
Abstract
Machine learning plays an increasingly important role in many areas of chemistry and materials science, being used to predict materials properties, accelerate simulations, design new structures, and predict synthesis routes of new materials. Graph neural networks (GNNs) are one of the fastest growing classes of machine learning models. They are of particular relevance for chemistry and materials science, as they directly work on a graph or structural representation of molecules and materials and therefore have full access to all relevant information required to characterize materials. In this Review, we provide an overview of the basic principles of GNNs, widely used datasets, and state-of-the-art architectures, followed by a discussion of a wide range of recent applications of GNNs in chemistry and materials science, and concluding with a road-map for the further development and application of GNNs.
Similar content being viewed by others
Introduction
Data science and machine learning have become an integral part of natural sciences, discussed as the fourth pillar in science, next to experiment, theory, and simulation1. Machine learning methods are increasingly applied in all steps of the materials development cycle, from finding initial candidate materials using property prediction2,3, database screening4,5 or even inverse materials design6,7, over the detailed analysis of materials in machine learning accelerated simulations8,9, to the prediction of synthesis conditions10,11 and automated experimental data analysis12,13 and experimental planning14. Machine learning models applied in chemistry and materials science cover a wide spectrum of methods, ranging from classical machine learning models such as decision tree ensembles to modern deep learning methods such as convolutional neural networks15 and sequence models16 originally developed for challenges in computer vision and natural language processing.
A recent addition to the toolbox of machine learning models for chemistry and materials science are graph neural networks (GNNs), which operate on graph-structured data and have strong ties to the field of geometric deep learning17,18,19. Aside from research on social and citation networks as well as knowledge graphs, chemistry has been one of the main drivers in the development of GNNs20,21. Graph neural networks can be interpreted as the generalization of convolutional neural networks to irregular-shaped graph structures. While other machine learning methods, e.g., convolutional neural networks are at the peak of publication activity, GNNs are still rising exponentially, with hundreds of papers per year since 2019. Their architecture allows them to directly work on natural input representations of molecules and materials, which are chemical graphs of atoms and bonds, or even 3D structures or point clouds of atoms. Therefore, GNNs have access to a complete representation of materials on the atomic level22, with a lot of flexibility to incorporate physical laws23, as well as phenomena on larger scales, such as doping and disorder. Using that information, GNNs can learn internal materials representations that are useful and informative for specific tasks such as the prediction of given materials’ properties. Therefore, GNNs can complement or even replace hand-crafted feature representations which were and are widely used in the context of natural sciences in general. A similar trend toward representation learning methods has also been observed in other application areas during the last years, where end-to-end trainable models show a systematic advantage over traditional feature-based methods24. However, despite promising recent developments toward higher sample efficiency25,26, this often comes at the cost of higher data requirements27, potentially limiting the applicability of existing GNNs to applications where large amounts of data are available. Overall, GNNs outperformed conventional machine learning models in predicting molecular properties throughout the last years22,28,29. While GNNs are not as widely applied (yet) in materials science as they are in chemistry, there are advantages and the potential to outperform other machine learning methods and thus boost virtual materials design and materials science in general, which will be discussed in this article.
In section ‘Graph neural networks in materials science and chemistry’, we will introduce the general formalism of GNNs and discuss the way they transform the atomic structure of materials and molecules and use it to predict materials’ properties. We will present and compare state-of-the-art architectures and benchmark datasets, as well as summarize initial efforts toward inverse materials design based on GNNs. The section ‘Applications’ covers a wide range of current application areas but also open challenges for GNNs in chemistry and materials science. In section ‘Outlook’ we conclude with a perspective on necessary and expected future developments and so far unused potential of GNNs in materials science.
Graph neural networks in materials science and chemistry
Basic principles
In the most general sense, graphs are used to describe abstract structures consisting of entities or objects represented as vertices (or nodes) and their connections, called edges. Formally, a graph is a tuple G = (V, E) of a set of vertices v ∈ V and a set of edges ev,w = (v, w) ∈ E, which defines the connection between vertices. Potential tasks that can be solved using graph neural networks (GNNs) include classification or regression of graph properties on graph level (molecular property prediction), node level (classification of members, i.e., nodes, of a social graph), or edge level (prediction of relations, i.e., edges, between objects in a scene graph). In materials science and chemistry, most tasks involve graph-level predictions, which will be the focus of this paper.
The concept of graphs is used in mathematical chemistry to represent the structure of compounds. The molecular structure is represented by an undirected graph, where nodes correspond to atoms and edges correspond to chemical bonds. In fact, chemical graphs were first considered as early as in 187430 and their idea traces back further31, which may place them even before the advent of the term graph in modern graph theory32. The description of molecules as graphs can also be transferred to solid-state materials, even though bonds might not be uniquely defined in crystals, and the exact three-dimensional arrangement of atoms plays a more decisive role.
