
Abstract
Herein, we theoretically demonstrate that simple metal (Ga and Al) substitutional atoms, rather than the conventional transition metal substitutional elements, not only stabilize the ThMn12-type SmFe12 and Sm(Fe,Co)12 phases thermodynamically but also further improve their intrinsic magnetic properties such that they are superior to those of the widely investigated SmFe11Ti and Sm(Fe,Co)11Ti magnets, and even to the state-of-the-art permanent magnet Nd2Fe14B. More specifically, the quaternary Sm(Fe,Co,Al)12 phase has the highest uniaxial magnetocrystalline anisotropy (MCA) of about 8 MJ m−3, anisotropy field of 18.2 T, and hardness parameter of 2.8 at room temperature and a Curie temperature of 764 K. Simultaneously, the Al and Ga substitutional atoms improve the single-domain size of the Sm(Fe,Co)12 grains by nearly a factor of two. Numerical results of MCA and MCA-driven hard magnetic properties can be described by the strong spin-orbit coupling and orbital angular momentum of the Sm 4f-electron orbitals.
Similar content being viewed by others
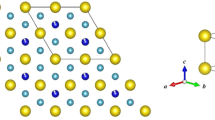


Introduction
ThMn12-type SmFe12 alloy is known for its attractive intrinsic hard magnetic properties1,2,3,4,5,6,7,8, which make it a potential high-performance permanent magnet. Despite its high saturation magnetization Ms and strong uniaxial magnetocrystalline anisotropy (MCA) Ku, obtaining a single-crystalline phase of the ThMn12 structure is extremely difficult9,10,11,12,13. In particular, thermodynamically stable large (bulk)-scale production of SmFe12 single crystals is in high demand for industrial applications such as electric motors and generators. To stabilize the ThMn12 structure, a third substitutional metal element, including Ti or V14,15,16,17,18,19,20,21, is essential. However, inclusions of these early transition metal (TM) elements are severely detrimental to intrinsic magnetic properties. For example, SmFe11Ti and SmFe11V (SmFe10V2) exhibit μ0Ms of 1.14−1.16 and 1.12 (0.81) T and Ku of 3.9−4.8 and 1.92 (1.58) MJ m−3 at room temperature15,16,17,21, respectively, which are much lower than the corresponding values (1.64 T and 5.4−5.67 MJ m−3) of the parent SmFe1222,23. In contrast, the replacement of 20 at.% Fe at the 8f site with Co enhances μ0Ms up to 1.78 T and Ku up to 6.2 MJ m−3 at room temperature22,23,24. These findings, in addition to the enhanced Curie temperature (Tc = 859 K), make ternary Sm(Fe0.8Co0.2)12 superior to the state-of-the-art permanent magnet Nd2Fe14B (μ0Ms = 1.57 T, Ku = 4.5 MJ m−3, and Tc = 585 K)25,26,27. However, the ThMn12 structure of this ternary compound could only be synthesized with a limited film thickness of no more than 0.5 μm22,23. In addition to the structural instability, to maximize the permanent hard magnetic properties, the grain size of SmFe12-based magnets must be prepared close to the single-domain (SD) size (~51−54 nm)28,29. However, preparing nanometre-sized ThMn12-type SmFe12 in a practical sample is quite difficult, which must be resolved to fully utilize SmFe12 as a practical high-performance permanent magnet. The development of thermodynamically stable bulk-scale SmFe12 single crystals with improved SD grain sizes and desirable intrinsic magnetic properties is thus essential and of timely importance.
In this paper, we propose a possible solution to realize an otherwise unstable ThMn12-type SmFe12 permanent magnet through systematic first-principles density functional theory (DFT), density functional perturbation theory (DFPT), and Monte Carlo (MC) simulations on ternary Sm(Fe,M)12 and quaternary Sm(Fe,Co,M)12 alloys (M is a 3d or 3p metal substitutional atom). In contrast to conventional TM substitutional elements, simple metal (SM) Al and Ga substitutional atoms stabilize the ThMn12-type Sm(Fe,Co)12 structure thermodynamically with an improved SD grain size and simultaneously enhanced Ku. We further predict that intrinsic hard magnetic properties at an elevated temperature of the proposed quaternary Sm(Fe,Co,Al)12 and Sm(Fe,Co,Ga)12 compounds in the present study are superior to those of the widely investigated SmFe11Ti and Sm(Fe,Co)11Ti compounds, and even to the present and reported values by the experimental25,26,27 and previous theoretical studies30,31 for the state-of-the-art permanent magnet Nd2Fe14B, including a higher Tc. We attribute the physical origin of the large MCA and MCA-driven hard magnetic properties to the strong spin-orbit coupling (SOC) and orbital angular momentum of the Sm 4f-electron shells.
Results
Structural stability
We first inspected the structural stability of ThMn12-type SmFe12 structure upon M (M = Ti − Ga and Al) replacement. The formation enthalpy is defined as Hf = (E0 − ∑iμiNi)NA/N, where E0 is the total energy of the system and μi and Ni are the chemical potential and the number of decomposable component i, respectively. NA is the Avogadro number and N is the total number of atoms in the computed unit cell. Our computed unit cell is composed of 2 Sm atoms at the 2a site and 24 Fe atoms at the inequivalent crystallographic 8f, 8i, and 8j sites (Wyckoff positions). Their optimized atomic coordinates along with the lattice parameters a, b, and c of SmFe12 are given in Table 1. The present a = b and c values are 8.481 and 4.661 Å, respectively, in agreement with the experimental (8.44 and 4.81 Å)5,22 and previous theoretical results (8.46−8.49 and 4.68−4.81 Å)8,32. Correspondingly, the three different substitution sites of M atoms for SmFe12−xMx (x = 0.5, 1, and 2) were considered and denoted as M(8f), M(8i), and M(8j). For each configuration, from the total energy minimization, the M atoms were identified to prefer a uniform distribution24, as indicated in Fig. 1a–c. The optimized lattice parameters of SmFe10M2 with the preferred M site are listed in Table 2. In agreement with an experiment33, ThMn12-type SmFe12 is not a stable phase and decomposes into bulk-Sm and α-Fe phases, as expected from the calculated positive Hf values of 2.9 kJ mol−1 from the standard DFT and 6.5 kJ mol−1 from the DFT plus U (DFT + U) with Ueff = 6 eV. A slightly smaller value of Hf = 1.8 kJ mol−1 was reported in the previous DFT calculations34. Nevertheless, the single-crystalline ThMn12 phase can be obtained in SmFe11Ti form15,16,17. For a ternary system, simply considering decomposition into the final constituent elements is not sufficient for predicting the structural stability35,36,37,38. The most competitive binary decomposable phases for Sm−Fe−M are identified by constructing the convex hull phase diagram35,36,37,38.

