
Yu, D. et al. Direct evidence of Ni magnetic moment in TbNi2Mn-X-ray magnetic circular dichroism. J. Magn. Magn. Mater. 370, 32–36 (2014).
Sapozhnik, A. A. et al. Experimental determination of exchange constants in antiferromagnetic Mn2Au. Phys. Rev. B 97, 184416 (2018).
Nguyen, T. L. et al. Hard x-ray photoemission spectroscopy of the ferrimagnetic series Gd6(Mn1−xFex)23. Phys. Rev. B 106, 045144 (2022).
Jaakkola, S., Korventausta, I., Hovi, V. & Lakkisto, M. Thermal expansion of Y6Mn23, Gd6Mn23, and Tb6Mn23 between 290 and 610 K, Hki: Suomalainen tiedeakatemia, Akateeminen kirjakauppa, 1980.
Kripyakevich, P., Frankevich, D. & Voroshilov, Y. Compounds with Th6Mn23 -type structures in alloys of the rare-earth metals with manganese and iron. Powder Metallurgy Metal Ceramics 4, 915–919 (1965).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron- energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Harmon, B. N., Antropov, V. P., Liechtenstein, A. I., Solovyev, I. V. & Anisimov, V. I. Calculation of magneto-optical properties for 4f systems: LSDA + Hubbard U results. Journal of Physics and Chemistry of Solids 56, 1521–1524 (1995).
Anisimov, V. I., Zaanen, J. & Andersen, O. K. Andersen, Band theory and Mott insulators: Hubbard U instead of Stoner I. Phys. Rev. B 44, 943–954 (1991).
Lang, J. K., Baer, Y. & Cox, P. A. Study of the 4f and valence band density of states in rare- earth metals. II. Experiment and results. Journal of Phys. F: Metal Physics 11, 121–138 (1981).
Trzhaskovskaya, M., Nikulin, V., Nefedov, V. & Yarzhemsky, V. Non-dipole second order parameters of the photoelectron angular distribution for elements Z=1–100 in the photoelectron energy range 1–10 keV. At. Data and Nucl. Data Tables 92, 245–304 (2006).
Kaul, S. N. Static critical phenomena in ferromagnets with quenched disorder. J. Magn. Magn. Mater. 53, 5–53 (1985).
Kouvel, J. & Fisher, M. Detailed Magnetic Behavior of Nickel Near its Curie Point. Phys. Rev. B 136, A1626 (1964).
Herrero-Albillos, J. et al. Observation of a different magnetic disorder in ErCo2. Phys. Rev. B 76, 094409 (2007).
Kovnir, K. et al. Modification of magnetic anisotropy through 3d−4f coupling in La0.75Pr0.25Co2P2. Phys. Rev. B 88, 104429 (2013).
Cid, R. et al. Perpendicular magnetic anisotropy in amorphous NdxCo1−x thin films studied by x-ray magnetic circular dichroism. Phys. Rev. B 95, 224402 (2017).
Krishnamurthy, V. V. et al. Robertson, Temperature dependence of Eu 4f and Eu 5d magnetizations in the filled skutterudite EuFe4Sb12. Phys. Rev. B 79, 014426 (2009).
Thole, B. T., Carra, P., Sette, F. & van der Laan, G. X-Ray Circular Dichroism as a Probe of Orbital Magnetization. Phys. Rev. Lett. 68, 1943–1946 (1992).
Chen, C. T. et al. Experimental Confirmation of the X-Ray Magnetic Circular Dichroism Sum Rules for Iron and Cobalt. Phys. Rev. Lett. 75, 152–155 (1995).
Kresse, G. & Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using A Plane-Wave Basis Set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Acknowledgements
This work was granted access to the HPC resources of TGCC, CINES, and IDRIS under the allocation 99,642 attributed by GENCI (Grand Equipement National de Calcul Intensif), France. High Performance Computing resources were also partially provided by the EXPLOR Centre hosted by the University de Lorraine (project 2017M4XXX0108), France. Y.C.T. thanks the National Science and Technology Council, Taiwan, Republic of China, for financially supporting this research under Contract No. NSTC 112-2622-8-A49-013 -SB. A.C. thanks the National Science and Technology Council, Taiwan, Republic of China, for financially supporting this research under Contract No. NSTC 111-2112-M-213-031.
Author information
Authors and Affiliations
Contributions
T. Ly Nguyen: Conceptualization, Data curation, Formal analysis, Investigation, Validation, Visualization; Roles/Writing - original draft, Writing - review & editing. Th. Mazet: Investigation, Methodology, Resources, Validation, Writing - review & editing. E. Gaudry: software, Formal analysis, Writing - review & editing. D. Malterre: Validation, Writing - review & editing, Validation, Supervision. F. H. Chang: Methodology. H. J. Lin: Methodology, Supervision. C. T. Chen: Methodology. Y. C. Tseng: Validation. A. Chainani: Conceptualization, Funding acquisition, Supervision, Project administration, Data curation, Formal analysis, Investigation, Validation, Visualization, Roles/Writing - original draft, Writing - review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Materials thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Alannah Hallas and Aldo Isidori. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, T.L., Mazet, T., Gaudry, É. et al. Element-specific Curie temperatures and Heisenberg criticality in ferrimagnetic Gd6(Mn1−xFex)23 via Kouvel-Fisher analysis. Commun Mater 5, 68 (2024). https://doi.org/10.1038/s43246-024-00496-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-024-00496-2