Since their proposal17,18,19, GNNs have become a popular machine learning method for processing irregularly shaped data encoded as graphs. They can be seen as an alternative to approaches, where predefined feature representations of molecules or materials are used as input to conventional machine learning models such as densely connected neural networks, random forest models, or Gaussian process regression models. In the case of GNNs, the full molecular structure or even geometry is used as input and the GNN itself learns informative molecular representations to predict given target properties. Due to their popularity and wide applicability, a large number of different GNN architectures have been proposed18,20,21,33,34,35. While the exact architecture type can notably differ, ranging from the initially proposed recursive GNNs18 to spectral neural filters35,36 and finally to spatial or convolutional GNNs34, most GNNs designed for chemistry and materials science can be summarized under the framework of Message Passing Graph Neural Networks (MPNN) as suggested by Gilmer et al.21. In this section, we give an overview of ideas of the message passing framework and discuss how learned graph- or node-level embeddings can be used for materials property prediction.
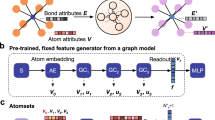
For MPNNs, associated node or edge information (e.g., atom and bond types) is commonly provided by node attributes \({h}_{v}^{0}\in {{\mathbb{R}}}^{d}\) and edge attributes \({h}_{e}^{0}\in {{\mathbb{R}}}^{c}\). Details about feature and structure representations are discussed in Section ‘Structure representation’. Using node and edge features in combination with the graph’s structure, GNNs are capable of deriving a node-level embedding of the graph, i.e., learned vectors representing each atom including its individual chemical environment. This is done in the so-called message passing phase, in which node information is propagated in form of messages mv through edges to neighboring nodes. The embedding of each node is then updated based on all incoming messages. The locality of the message passing is sought to be alleviated by repeating the message passing phase t = 1…K times, in principle allowing information to travel longer distances, i.e., within the K-hop neighborhood. In practice, however, information from long-range dependencies can be distorted in node bottlenecks, referred to as over-squashing37, or be washed out, leaving indistinguishable representations of neighboring nodes, known as over-smoothing38. Note that for typical (not fully linear) molecules and crystal unit cells with n atoms, only approximately \(\log n\) message passing steps are required to pass information to all other atoms. The information processing is facilitated by the learnable functions Ut(⋅) for node update and Mt(⋅) for the message generation. Finally, in the readout phase, a graph-level embedding y is obtained by pooling node embeddings of the entire graph via a parametric readout function R(⋅). The final representation of the graph is used for training both regression and classification tasks. In summary, the MPNN scheme reads21:
where N(v) = {u ∈ V∣(v, u) ∈ E} denotes the set of neighbors of node v. Note that readout and aggregation can be in principle any mathematical operation that is permutation invariant, e.g., a sum, mean or maximum operation similar to Eq. (1) or learnable such as the Set2Set encoder proposed by Vinyals et al.39, which was originally used for the readout R(⋅). The learnable functions are mostly neural networks and eventually determine the performance characteristics of the GNN, both in prediction accuracy and computational cost. Figure 1a shows a schematic of the message passing scheme for the example of a molecular graph. Message passing can also be understood as a convolution operation running over each node in a graph. Different extensions and modifications of the message passing schemes are discussed in Section ‘State-of-the-art architectures and benchmarks’ and include edge updates40, skip connections41, and geometric information26,42,43.

a Schematic depiction of the message passing operation for molecules and crystalline materials. b QM9 benchmark. Mean absolute error of the prediction of internal (red circles), highest occupied molecular orbital (HOMO, orange triangles), and lowest unoccupied molecular orbital (LUMO, inverted blue triangles) energies for different GNN models since 2017.
A main open research question of GNNs revolves around their limited expressive performance for specific tasks44 and how GNNs compare with Weisfeiler–Lehman hierarchy for graph isomorphism testing45,46. With regard to this topic, there are many promising extensions to GNNs proposed in literature, such as hypergraph representations47,48, universal equivariant models49 or higher-order graph networks50. Furthermore, the challenge of over-smoothing due to commonly used aggregation functions38, transfer and multitask learning51, as well as training (in)stability52 are subject of current research.
Structure representation
Many graph networks directly use the chemical graph as input, representing both molecules21 and inorganic compounds24,53, and offering advantages over compositional or fixed-sized vector representations in terms of flexibility and scalability. Consequently, GNNs can be applied for tasks such as drug design or material screening54, which require knowledge about functional groups, scaffolds55 or the full chemical structure and its topology. In molecular applications, the chemical graph is often extracted from SMILES codes and augmented with features that can be readily obtained from cheminformatics software such as RDKit56 or OpenBabel57. Common features for atoms, bonds, and the molecule itself are listed in Table 1. Besides hand-crafted input features, learned embeddings of molecules and materials motivated by word embedding techniques in natural language processing have been explored which can be used for downstream tasks48,58,59,60. For specific tasks in chemistry, the connectivity of atoms in molecules (i.e., the molecular graph) contains sufficient and complete information to predict given molecular properties which do not depend on the exact geometry. Geometry or stereochemical information can be taken into account e.g., in form of additional edge features representing the distance between atoms61. In contrast to that, in materials applications, atom connectivity is not well defined in most cases (apart from e.g., covalently linked frameworks) and graphs have to be extracted from crystal structures based on distance heuristics.