ThMn12-type atomic structures of SmFe12−xMx (when x = 2) for the a 8f, b 8i, and c 8j site M substitutes. Only the most stable configuration of the distribution patterns of the M atoms is shown for each substitution site.
We show heat map of the calculated formation enthalpy of the ternary Sm−Fe−Ti system in Fig. 2a. The solid nodes on the phase diagram represent the present ternary SmFe12−xTix phases (x = 0.5, 1, and 2), while the known binary and elemental phases are indicated by the open nodes. In the Sm−Fe−Ti diagram, since all Sm−Fe binary phases are unstable against their elemental bulk-Sm and α-Fe phases, we have chosen the bulk-Sm and α-Fe as decomposable phases rather than a Sm−Fe binary phase. We find that the Hf values of the known binary Sm2Fe17, Sm3Fe29, Sm6Fe23, and SmFe2 phases are 8.4, 9.2, 4.9, and 0.9 kJ mol−1, respectively. In the Ti-poor region (i.e., x = 0.5), Fe2Ti is chosen as the decomposable binary phase for Sm−Fe−Ti, as indicated by the dotted lines in Fig. 2a. In the Ti-rich region (i.e., x = 1 and 2), Fe2Ti + FeTi decomposition is taken into account (solid lines). The numerical values of the obtained \({H}_{{{{\rm{f}}}}}^{{{{\rm{elm}}}}}\) (against the elemental decomposition) and \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) (against the binary decomposition) of SmFe12−xTix phases are shown in Fig. 2b for x = 0, 0.5, 1, and 2. Both \({H}_{{{{\rm{f}}}}}^{{{{\rm{elm}}}}}\) and \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) decrease as x increases and reach the negative values of −10.7 and −4.2 kJ mol−1 at x = 2, respectively. For SmFe11Ti, \({H}_{{{{\rm{f}}}}}^{{{{\rm{elm}}}}}=-2.6\) kJ mol−1 and \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}=0.2\) kJ mol−1. Since the single-crystalline SmFe11Ti phase is practically achieved in the bulk form15,16,17, the latter value (0.2 kJ mol−1) of SmFe11Ti is referred to as a threshold of the ThMn12-phase stability in the present study. The aforementioned experimental observations of the reduced μ0Ms and Ku upon the Ti substitution15,16,17 have also been obtained in our calculations, as shown in Fig. 2c, which will be discussed more explicitly in the following paragraphs.

a Heat map of the formation enthalpy of the ternary Sm−Fe−Ti system. Open nodes mark the known elemental and binary phases. Solid nodes represent the present ternary SmFe12−xMx (x = 0.5, 1, and 2) phases. Dotted and solid lines are the projections of the convex hull construction into compositional space. b Formation enthalpy Hf and c saturation magnetization μ0Ms (left) and uniaxial magnetocrystalline anisotropy Ku (right) of SmFe12−xMx (x = 0, 0.5, 1, and 2). The corresponding experimental results for SmFe12 measured at 0−300 K22,23 and SmFe11Ti at 4.2−300 K15,16,17 are indicated by the open symbols, where the result at 0 K is determined by fitting with low-temperature measurements22.
Figure 3a presents the \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) values of SmFe12−xMx (M = Ti − Ga and Al; x = 0.5 and 2) for M(8f), M(8i), and M(8j). Early TMs, Ti to Mn, prefer the 8i site rather than the 8f and 8j sites for both x = 0.5 and 2. A similar trend is also observed for SmFe11M, which, except for SmFe11Ti, is not shown for simplification of the figure. Hereafter, we thus refer to the results corresponding to the practically acceptable stoichiometry SmFe10M2, unless specifically mentioned. These results are supported by the experiment in which Ti and V dopants occupied the 8i site39. Late TMs, except for Co(8f), prefer the 8j site. The different site preferences can be interpreted in correlation with the atomic radii and electronegativity. In the SmFe12 structure, the 8i site (3.06 and 4.98 Å) is farther from Sm than the 8j (3.04 and 4.82 Å) and 8f (3.21 and 3.21 Å) sites (Table 1). Ti − Mn with large atomic radii thus naturally prefer the 8i site. In contrast, Ni − Ga attract Sm more than Ti − Mn, as Ni − Ga (1.65−1.91) have higher electronegativities than Ti − Mn (1.54−1.66).

a Formation enthalpy \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) of SmFe12−xMx with x = 0.5 (open) and 2 (solid symbols) for M = Ti−Zn (transition metals), Ga and Al (simple metals) for the 8f, 8i, and 8j sites. Horizontal dashed line corresponds to the calculated \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) value of SmFe11Ti, a threshold of the ThMn12-phase stability. b Saturation magnetization μ0Ms and c uniaxial magnetocrystalline anisotropy Ku of SmFe10M2 with the preferred M site. Solid and open circles indicate the experimental data for SmFe11M and SmFe10V2/SmFe9.6Co2.4 measured at 0−300 K14,15,16,17,20,21,22,23, respectively, where the result at 0 K is determined by fitting with low-temperature measurements22.
As predicted from the \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) values (Fig. 3a), the Ga (\({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}=-1.5\) kJ mol−1) and Al (−3.1 kJ mol−1) substitutional atoms stabilize the ThMn12 structure. These SMs improve the stability comparably to the conventional stabilizing early TM substitutional elements Ti (−4.2 kJ mol−1) and V (0.2 kJ mol−1). On the other hand, the dopant amount of x = 0.5 is not sufficient for stabilizing the ThMn12 phase as the aforementioned basic requirement of the crystal formation (\({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}} \,<\, 0.2\) kJ mol−1; as indicated by the horizontal dashed line in Fig. 3a) is not fulfilled. Practically, SmFe12−xTix and SmFe12−xVx phases are synthesized with a nearly perfect ThMn12 structure when x ≥ 114,15,16,17,20,21. Furthermore, a very recent experiment reported that the stability of SmFe8.8Co2.2Ti was greatly improved by Ga addition33. The authors also predicted theoretically that Ga is the most effective stabilizing elements with higher μ0Ms over Ti, V, and Cr dopants. Our prediction in support with these experimental and theoretical studies suggests a possible synthesis of the single-crystalline ThMn12 structure, including a series of Sm(Fe,Co)12 and Sm(Fe,Co,Ti)12 compounds, with Al and/or Ga, at least at a low temperature.