The sole chemical graph and its connectivity are often not sufficient to accurately predict quantum-mechanical or electronic-structure properties62 that strongly depend on the exact molecular geometry, even though ground-state or equilibrium geometries can in principle be inferred from the molecular graph alone. In tasks that intrinsically involve geometric dependencies, e.g., predicting the potential energy surface of molecules and materials, it becomes obvious that geometric information is required. The representation of positional and geometric information to learn quantum properties has been explored among others in the work of Lilienfeld et al.63 and Behler et al.64 and lead to a large variety of descriptors. Some examples of descriptors are atomic centered symmetry functions (ACSF)65,66, angular Fourier series (AFS)67, the smooth overlap of atomic orbitals (SOAP)67, partial radial distribution function (PRDF)68, many-body tensor (MBTR)69, Spectral London Axilrod-Teller-Muto (SLATM)70 and the Faber-Christensen-Huang-Lilienfeld (FCHL)71 representation. Many of those descriptors expand geometric information into symmetry or basis functions. The resulting vector representation is typically used as input for conventional machine learning models such as neural networks or Gaussian Processes. Geometric information can also be used for node or edge representations in graph neural networks. Graph networks have been adopting distances61, bond72 and even dihedral angles26,73, motivated by the comparison to force fields74. Angles or distances are similarly expanded into Gaussian-like22, radial23 and spherical Fourier-Bessel functions28. Although architectures such as the Behler-Parinello (BP) neural network potentials8 or SchNet22 are not strictly graph networks in terms of the chemical graph, and often do not refer to themselves as such, they can be summarized within the term geometric deep learning75,76.
Under the term geometric deep learning, architectures and descriptors are summarized that focus on manifolds, geometric data, or structured data77,78. This includes the work on 3D point clouds79, which aims at learning segmentation and object detection of a large number of 3D points. In the case of PointNet++80 a graph is constructed which reduces the point set from learned descriptors using the points’ features. Commonly, adjacency matrices are defined by using distance cutoffs between points in 3D clouds, while edges carry explicit information about distances between nodes, i.e., points. Graph pooling or coarsening algorithms81,82 that reduce the input representation and condense structure information are also promising for GNNs to tackle larger molecules such as proteins or polymers.
Eventually, the representation of materials for graph networks can be structural or geometric but must follow certain symmetry considerations83,84. For example molecules without external fields have rotational and translation symmetries. If they are incorporated into the model and its representation, less data are required and overall performance can be improved. This concept can be extended to equivariant representations85,86, which in combination with equivariant filters, enable equivariant graph neural networks29,87. Scalar features are extended to (directional) features, like vectors and higher-order tensors (in the geometric context of the term tensor), since, embedded in 3D Euclidean space, they can transform predictably under geometric rotation, reflection, and translation25,87.
For solid crystals and periodic structures, the periodicity and space group symmetries are additional symmetries to be added to the representation for GNNs. Periodic extensions of the crystal graph2,53 of the unit cell have been introduced61 and their representation builds on the idea of a k-point mesh of Monkhorst-Pack grid points to sample the Brillouin zone88.
State-of-the-art architectures and benchmarks
Different architectures have been proposed in the literature to improve the performance of GNNs on typical tasks arising in chemistry and materials sciences. Table 2 shows a list of popular benchmark datasets for different materials classes, i.e., molecules62,89,90,91,92,93,94,95,96,97,98,99 or crystals100,101,102, and respective supervised tasks, i.e., regression or classification. While some datasets contain experimental data, the largest datasets typically use computational methods to generate labels. Most datasets in this table can be downloaded from data collections such as TUDatasets103 and MoleculeNet95. While a large variety of datasets and tasks exist for chemistry, there are only a few large datasets for materials, limited to crystalline structures. Recent datasets were constructed by filtering the Materials Project (MP)100 and Open Quantum Materials Database (OQMD)101 for specific targets such as electronic band-gap or formation energy while removing incomplete data88.
In Section ‘Basic principles’, the message passing framework for GNNs has been illustrated. Here, we will discuss modified and extended GNN models, which are relevant for materials science and chemistry. However, listing all graph network architectures would be beyond the scope of this review.
Some of the earliest work on neural networks for molecular graphs dates back to the 90s and 2000s, without explicitly referring to the term graph neural network8,33. In 2017, a graph convolutional network was proposed by Kipf et al.34 for semi-supervised learning, which can be interpreted as a first-order approximation of spectral graph convolutions35,36. In Table 3, we distinguish GNN architectures that operate on the graph’s spectrum in category spectral convolution and GCN models that process the structure of the graph in spatial convolution, which in principle includes message passing schemes that pass information along edges in the graph. We added a soft distinction in this category to separate models that follow the convolution naming and models that explicitly refer to the term message passing and its extension.
The addition of more complicated node aggregation functions such as gated recurrent units104 or long short-term memories105 has been employed by GraphSAGE for inductive learning106. For graph embedding tasks, a state- or super-node21, which is connected to all nodes, extends the message passing framework to help extract global graph information, in addition to the final node aggregation step. A message passing neural network (MPNN) with edge features capturing bond information was applied to molecular graphs21 and crystal graphs53. A variant of the original MPNN involves directed edge embeddings and message passing between edges in D-MPNN40. Known from models in natural language processing107, masked self-attention layers which attend over the node’s neighborhood have been suggested for graph attention networks108 and used explicitly for molecules in Attentive Fingerprint models54. More attention-based architectures are listed in Table 3.
Besides graph models which focus on the chemical graph, there is a large group of models explicitly designed for learning quantum properties. They commonly take atomic numbers and positions as input and train on data derived from (approximate) solutions of the steady-state Schrödinger equation. A popular benchmark dataset is the QM9 dataset62 with 13 quantum properties of small molecules with up to nine atoms apart from hydrogen. The improvement of graph networks on QM9 property prediction over the past few years is highlighted in Fig. 1b. Among the first graph networks that reached chemical accuracy on QM9 is SchNet22, which makes use of convolutional filters for inter-atomic distances and applies skip connections between node updates. One improvement to SchNet was to update the positional features along the graph edges as seen in Jørgensen et al.109. The application of GNNs to crystals using geometric information has been explored by MEGNET61, which further leverages global properties such as temperature, which is of importance for solid-state crystalline systems. The potential energy of molecules depends on bond angles and therefore, in DimeNet28,72, edge embedding uses messages passing steps from atomic triplets and bond pairs in order to incorporate angular features. This formalism has been adopted in other recent GNNs110,111 and can be further extended to include dihedral (or torsion) angles26,112,113.
For explicit angle plus node information in directed edge updates as in DimeNet28,72, the message passing essentially operates on higher order paths73 or k-pairs of atoms50. This is unfeasible for fully connected larger graphs because the number of multi-node interactions that need to be computed is dramatically increasing. To reduce the computational costs, models like MXMNet111 make use of multiplex graphs, which selectively consider only specific edges when going to higher order pathways for calculating bond angles110.
Note that the GNNs mentioned previously are invariant to the translation and rotation of the molecules throughout space. Recently, equivariant GNNs have been proposed29,43,85,114, which transform equivariantly under symmetry operations of its (positional) input, meaning the GNN’s features or its output undergoes the same operations as well. The type of symmetry operations is commonly specified in 3D-space by the corresponding Euclidean group E(3). The special (isometric) subgroup of E(3), which preserves handedness in transformation, is denoted by SE(3). Symmetry operations preserving distance and a fixed point form the orthogonal euclidean group. Analogously, the orthogonal group is denoted by O(3) and its composable special group by SO(3). For example, SO(3) contains rotations, O(3) includes rotations plus reflections, SE(3) allows rotations plus translations, and E(3) encompasses both rotations, translations and reflections. For GNNs also the permutation equivariance with respect to the input set of nodes can be considered and is characterized the by symmetric group Sn. In TFN87 equivariant filters are constructed based on learnable radial functions and spherical harmonics for (geometric) feature vectors of different order, namely scalars (type-0), vectors (type-1) and higher-order tensors, which comprise a direct sum of irreducible representations of the O(3) symmetry group. Equivariant convolution layers can be obtained by tensor-products thereof using Clebsch-Gordan coefficients87. Simplifying to type-1 representations, EGNN85 uses the relative squared distance and updates the position of each particle as a vector field in a radial direction to preserve E(3) equivariance. Equivariant graph models convey directional information between atoms29 without higher-order pathways and enable the prediction of tensorial properties114,115. We have listed popular equivariant GNNs is a separate category in Table 3.
Further adapted message passing steps allow for the determination of the molecular orbitals2,116,117,118. Molecular orbital interactions can in turn be used for improving the prediction performance2. Lastly, the mapping of atoms to non-Euclidean space such as in the proposed hyperbolic GNNs119 can lead to gains in representational efficiency. For a more in-depth discussion of graph variants120 and graph taxonomy121 that goes beyond Table 3, we refer to more general articles about GNNs122,123, e.g., Zhou et al.121 and Wu et al.124.
With regard to the QM9 benchmark in Fig. 1b some models have slightly lower performance for the total energy but can be superior in other QM9 properties or achieve similar results with much less computational effort. Some other factors that complicate a stringent comparison are differences in train-test splits, cleaning steps, e.g., of ill-converged molecules in QM9, multi-task vs. single task settings, where a separate model for each QM9 target is usually trained61, and differences in used loss metrics (a mean absolute error loss was found to yield lower overall test errors23 than the mean squared error loss used in previous models22, although the mean absolute error is typically given as a benchmark reference). It has to be noted that hyperparameters are generally very important and are often not exhaustively optimized for GNNs which can cause differences in performance apart from the model architecture125,126,127.