Intrinsic magnetic properties
The Slater − Pauling-like behavior of μ0Ms is evident in Fig. 3b for the late TMs (Mn − Zn). For each M, only the result obtained by the full-potential calculations for the preferred substitution site is presented. For SmFe12, the experimental μ0Ms of 1.94 T at 0 K determined by fitting with low-temperature measurements22 is underestimated in the present theory (1.64 T). This discrepancy might be attributed to the existence of the α-Fe phase and surface/interface effect in a real thin-film sample, which are fully ignored in the present simulation. To obtain microscopic insight, we decompose μ0Ms into the atomic-level spin magnetic moment (μS) in Table 2. The spin moment contributions of the Fe(8f), Fe(8i), and Fe(8j) atoms to μ0Ms are 1.78, 2.53, and 2.34 μB, respectively. The Sm 4f spin moment (−5.44 μB) is antiparallel to the Fe 3d spin moment, which is mediated by the Sm 5d orbitals40,41. The magnitude of μS adheres to the high-spin state. Nevertheless, the orbital moment contribution (μL = 2.25 μB) of the 4f electrons of Sm (antiparallel to its spin moment according to the 3rd Hund’s rule, thus parallel to the Fe 3d spin moment) to μ0Ms is substantial, whereas those from the Fe 3d orbitals are <0.1 μB.
In the Λ-shape curve (Slater − Pauling-like) shown in Fig. 3b, a peak occurs for M = Co; μ0Ms increases from 1.64 T for SmFe12 to 1.68 T for SmFe10Co2. A similar trend was found in the room-temperature measurement22, as indicated by the circle symbols in Fig. 3b; 1.78 T for SmFe9.6Co2.4 and 1.64 T for SmFe12. But this is not the case in the extrapolated zero-temperature measurement22; μ0Ms decreases from 1.94 T for SmFe12 to 1.88 T for SmFe9.6Co2.4. The difference could be attributable to the aforementioned shortcomings as well as to dissimilar Co concentrations in the present theory (i.e., SmFe10Co2) and experiment (i.e., SmFe9.6Co2.4)22. Both the theory and experiment reveal that the early TMs (Ti − Mn) greatly lower μ0Ms (0.75 − 0.95 T)16,17,21 because of their antiparallel spin coupling to Fe (Table 2). Thus, the concentration of these early TMs should be kept at a minimal. For SmFe11Ti, the present μ0Ms is 1.31 T, which is comparable with 1.28−1.14 T at 4.2−300 K in the experiments15,16,17. In contrast to the early TMs, the nonmagnetic late TMs and SMs are not as detrimental to μ0Ms (1.2−1.3 T). Overall, μ0Ms is mainly affected by the magnetic moments of the M atom and its neighboring Fe atoms, as addressed explicitly in Table 2.
Figure 3c shows the computed Ku of SmFe10M2 (M = Ti − Ga and Al). We recall from ref. 24 that the Ku values of SmFe12 are 12.8 MJ m−3 in the DFT calculation and 10.6 MJ m−3 in the DFT + U calculation (with Ueff = 6 eV). Both values are in reasonable agreement with the low-temperature experimental values of 10.76−11.1 MJ m−3 at 100−10 K23. The Ku results thus refer to those from DFT calculations without the Ueff parameter to minimize computational complications. From a theoretical point of view, we here would like to remind that direct comparison with experiment, particularly on the precise magnitude of MCA, requires some caution, as an accurate treatment of f-electron systems by first-principles is quite challenging. Nevertheless, our theory for M = Co (Ku = 11.2 MJ m−3) further supports the low-temperature experiment in which the substitution of Co atoms for 20 at.% Fe at the 8f site reduced Ku to 9.73 MJ m−3 at 10 K23. According to Fig. 3c, substantial reductions obviously appear for the other TM substitutions, including the reference SmFe11Ti system (Ku = 9.8 MJ m−3). Similar reductions were also observed for SmFe11Ti (7.2−3.9 MJ m−3 at 77−300 K)15,16,17 and SmFe10V2 (1.58 MJ m−3 at 300 K) in the experiments21. Cr and Mn even turn the magnetic easy axis from the c axis to the ab plane (which is improper for permanent magnets). In contrast, the SM substitutes (Ga and Al), which stabilize the ThMn12 structure, still preserve the MCA uniaxiality. In particular, Ku reaches as high as 7.6 MJ m−3 for M = Al, which is 6.3 MJ m−3 for M = Ga. Hence, in the discussion below we focus mainly on Al rather than Ga. Note that this uniaxial MCA makes Sm(Fe,M)12-based magnets a potential high-performance permanent magnet beyond the other ThMn12-type Nd(Fe,M)12 magnets with a biaxial MCA12,13,18,19.
Recalling the favorable formation (\({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}} \,<\, 0\) in Fig. 3a), in a real sample, Al (which has the smallest atomic radius among the M) could occupy either the 8j or 8i site, or both. These two sites are energetically competitive and differ in \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) by only within 1.4 kJ mol−1. For a given temperature, we estimate the occupation probability \(\left\langle {N}_{\nu }\right\rangle\) of the Al substitution sites (ν = 8f, 8i, and 8j) by using Maxwell-Boltzmann statistics. As shown in Fig. 4, the occupation probability of the Al substitute at the 8j (8i) site decreases (increases) with temperature; \(\langle {N}_{{{{\rm{8i}}}}}\rangle :\langle {N}_{{{{\rm{8j}}}}}\rangle =0.30:0.57\) at 300 K and 0.33:0.48 at 500 K. A certain amount of the 8f site could also be occupied by the Al atoms in a high-temperature sample. We further find that Ku is almost independent of the stable 8i (10.9 MJ m−3) and 8j (7.6 MJ m−3) sites. From a practical viewpoint, this substitution-site-independent uniaxial feature of the MCA is worth noting, in addition to the abundance of Al on earth.