GNNs in generative models and reinforcement learning
An important challenge in materials science is inverse materials design, aiming to generate new materials or molecules that possess required properties and fulfill specific criteria128. GNN-based generative methods have been suggested to deal with this challenge in the context of chemistry, e.g., for drug discovery129 and retrosynthesis130. In most cases, only the chemical structure of molecules is generated, i.e., the connectivity of the molecular graph, without additional information on specific 3D geometry.
Initial graph generative models were designed to generate graphs based on simplified theoretical assumptions, such as the random Erdös-Renyi (ER) model131, and improvements thereof using small-world approaches132 or the Kronecker graph model133. While these traditional approaches attempt to model real-world graphs, they are based on several assumptions about the nature of graphs generated and are thus inflexible for many data-related applications. Machine learning approaches for graph generation are promising because they can directly learn to generate realistic graphs from the distribution of observed data while accommodating goal-directed tasks such as property optimization. Examples include variational autoencoders (VAEs)134, generative adversarial networks (GANs)135, reinforcement learning136, recurrent neural networks (RNNs)137, and flow-based generative models138,139,140.
Several architectures of VAEs have been developed to work with different types of input data, such as images141, text-based data142,143, or graphs144,145. Kipf et al. introduced a variational graph auto-encoder (VGAE) to learn latent representations of undirected graphs and applied it to a link prediction task in citation networks144. Liu et al. introduced a Constrained Graph Variational Autoencoder (CGVAE), in which node type information is learned from and added to the latent vectors146. Starting from a set of these latent node representations, CGVAE iteratively forms valid molecules following hard valency constraints derived from the explicit node types. Jin et al. introduced Junction Tree Variational Autoencoder (JT-VAE) to work directly on molecular graphs and achieved an improvement over baseline methods in molecular design tasks147. The JT-VAE approach encodes and decodes molecules in two steps: First, tree-structured objects called junction trees are generated which represent trees of molecular subgraphs and their arrangements. GNN-based encoders and decoders are used to generate latent embeddings of the junction trees. In parallel, molecular graph embeddings are generated using GNNs, and junction trees are decoded into molecular representations21. These are then encoded to a latent vector and decoded back to their original representations using graph and tree-based encoders and decoders. While this scheme works well for molecules, it is hard to adapt for crystalline materials, where the graphs are less tree-like and the definition of scaffolds is not as straightforward.
GANs have shown promising results in a number of fields, such as image148 or sequence149 generation, and have also been applied to 3D grid representations of materials150 and graphs151. De Cao et al. introduced MolGAN152 as a framework for generating molecular graphs using GNNs. The generator learns to directly output the graph’s representation. While the standard GAN loss forces the generator to generate molecules following a particular prior distribution, the authors add a reinforcement learning (RL) objective to generate molecules with optimized properties. The generation of invalid molecules is avoided by the assignment of zero reward to them. While direct prediction of outputs is appealing in methods using VAEs or GANs, they usually predict outputs of small, fixed sizes. Therefore, another branch of deep graph generative models employs sequential decision-making procedures to overcome these limitations.
You et al. identify three challenging aspects when graphs are generated directly137. First, as these methods need to model the adjacency matrix in some form, the output size grows as \({{{{{{{\mathcal{O}}}}}}}}({n}^{2})\) for graphs with a maximum number of n nodes. This is especially undesirable for large and sparse graphs as the model dedicates much of its capacity to learn which nodes are not connected. Second, graph isomorphism complicates the calculation of reconstruction losses for VAEs and usually involves expensive graph matching procedures. Third, VAEs assume the outputs, e.g., the entries of an adjacency matrix, to be i.i.d., which is not true in practice. As a solution, the authors propose GraphRNN, a framework in which graphs are represented as sequences of node and edge additions. By using recurrent networks, graph constituents are generated conditioned on previous additions, thus taking into account the history of modifications. Another sequential graph generation scheme was proposed by Li et al.153. In this framework, the generation process is decomposed into modular steps, e.g., whether to add a node or which nodes to connect. Each module is a fully-connected neural network modeling probabilities of executing particular types of modifications.
You et al. suggested a purely RL-based approach based on Graph Convolutional Policy Networks (GCPN)154 (see Fig. 2b). In this setting, the agent explores the chemical space and generates new molecular graphs by modifying the starting molecules according to the reward function, representing the molecular property to optimize. Atance et al. recently introduced a similar RL approach based on Gated GNNs155, outperforming other GNN-based approaches in molecular graph generation tasks156. Another sequential approach based on conditional graph generative models has been used by Li et al. on drug design tasks157. The previous two works inspired recent molecular graph generation frameworks such as GraphINVENT156.

a prediction of ADMET properties (adapted with permission from Feiberg et al.165, Copyright 2020 American Chemical Society), GNNs accounting for environment effects of molecules (reproduced from ref. 179 with permission from the Royal Society of Chemistry.), GNNs to predict the toxicity of molecules for bees (this illustration was published in ref. 184, Copyright Elsevier), b RL-based approach for inverse molecular design based on Graph Convolutional Policy Networks (GCPN) (adapted from ref. 154), c template-free retrosynthesis (adapted from ref. 221), d transferable excited states dynamics (reproduced from ref. 199 with permission from Springer Nature), coarse graining (reproduced from ref. 194 with permission from the Royal Society of Chemistry), e explainable GNNs (adapted from ref. 311), f Crystal GNN to predict methane adsorption volumes in metal organic frameworks (MOFs) (this illustration was published in ref. 234, Copyright Elsevier), doped structures (this illustration was published in ref. 244, Copyright Elsevier), point defects (adapted with permission from Frey et al.245, Copyright 2020 American Chemical Society) g reactions of Al2O3 surface in contact with HF gas (reproduced from ref. 190 with permission from Springer Nature), GNNs to predict magnetostriction of polycrystalline systems (reproduced from ref. 240 with permission from Springer Nature), h a GNN classifier to predict if a system is in a liquid or a glassy phase only by the positions of the atoms (reproduced from ref. 249 with permission from the Royal Society of Chemistry).
More recently, attention has turned to generative modeling based on normalizing flows (NFs)158, capable of modeling complex target distributions by directly mapping them to simpler ones using revertible bijective transformations. NFs potentially offer significant improvements in the field of graph generation because they allow exact evaluation and inference of the probability distribution. This approach has been applied in a number of molecular tasks129,159,160,161.
Overall, graph generative models have been extensively applied for molecular materials and stayed up to date with recent developments in the field of graph generation. However, these remain under-explored for crystalline materials, mainly due to graph representation challenges162. While finding such a reliable graph representation is still an open question128 and will likely remain case-specific, we believe that using generative models based on GNNs is a promising research direction in inverse design, especially given current breakthroughs such as normalizing flows.
Applications
After introducing the basic principles of GNNs as well as selected GNN architectures and benchmark datasets, we will provide a structured overview of GNN applications in chemistry and materials science. GNNs were successfully applied to a rich variety of different challenges, ranging from property prediction of molecules and materials over accelerated atomistic simulations to predicting reactivity and synthesis routes of molecules and materials. While other machine learning models such as densely connected neural networks were successfully applied to these tasks as well, state-of-the-art GNNs in many cases currently outperform other models. However, there exists a range of open challenges, including data requirements and data efficiency, as well as a lack of fully GNN-based generative models for molecular and materials design.
Molecular systems
Some of the first applications of GNNs and probably also one of the main driving forces for the ongoing development of GNN models are challenges in the area of molecular chemistry. Most prevalent is the task of predicting molecular properties, i.e., a regression or classification task which is challenging to solve with conventional machine learning models, as they typically require predefined molecular representations (e.g., molecular feature vectors or molecular fingerprints) which are informative for the label to predict. GNNs have access to the full chemical graph or even molecular geometry and learn to extract feature representations, which yields an advantage over other machine learning models. Compared to domain knowledge-informed feature representations combined with conventional machine learning models, e.g., Gaussian process regression, GNNs often have comparably high data requirements but outperform conventional models when enough data is available. Once trained, accurate machine learning models can then be used to accelerate the high-throughput virtual screening of molecules163,164 to find promising candidate molecules for many different applications. However, property prediction is not the only application of GNNs. They were also successfully applied to provide trainable interatomic potentials to accelerate costly ab initio molecular dynamics simulations, as well as to predict the outcome of chemical reactions and synthetic routes.
Molecular property prediction
Among the most relevant molecular properties in the area of drug discovery are the ADMET (absorption, distribution, metabolism, exclusion, and toxicity) properties of potential drug-like molecules (see Fig. 2a)165,166,167,168. A review on GNNs for drug design can be found in Xiong et al.169. In recent years, one application focus were Covid 19 related challenges, where GNNs were used for e.g., finding new drug candidates170 or detecting infections in medical images171,172. Similar methods are also applicable to other challenges in drug design and medicine.
Furthermore, GNNs were applied to predict electronic and optical properties of molecules. For many applications such as organic electronics, organic photovoltaics, and organic light-emitting diodes, the energy of the highest occupied molecular orbital (HOMO), the lowest unoccupied MO (LUMO), and the optical gap are of high importance for the device efficiency. These properties can therefore be found in numerous databases62,90,173,174,175,176,177. Related properties include (transition) dipole moment62,178, ionization potential, and electron affinity90. In devices, these properties often depend on the molecular environment, which can be modeled and accounted for with GNNs (see Fig. 2a)179. For certain applications such as opto-electronic materials174,175, GNNs have been proposed to complement scalar molecular properties with spectroscopic properties112.
The aforementioned properties are often determined using computationally expensive simulation methods. Cheaper, mostly semi-empirical methods can provide fast but potentially less accurate estimates. GNNs have been used to represent Kohn-Sham wavefunctions and energy levels in a minimal basis representation180, as well as for delta-learning from semi-empirical methods to DFT computed molecular properties177. For applications where not enough data is available to train GNNs, the representations learned by GNNs on large generic datasets can also be transferred to supervised tasks with little data20,181,182, where they are used as input for other machine learning models such as gradient-boosted models183.