Occupation probability \(\left\langle {N}_{\nu }\right\rangle\) of the Al substitution sites (ν = 8f, 8i, and 8j) at an elevated temperature in SmFe10Al2.
Impact of multielement substitution
We next explored the effects of multielement substitution on the intrinsic permanent magnetic properties. According to Fig. 3, only Co substitution maximizes μ0Ms, while Ti, V, Ga, and Al improve the SmFe12 stability. To support this scenario, we extended our calculations to the quaternary stoichiometries SmFe10CoM and SmFe9Co2M (M = Ti, V, Ga, and Al). For each M, we considered several different substitutional configurations of the M atoms while the Co atoms were kept fixed at their optimized 8f sites, as shown in Fig. 1a. As examples, we illustrate the most favorable structures of SmFe10CoAl and SmFe9Co2Al compounds obtained from our total energy minimization in Fig. 5a and b, respectively.

Optimized atomic structures of a SmFe10CoAl and b SmFe9Co2Al. Blue and orange spheres represent the Co and Al substitutional atoms, respectively. Other atomic symbols are the same as those used in Fig. 1. c Formation enthalpy \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\), d saturation magnetization μ0Ms, and e uniaxial magnetocrystalline anisotropy Ku of SmFe10CoM and SmFe9Co2M for M = Ti, V, Ga, and Al. Horizontal dashed lines correspond to the calculated results of SmFe11Ti for reference. f Atom-by-atom contribution to magnetocrystalline anisotropy energy EMCA and g orbital magnetic anisotropy ΔμL of SmFe12 (denoted as 1:12), SmFe10Co2 (1:10:2), SmFe9Co2Al (1:9:2:1), and SmFe10CoAl (1:10:1:1).
Figures 5c, d and e show the \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\), μ0Ms, and Ku values, respectively, of SmFe10CoM and SmFe9Co2M for M = Ti, V, Ga, and Al. Here, the formation enthalpy \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) is calculated against Fe3Co + Fe3M + FeCo + FeM decomposition. All the quaternary phases considered here, except for M = V, are stable as their \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\) values are <0.2 kJ mol−1 for SmFe11Ti. The obtained μ0Ms ranges from 1.3 T (M = Ti and V) to 1.45 T (M = Ga and Al), which are sufficient values for practical permanent magnet applications. In our further prediction, as shown in Fig. 5e, the main trend of the Ku of SmFe10M2 (Fig. 3c) is roughly preserved for both SmFe10CoM and SmFe9Co2M; Ku reaches the largest values of 12.5 MJ m−3 for SmFe9Co2Al and 14.1 MJ m−3 for SmFe10CoAl. We emphasize that the latter value is the highest Ku achieved in the present study, which is 1.3−4.3 MJ m−3 larger than those of SmFe12 (12.8 MJ m–3) and SmFe11Ti (9.8 MJ m−3). More remarkably, these Ku values (12.5−14.1 MJ m−3) are even superior to those obtained in the present (5.4 MJ m−3) and previous theoretical (4.31 MJ m−3)31 and experimental studies (4.5 MJ m−3)27 for the state-of-the-art permanent magnet Nd2Fe14B.
Microscopic origin of magnetic anisotropy
The microscopic origin of MCA is the SOC interaction between the atomic orbital angular momentum (L) and the atomic spin angular momentum (S), given by HSOC = λL ⋅ S, where λ is the atomic SOC parameter. We provide the atom-by-atom contributions to MCA energy (EMCA) for the selected SmFe12, SmFe10Co2, SmFe10CoAl and SmFe9Co2Al compounds in Fig. 5f. Here, EMCA is scaled down to the microscopic atomic level (meV atom−1), rather than the macroscopic energy density (MJ m−3). For SmFe12 and SmFe10Co2, Sm dominates EMCA, whereas the contributions from the Fe and Co 3d orbitals are 2 orders of magnitude smaller. The two equivalent Sm sites in the unit cells of SmFe12 and SmFe10Co2 are no longer symmetrically equivalent in the quaternary compounds due to the presence of the 8j-site substitutional atoms. In the optimized structure, the 2 Sm atoms are also not equally separated from the Al(8j) sites. As described in Fig. 5a, the Sm1 − Al1 (Sm1 − Al2) and Sm2 − Al1 (Sm2 − Al2) separations are 3.08 (4.79) and 4.88 (3.11) Å, respectively. We find that the Sm1 site predominantly contributes to the large EMCA and that Sm2 undermines EMCA. The same trend holds for the orbital magnetic anisotropy (ΔμL) in Fig. 5g, defined as \({{\Delta }}{\mu }_{{{{\rm{L}}}}}={({\mu }_{{{{\rm{L}}}}})}_{c}-{({\mu }_{{{{\rm{L}}}}})}_{a}\), consistent with the Bruno theory42; ΔμL is larger by 1 − 2 orders of magnitude in the Sm 4f orbitals than in the 3d orbitals. The contribution from the Al 3p orbitals to EMCA (ΔμL) is essentially zero.
Thermodynamic phase stability
The Helmholtz free energy can be written as F(T, V) = E0(V) + Fel(T, V) + Fvib(T, V) + Fmag(T, V), where E0(V) is the zero-temperature total energy of the system, and Fel(T, V), Fvib(T, V), and Fmag(T, V) are the electronic, vibrational, and magnon contributions of the free energy, respectively. The phase stability against phase decomposition into the most competitive decomposable compounds can be described thermodynamically by the change in the free energy43,44: ΔF(T, V) = F(T, V) − ∑iNiFi(T, V), where Ni and Fi(T, V) are the number and free energy of decomposable compounds i, respectively. The temperature-dependent free energy contributions of Fel(T, V), Fvib(T, V), and Fmag(T, V) are shown in Fig. 6a for SmFe12, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. In Fig. 6a, the same for SmFe11Ti is also shown as a reference. All systems exhibit similar trends in the free energy against temperature; Fel(T, V) and Fvib(T, V) decrease as temperature increases, whereas Fmag(T, V) increases with temperature. Obviously, the former two contributions, particularly the vibrational, mainly determine the temperature-induced changes in the free energy.