Further application areas of GNNs spread across all application domains of molecular materials, including the prediction of toxicity of molecules to bees (see Fig. 2a),184 determining the quality of a material for fuel ignition185 and the classification of different phases of materials, in particular water186. It should be noted that another review paper on molecular property prediction utilizing GNNs exists by Wieder et al.120. In many application areas, tools such as GNNExplainer187 are used to validate and analyze GNN predictions, e.g., in the prediction of scents188 and for porous organic cages189.
Dynamics simulations
Molecular dynamics simulations are an important tool for understanding dynamic processes and mechanisms on a microscopic level in various areas of chemistry, biology, and materials science. Besides the prediction of equilibrium properties of molecules and materials, they also offer the possibility to simulate excited states and non-equilibrium dynamics, as well as slow processes and rare events.
In molecular dynamics simulations, total energy and forces are needed in every time step to propagate the system. Computationally demanding ab initio methods that calculate the energy and forces of a particular atomic configuration of a system at every time step are therefore often too costly. Machine learning methods can replace ab initio calculations to speed up simulations while ideally retaining their accuracy9. Therefore, long and highly accurate MD simulations can be performed based on machine-learned potentials, which have not been possible using classical force fields nor ab initio methods. GNNs are perfectly suited for this task, as atomic forces depend on the (local) atomic environment and global aggregation is not needed.
The concept of integrating machine learning models in atomistic simulations was demonstrated multiple times using for example SchNet22, PhysNet23, DimeNet28, or DimeNet++72. However, there are several open challenges that need to be overcome in order to move to larger systems, longer time scales, higher data efficiency, better generalization and transferability, and eventually more accurate and realistic applications. Usually, machine learning models learn the potential energy surface and calculate forces using derivatives of the energy predictions. This ensures that energy predictions and forces are consistent. Since only forces are required in MD simulations, architectures are being developed in which these forces are predicted directly—so that the costly derivative calculations are omitted. In the GNN framework (GNNFF190), a message passing step builds upon an embedding step in which node and edge features include atom type and interatomic distances respectively. Force magnitudes per atom are then calculated from the sum of the forces of the neighboring atoms. Evaluation on the ISO17 database reveals higher force accuracies compared to SchNet while being 1.6 × faster. The approach is also shown to be scalable to larger systems. Due to the direct prediction of the forces, the energy of the system is however not necessarily preserved, making the model not suitable to predict energy-related properties.
ForceNet191 is based on an encoder-decoder architecture and tries to completely capture 3D geometry through a specific design of the message passing structure. In contrast to models such as SchNet and DimeNet, ForceNet encodes physical information without constraining the model architecture to enforce physical invariances. Instead, physics-based data augmentation is performed on the data level to achieve rotational invariance. The evaluation was performed on the OC20 dataset which contains DFT calculated energies and per-atom forces of more than 200 million large atomic structures (20–200 atoms) including non-equilibrium structures from optimization trajectories. The resulting mean absolute force errors are comparable to DimeNet++, while being faster in training and prediction.
A promising approach to encoding more physical information about a system is the design of equivariant models. Models that are based on equivariant message passing, e.g., PaiNN29, NequIP25, NewtonNet192, are shown to significantly increase data efficiency and predictive performance compared to models that are based on invariant convolutions. The Neural Equivariant Interatomic Potential (NequIP25) predicts both energy and forces utilizing E(3)-equivariant convolutions over geometric tensors. Evaluated on the MD17 data set its accuracy exceeds those of existing models while needing up to three orders of magnitude less training data. Due to its data efficiency, it was also used with coupled cluster (CCSD(T)) based training data, showing great potential for applications where a prediction accuracy beyond density functional theory (DFT) is needed. In order to further improve data efficiency, GNNFF190 and NewtonNet192 introduce more physical priors in the form of latent force vectors as well as operators containing physical information. This leads to good prediction accuracy with higher computational efficiency at only 1–10% of the training data compared to other models.
To improve generalization, hybrid models such as SpookyNet193 explicitly include electronic structure information such as total charge or spin state, not included in most machine-learned potentials, by applying self-attention in a transformer architecture. Empirical augmentations to include non-local and long-range contributions such as electrostatic interaction and dispersion improve transferability and at the same time enable interpretability193.
GNN for large-scale MD simulations. Many applications require models that scale to large system sizes. For example, simulations of biological systems, e.g., to study protein dynamics or drug binding mechanisms involve orders of magnitude more atoms than many other applications, while configuration changes occur on much longer timescales (10−3−103 s) than a typical MD timestep (10−15 s). One way to address this enormous challenge is the development of models in a QM/MM-inspired approach, where only a small relevant subsystem, e.g., a reaction site, needs to be simulated at ab initio accuracy, while the rest of the system can be described using classical force fields.
GNNs also have the potential to support (adaptive) coarse-graining methods. They were shown to be useful in mapping atoms of a molecule into coarse-grained groups needed for large-scale simulations. The Deep Supervised Graph Partitioning Model (DSGPM)194 treats mapping operators as a graph segmentation problem. It predicted a coarse-grained mapping nearly indistinguishable from human annotations (see Fig. 2d). Furthermore, the machine learning framework by Wang et al.195, which generates a coarse-grained force field, was further improved by Husic et al.196 replacing manual input features with a GNN architecture making the models transferable across different molecular systems such as small proteins.