Temperature-dependent a free energy contributions of the electronic Fel(T, V), vibrational Fvib(T, V), and magnon Fmag(T, V), and b free energy change ΔF(T, V) of SmFe12, SmFe11Ti, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. In b the contributions of the electronic ΔFel(T, V), vibrational ΔFvib(T, V), and magnon ΔFmag(T, V) to ΔF(T, V) are also presented for each compound. Open symbols at 0 K represent the formation enthalpy \({H}_{{{{\rm{f}}}}}^{{{{\rm{bin}}}}}\).
Figure 6b shows the temperature-dependent free energy change ΔF(T, V) and its contributions, i.e., ΔFel(T, V), ΔFvib(T, V), and ΔFmag(T, V), for SmFe12, SmFe11Ti, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. As a generic for all compounds, ΔF(T, V) decreases as temperature increases, which is almost entirely accounted for by the vibrational contribution. The other two contributions, ΔFel(T, V) and ΔFmag(T, V), are insignificant. For SmFe12, ΔF(T, V) changes its sign from positive to negative at temperature around 1000 K. Similar results were reported in previous calculations although the sign change in ΔF(T, V) occurs much earlier around 200 K34. The main cause for this discrepancy is the choice of the exchange-correlation in the computation, as mentioned early; DFT + U in the present study and DFT in the previous study34. The Ti and Co substitutional atoms reduce the onset temperature significantly; around 200 K for SmFe11Ti and 500 K for SmFe10Co2. This indicates that SmFe11Ti is thermally stable as observed in the experiments15,16,17. For SmFe10CoAl and SmFe9Co2Al, ΔF(T, V) remains negative over the temperature range, which suggests an acceptable thermodynamic stability of Sm(Fe,Co,Al)12 magnets under realistic conditions.
Exchange interaction
The exchange interaction in the Heisenberg model is calculated to determine the intrinsic hard magnetic properties at an elevated temperature. When an external magnetic field is not applied, the Heisenberg spin Hamiltonian is expressed as \(H=-(1/2){\sum }_{i\ne j}{J}_{ij}{{{{\bf{S}}}}}_{i}\cdot {{{{\bf{S}}}}}_{j}-{K}_{{{{\rm{u}}}}}{\sum }_{i}{({{{{\bf{S}}}}}_{i}\cdot {{{\bf{e}}}})}^{2}\), where Jij is the exchange coupling interaction between two spins Si (at the i site) and Sj (at the j site), and e is the unit vector along the magnetic easy axis. The exchange interaction parameters were estimated by the constrained local moment approach in the DFT calculations through Jij = (Δij − Δi − Δj)/4nizijSiSj45,46, where Δij is the energy difference between the magnetic ground state and the excited state (i.e., the magnetic moments at sites i and j are inverted). Δi (Δj) refers to the inverted magnetic moment at the i (j) site. ni is the number of atoms in sublattice i, and zij is the number of nearest neighbor sites in sublattice j relative to sublattice i. The exchange coupling integral Jex is the sum of Jij, i.e., Jex = ∑i,jzijJij. In this approach, the spin in the ith sublattice is assumed to interact with the jth neighbor only while the other possible intrasublattice exchange interactions are disregarded. The present results of the exchange interaction parameters in the analyses below have to be thus double checked with a more precise state-of-the-art approach.
Figure 7a shows the calculated Jij values for the different atomic couplings in SmFe12, SmFe11Ti, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. The positive (negative) exchange parameter reflects the preference for spin parallel (antiparallel) coupling between the two magnetic moments. To inspect the validity of our results, we first calculated the exchange interactions of the prototypical ferromagnetic systems, including α-Fe, α″-Fe16N247, and B2-FeCo46. For α-Fe, the nearest- and next-nearest-neighbor Fe−Fe interactions are 16.8 and 7.6 meV, respectively. These values are in good agreement with previous calculations48,49. The third-nearest-neighbor interactions are found to be rather small, although Jij is a long-range interaction. As shown in Fig. 7a, the Fe−Fe exchange parameters are more or less retained in SmFe12 and SmFe11Ti, while the Sm−Fe interactions prefer antiparallel coupling. The Fe(8i)−Fe(8i) interaction is the strongest among the Fe−Fe interactions since the Fe(8i) atom has the largest spin magnetic moment compared to the other two sites (Table 2). In the Sm−Fe−Co and Sm−Fe−Co−Al systems, the Co substitute enhances all the first-nearest-neighbor exchange parameters, which indeed occurs quite often in Fe−Co magnetic systems46,49. The second-nearest-neighbor Fe−Fe and Sm−Fe interactions are not much altered from those of SmFe12, and the Fe−Al interactions are negligible (thus not shown).

a Exchange interaction parameters Jij as a function of the interatomic distance, b saturation magnetization μ0Ms versus temperature, and c uniaxial magnetocrystalline anisotropy Ku versus temperature of SmFe12, SmFe11Ti, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. In a the nearest- and next-nearest-neighbor exchange interactions of the α-Fe structure are shown in the open circles. Open symbols in b and c denote the experimental data for SmFe12, SmFe11Ti, and SmFe9.6Co2.4, taken from refs. 15–17,22,23.
Temperature-dependent intrinsic magnetic properties
We plot the temperature-dependent μ0Ms and Ku obtained from the constrained MC simulation in Fig. 7b and c, respectively. In the MC simulation, we adopted an 8.47 nm × 8.47 nm × 4.64 nm cell with 26,000 atoms under periodic boundary conditions. One thousand equilibrations, a critical damping of 0.1, and a time step of 1 fs were imposed. The open symbols in Fig. 7b and c denote the available experimental data for SmFe12, SmFe11Ti, and SmFe9.6Co2.415,16,17,22,23. For both SmFe12 and Sm(Fe,Co)12, the present theory reproduces the experimental trends of μ0Ms and Ku versus temperature, even though small discrepancies in the absolute values are detected. The main possible causes of such small discrepancies have already been addressed in previous paragraphs. Moreover, the Ku values from the present theory become larger in SmFe10Co2 than in SmFe12 at T > 200 K, which occurs at a slightly higher temperature of ~300 K in the experiment22,23. In our prediction, the room-temperature Ku values are 7.8 MJ m−3 for SmFe10CoAl and 8 MJ m−3 for SmFe9Co2Al (Fig. 7c).
Table 3 summarizes the present theoretical Curie temperature Tc, anisotropy field μ0Ha( = 2Ku/Ms), and hardness parameter \(\kappa (={({K}_{{{{\rm{u}}}}}/{\mu }_{0}{M}_{{{{\rm{s}}}}}^{2})}^{1/2})\) at 0, 300, and 500 K in comparison with the available experimental data. We predict Tc to be about 554, 568, 885, 665, and 764 K for SmFe12, SmFe11Ti, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al, respectively. The fairly good consistency of the predicted values of SmFe12 (554 K), SmFe11Ti (568 K), and SmFe10Co2 (885 K) with the experimental values (555, 597, and 859 K)22,50,51 conforms the reliability of our calculations. Notably, the Tc (665−764 K) of Sm(Fe,Co,Al)12 fulfils the basic requirement of high-performance permanent magnets (i.e., Tc ≥ 550 K), as suggested in ref. 9. For μ0Ha and κ, overall the agreement between the present theory and the experiment22 is satisfactory. In order to further justify a potential replacement of Sm(Fe,M)12-based magnets for the currently best permanent magnet Nd2Fe14B, we also compare our results for the present Sm(Fe,M)12 systems to the results reported by the previous theoretical and experimental studies for Nd2Fe14B in Table 3. The obtained results of Tc and μ0Ha as well as κ of the present quaternary Sm(Fe,Co,Al)12 systems are superior to the previously reported theoretical (602 K and 2.4−7 T)30,31 and experimental values (585 K, 4.5−8.8 T, and 1.5)25,26,27 of Nd2Fe14B in the entire temperature range up to 500 K.
Magnetic grain size
The magnetic grain size plays an important role in practice to maximally utilize the intrinsic magnetic properties of a permanent magnet. A permanent magnet exhibits maximal coercivity when the magnet domain grains reach the SD size. Here we estimate the minimal and maximal limits of the stable SD size. The SD size ranges from the minimal stable particle diameter \({D}_{{{{\rm{sp}}}}}={(60{k}_{{{{\rm{B}}}}}T/{K}_{{{{\rm{u}}}}})}^{1/3}\) to the domain threshold diameter \({D}_{{{{\rm{sd}}}}}=72{({A}_{{{{\rm{ex}}}}}{K}_{{{{\rm{u}}}}})}^{1/2}/{\mu }_{0}{M}_{{{{\rm{s}}}}}^{2}\) (Aex is the exchange stiffness constant and \(={J}_{{{{\rm{ex}}}}}\left\langle {S}^{2}\right\rangle /{V}^{1/3}\))52,53. If grain sizes are smaller than Dsp, then the particle behaves like a superparamagnet, and if grain sizes are larger than Dsd, then the grains are energetically favorable for splitting into multiple domains. The estimated temperature-dependent Dsp and Dsd are reported in Fig. 8a and b, respectively. For all compounds, both Dsp and Dsd increase with increasing temperature up to Tc. The increase is primarily a reflection of the reduced μ0Ms and Ku with temperature, as discussed regarding Fig. 7. For SmFe12, we find Dsp = 3.5 nm and Dsd = 48.6 nm at room temperature. These values at room temperature do not change much upon Co substitution, in reasonable agreement with the experimental SD size of 51 nm in SmFe8.8Co2.2Ti29. For SmFe10CoAl and SmFe9Co2Al, Dsp = 3 and 4.5 nm and Dsd = 90.5 and 86.3 nm at room temperature, respectively. We note that their stable SD regimes are approximately two times wider than those of the other two compounds (SmFe12 and SmFe10Co2) in the entire temperature range up to 500 K.

a Minimal stable grain size Dsp and b single-domain threshold size Dsd versus temperature of SmFe12, SmFe10Co2, SmFe10CoAl, and SmFe9Co2Al. Vertical dotted lines denote the Curie temperatures Tc. Open circle in b indicates an experimental single-domain size (51 nm) of SmFe8.8Co2.2Ti at room temperature, taken from ref. 29.
Discussion
Using systematic DFT, DFPT, and MC simulations, we reveal that the substitutes of simple metal atoms (Ga and Al) for the Fe sites, rather than the conventional TM substitutional elements, stabilize the ThMn12-type Sm(Fe,Co)12 structure with desirable intrinsic magnetism. In particular, the optimal intrinsic magnetic properties of the quaternary Sm(Fe,Co,Al)12 compounds, including the highest Ku of 8 MJ m−3, μ0Ha of 18.2 T, κ of 2.8 at room temperature and a Tc of 764 K, are predicted to be superior to those of the widely investigated SmFe11Ti and Sm(Fe,Co)11Ti magnets, and even to the present (Ku = 5.4 MJ m−3) and previously reported theoretical (4.31 MJ m−3, 2.4 − 7.0 T, and 602 K)30,31 and experimental results (4.5 MJ m−3, 8.8 T, and 1.5 at room temperature and 585 K)25,26,27 and for the state-of-the-art magnet Nd2Fe14B. We further predict that the Ga and Al substitutional atoms also significantly improve the SD size of the Sm(Fe,Co)12 grains. We hope that the present prediction may resolve the major problem of the structural and thermal instabilities of the ThMn12 structure, leading to practical realization of SmFe12-based high-performance permanent magnets.
Methods
We adopted the WIEN2k package54 with the generalized gradient approximation (GGA) for the exchange-correlation functional55. This method accurately deals with both the core and valence electrons and is suitable for f-electron magnetic systems. Herein, Ku is defined as Ku = (Ea − Ec)/volume, where Ea and Ec are the total energies with magnetization along the a and c directions, respectively. A total of 1271 k points (or a 11 × 11 × 21 k-point mesh) were used in the irreducible Brillouin zone56. The convergence of Ku with respect to the number of k points was seriously checked. The atom-by-atom contribution to EMCA was obtained by switching on/off the SOC of individual atom types in the WIEN2k calculations. The lattice and ionic coordinate relaxations were performed by the Vienna ab initio simulation package (VASP)57 version 5.4.4. 4s13d7, 4s13d8, and 6s25p64f6 are treated as valence electrons for Fe, Co, and Sm, respectively. The strongly correlated 4f electrons were treated with the Hubbard model in the DFT + U scheme. The effective onsite Ueff parameter (U − J) was chosen as 6 eV, which is sufficient to split the f-orbital bands into lower and upper Hubbard bands24. An energy cutoff of 600 eV, a 9 × 9 × 17 k-point mesh, and a high force convergence criterion of 10−3 eV Å−1 were imposed for the structure optimization. The phonon dispersion and thermodynamic properties were carried out by using the PHONOPY code58 within the harmonic approximation in the DFPT59 implemented in the VASP. For the phonon calculations, we employed a 2 × 2 × 1 supercell and a 3 × 3 × 3 k-point mesh in the interpolation of the force constants matrices. Numerical calculations of temperature-dependent intrinsic magnetic properties and Tc were carried out using MC simulation in the VAMPIRE software package60,61.
Data availability
The data that support the findings of this study are available upon reasonable request to the corresponding authors.
References
Ohashi, K., Tawara, Y., Osugi, R. & Shimao, M. Magnetic properties of Fe-rich rare-earth intermetallic compounds with a ThMn12 structure. J. Appl. Phys. 64, 5714 (1988).
Cheng, S. F. et al. Magnetic and structural properties of SmTiFe11−xCox alloys. J. Magn. Magn. Mater. 75, 330 (1988).
Yang, Y.-C. et al. Magnetic and crystallographic properties of novel Fe-rich rare-earth nitrides of the type RTiFe11N1−δ (invited). J. Appl. Phys. 70, 6001 (1991).
Coehoorn, R. Electronic structure and magnetism of transition-metal-stabilized YFe12−xMx intermetallic compounds. Phys. Rev. B 41, 11790 (1990).
Buschow, K. H. J. Handbook of Magnetic Materials Vol. 6 (Elsevier, 1991).
Cadieu, F. J., Hegde, H., Navarathna, A., Rani, R. & Chen, K. High-energy product ThMn12 Sm-Fe-T and Sm-Fe permanent magnets synthesized as oriented sputtered films. Appl. Phys. Lett. 59, 875 (1991).
Buschow, K. H. J. Permanent magnet materials based on tetragonal rare earth compounds of the type RFe12−xMx. J. Magn. Magn. Mater. 100, 79 (1991).
Kou, X. C. et al. Magnetic phase transitions, magnetocrystalline anisotropy, and crystal-field interactions in the RFe11Ti series (where R=Y, Pr, Nd, Sm, Gd, Tb, Dy, Ho, Er, or Tm). Phys. Rev. B 47, 3231 (1993).
Coey, J. M. D. Permanent magnets: plugging the gap. Scr. Mater. 67, 524 (2012).
Skomski, R. & Coey, J. M. D. Magnetic anisotropy − How much is enough for a permanent magnet? Scr. Mater. 112, 3 (2016).
Takahashi, Y., Sepehri-Amin, H. & Ohkubo, T. Recent advances in SmFe12-based permanent magnets. Sci. Technol. Adv. Mater. 22, 449 (2021).
Hirayama, Y., Takahashi, Y. K., Hirosawa, S. & Hono, K. NdFe12Nx hard-magnetic compound with high magnetization and anisotropy field. Scr. Mater. 95, 70 (2015).
Sato, T., Ohsuna, T., Yano, M., Kato, A. & Kaneko, Y. Permanent magnetic properties of NdFe12Nx sputtered films epitaxially grown on V buffer layer. J. Appl. Phys. 122, 053903 (2017).
Nan-xian, C., Shi-qiang, H., Yu, W. & Jiang, S. Phase stability and site preference of Sm(Fe,Ti)12. J. Magn. Magn. Mater. 233, 169 (2001).
Hu, B.-P., Li, H.-S., Gavigan, J. P. & Coey, J. M. D. Intrinsic magnetic properties of the iron-rich ThMn12-structure alloys R(Fe11Ti); R=Y, Nd, Sm, Gd, Tb, Dy, Ho, Er, Tm and Lu. J. Phys. Condens. Matter 1, 755 (1989).
Bodriakov, V. Y., Ivanova, T. I., Nikitin, S. A. & Tereshina, I. S. Magnetic anisotropy and magnetoelastic properties of SmFe11Ti. J. Alloy. Compd. 259, 265 (1997).
Isnard, O., Guillot, M., Miraglia, S. & Fruchart, D. High field magnetization measurements of SmFe11Ti and SmFe11TiH1−δ. J. Appl. Phys. 79, 5542 (1996).
Harashima, Y., Terakura, K., Kino, H., Ishibashi, S. & Miyake, T. First-principles study of structural and magnetic properties of R(Fe,Ti)12 and R(Fe,Ti)12N (R=Nd,Sm,Y). Proc. Comput. Sci. Workshop 5, 011021 (2015).
Harashima, Y., Terakura, K., Kino, H., Ishibashi, S. & Miyake, T. First-principles study on stability and magnetism of NdFe11M and NdFe11MN for M=Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn. J. Appl. Phys. 120, 203904 (2016).
De Boer, F. R., Yung-Kai, H., De Mooij, D. B. & Buschow, K. H. J. Magnetic properties of a series of novel ternary intermetallics (RFe10V2). J. Less. Common. Met. 135, 199 (1987).
Schönhöbel, A. M. et al. Intrinsic magnetic properties of SmFe12−xVx alloys with reduced V-concentration. J. Alloy. Compd. 786, 969 (2019).
Hirayama, Y., Takahashi, Y. K., Hirosawa, S. & Hono, K. Intrinsic hard magnetic properties of Sm(Fe1−xCox)12 compound with the ThMn12 structure. Scr. Mater. 138, 62 (2017).
Ogawa, D. et al. Magnetic anisotropy constants of ThMn12-type Sm(Fe1−xCox)12 compounds and their temperature dependence. J. Magn. Magn. Mater. 497, 165965 (2020).
Odkhuu, D., Ochirkhuyag, T. & Hong, S. C. Enhancing energy product and thermal stability of SmFe12 by interstitial doping. Phys. Rev. Appl. 13, 054076 (2020).
Durst, K.-D. & Kronmuller, H. Determination of intrinsic magnetic material parameters of Nd2Fe14B from magnetic measurements of sintered Nd15Fe77B8 magnets. J. Magn. Magn. Mater. 59, 86 (1986).
Yamamoto, H., Matsuura, Y., Fujimura, S. & Sagawa, M. Magnetocrystalline anisotropy of R2Fe14B tetragonal compounds. Appl. Phys. Lett. 45, 1141 (1984).
Herbst, J. F. R2Fe14B materials: Intrinsic properties and technological aspects. Rev. Mod. Phys. 63, 819 (1991).
Sun, H., Otani, Y., Coey, J. M. D., Meekison, C. D. & Jakubovics, J. P. Coercivity and microstructure of melt-spun Sm(Fe11Ti). J. Appl. Phys. 67, 4659 (1990).
Qian, H.-D. et al. Phase transformation and magnetic properties of fully dense Sm(Fe0.8Co0.2)11Ti bulk magnets. Scr. Mater. 193, 17 (2021).
Sepehri-Amin, H., Ohkubo, T. & Hono, K. Micromagnetic simulations of magnetization reversals in Nd-Fe-B based permanent magnets. Mater. Trans. 57, 1221 (2016).
Gong, Q., Yi, M., Evans, R. F. L., Xu, B.-X. & Gutfleisch, O. Calculating temperature-dependent properties of Nd2Fe14B permanent magnets by atomistic spin model simulations. Phys. Rev. B 99, 214409 (2019).
Delange, P., Biermann, S., Miyake, T. & Pourovskii, L. Crystal-field splittings in rare-earth-based hard magnets: an ab initio approach. Phys. Rev. B 96, 155132 (2017).
Dirba, I. et al. Thermal decomposition of ThMn12-type phase and its optimum stabilizing elements in SmFe12-based alloys. J. Alloy. Compd. 813, 152224 (2020).
Saengdeejing, A. & Chen, Y. Improving thermodynamic stability of SmFe12-type permanent magnets from high entropy effect. J. Phase. Equilib. Diffus. 42, 592 (2021).
Jain, A. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Barber, C. B., Dobkin, D. P. & Huhdanpaa, H. The quickhull algorithm for convex hulls. ACM Trans. Math. Softw. 22, 469 (1996).
Akbarzadeh, A. R., Ozolins, V. & Wolverton, C. First-principles determination of multicomponent hydride phase diagrams: application to the Li-Mg-N-H system. Adv. Mater. 19, 3233 (2007).
Emery, A. A. & Wolverton, C. High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites. Sci. Data 4, 170153 (2017).
Suski, W. Handbook on the Physics and Chemistry of Rare Earth Vol. 22 (North-Holland Physics Publishing, 1996).
Miyake, T. & Akai, H. Quantum theory of rare-earth magnets. J. Phys. Soc. Jpn. 87, 041009 (2018).
Bronova, A., Bredow, T., Glaum, R., Riley, M. J. & Urland, W. BonnMag: Computer program for ligand-field analysis of fn systems within the angular overlap model. J. Comp. Chem. 39, 176 (2018).
Bruno, P. Tight-binding approach to the orbital magnetic moment and magnetocrystalline anisotropy of transition-metal monolayers. Phys. Rev. B 39, 865(R) (1989).
Sözen, H. I. et al. Ab initio phase stabilities of Ce-based hard magnetic materials and comparison with experimental phase diagrams. Phys. Rev. Mater. 3, 084407 (2019).
Mediukh, N. R., Ivashchenko, V. I., Pogrebnjak, D. A. & Shevchenko, V. I. First-principles study of thermodynamic and stability properties of TiC-SiC alloys. Proc. NAP 1, 01NTF4 (2019).
Novak, P. & Rusz, J. Exchange interactions in barium hexaferrite. Phys. Rev. B 71, 184433 (2005).
Odkhuu, D. & Hong, S. C. First-principles prediction of possible rare-earth free permanent magnet of tetragonal FeCo with enhanced magnetic anisotropy and energy product through interstitial nitrogen. Phys. Rev. Appl. 11, 054085 (2019).
Odkhuu, D. & Hong, S. C. Simultaneous tuning of the magnetic anisotropy and thermal stability of α″-phase Fe16N2. Sci. Rep. 11, 7823 (2021).
Schilfgaarde, M. V. & Antropov, V. P. First-principles exchange interactions in Fe, Ni, and Co. J. Appl. Phys. 85, 4827 (1999).
Lezaic, M., Mavropoulos, P. & Blügel, S. First-principles prediction of high Curie temperature for ferromagnetic bcc-Co and bcc-FeCo alloys and its relevance to tunneling magnetoresistance. Appl. Phys. Lett. 90, 082504 (2007).
Wang, D. et al. SmFe12 and SmFe12Nx films fabricated by sputtering. J. Magn. Magn. Mater. 124, 62 (1993).
Sun, H., Tomida, T. & Hirosawa, S. Magnetic properties and microstructure studies of Sm-Fe magnetic thin films. J. Appl. Phys. 81, 328 (1997).
Bruck, E. Handbook of Magnetic Materials Vol. 1, (Elsevier, 2018).
Charap, S. H., Lu, P. L. & He, Y. Thermal stability of recorded information at high densities. IEEE Trans. Magn. 33, 978 (1997).
Blaha, P., Schwarz, K., Madsen, G. K. H., Kvasnicka, D. & Luitz, J. WIEN2k, An augmented plane wave + Local orbitals program for calculating crystal properties. (Technische Universitat Wien, 2001).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671 (1992).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558(R) (1993).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1 (2015).
Giannozzi, P., Gironcoli, S., Pavone, P. & Baroni, S. Ab initio calculation of phonon dispersions in semiconductors. Phys. Rev. B 43, 7231 (1991).
Asselin, P. et al. Constrained Monte Carlo method and calculation of the temperature dependence of magnetic anisotropy. Phys. Rev. B 82, 054415 (2010).
Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H. & Teller, E. Equations of state calculations by fast computing machines. J. Chem. Phys. 21, 1087 (1953).
Acknowledgements
This work is supported by the Future Materials Discovery Program (Grant No. 2016M3D1A1027831) and the Basic Research Program (Grant No. 2020R1F1A1067589) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT and the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (Grant No. 20192010106850, Development of magnetic materials for IE4 class motor).
Author information
Authors and Affiliations
Contributions
S.C.H. and D.O. conceived the project and wrote the paper. T.O. performed the calculations. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ochirkhuyag, T., Hong, S.C. & Odkhuu, D. Intrinsic hard magnetism and thermal stability of a ThMn12-type permanent magnet. npj Comput Mater 8, 193 (2022). https://doi.org/10.1038/s41524-022-00821-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00821-8