Excited states dynamics. GNNs were also shown as a very promising tool to tackle the challenging task of simulating excited state dynamics of complex systems197. Unlike ground-state dynamics, multiple potential energy surfaces as well as their crossings and couplings must be considered, leading to a higher dimensionality and complexity of the problem. Furthermore, even the generation of reliable training data using quantum mechanical calculations is challenging.
Westermayr et al. developed SchNarc198 for photodynamics simulations by adapting SchNet for excited states potentials, forces, and couplings, combining it with the MD framework SHARC (surface hopping including arbitrary couplings). While SchNarc is molecule specific and was applied to two compounds, CH2NH\({}_{2}^{+}\) and CSH2, Axelrod et al. developed the first transferable excited state potential (see Fig. 2d)199. The diabatic artificial neural network (DANN) is based on PaiNN combined with a physics-informed diabatic model for photodynamic simulations for virtual screening. The resulting machine-learned potential is transferable among azobenzene derivatives and estimated to be multiple orders of magnitude faster than the underlying quantum mechanical calculation method, even considering computational effort for transfer which required additional training data.
Reaction prediction and retrosynthesis
While reliable property prediction and simulation methods are crucial for virtual molecular design, synthesis is often one of the main bottlenecks in the overall development process of new molecules. Progress in reaction prediction and retrosynthesis, i.e., the prediction of a reaction outcome and the design of synthetic routes for a desired product, can help to accelerate and also automate200 the development of new molecules. However, the two problems are still considered challenging due to the vast chemical space and currently require skills and experience from well-trained chemists. Therefore, many machine learning algorithms, e.g., seq2seq models and transformers, have been proposed for synthesis prediction and retrosynthesis, aiming at reducing manual effort. In many cases, molecules are embedded as SMILES codes, and reaction predictions as well as retrosynthesis predictions are formulated as natural language processing tasks201,202,203,204. Furthermore, fingerprints are widely used as structure encodings and neural networks trained on them are able to predict the most probable transformations for given molecules205,206. GNN based graph embeddings have recently attracted growing attention, due to the natural representation of molecular structures as graphs.
Prediction of reactivity with GNNs has been formalized into reaction center identification and graph edit tasks. Jin et al. developed a GNN-based approach to scoring each pair of connected atoms with bond change likelihood, followed by the selection of bonds with the highest score and transformation to potential products using a Weisfeiler-Lehman Difference Network (WLDN)207,208. The WLN architecture is also adopted by Struble and coworkers209 to perform multitask prediction of aromatic C-H functionalization reactions with site selectivity, while Guan et al. predict reaction outcomes with regio-selectivity through combining molecule representations learned by the WLN with on-the-fly calculated quantum mechanical descriptors210. In 2020, Nikitin et al. introduced a strategy to treat reaction prediction as a node classification problem, where the role of each node in the reactant graphs is predicted by a GNN with a self-attention mechanism and pseudo-global nodes211.
Furthermore, predicting reaction products as the result of a sequence of graph editing actions on the reactant molecules has also been investigated. One example is the work by Do et al. where graph editing actions are predicted by a reinforcement learning algorithm based on reactant and reagent molecule representations generated by GNNs212. In 2019, Bradshaw et al. developed a generative model that uses GNNs to predict a series of electron movements for reactions, through which the products and reaction mechanisms are predicted at the same time213. Apart from product prediction, GNNs are also employed to predict important reaction properties including bond dissociation energies214,215, transition states216, and activation energies217.
For the retrosynthesis task, recent studies can be divided into template-based and template-free approaches. The former matches the graph embedding of the product molecule to a large number of reaction templates, which determine bond changes and thus predict possible reactants, while the latter bypass the templates and directly modifies input graphs to generate synthetic precursors. Examples of template-based retrosynthesis prediction include the work by Dai et al., who predict the probability distribution of reaction templates to be fitted to the reaction outcomes by a Conditional Graph Logic Network (GLN)218. The reactants are generated by the GLN as the result of a probability prediction given the reaction outcome and the selected template. Another example by Ishida et al. uses GCNs to classify single retrosynthesis steps into reaction templates, where integrated gradients are applied to visualize atom contributions toward the GCN prediction219. More recently, Chen and coworkers proposed a framework based on MPNNs with a global attention mechanism to predict the reaction center of a target molecule, as well as the corresponding “local” reaction template based on atoms and bonds in the reaction center, which is then applied to generate reactants220.
For template-free retrosynthesis, Yan et al. use an edge-enhanced graph attention network to locate the reaction center in product molecules, which are transformed through bond dissociation into molecular fragments called synthons221. The synthon graphs are converted to SMILES and expanded to reactants by a sequence-to-sequence algorithm. At the same time, Somnath et al. proposed an approach that uses GNNs to predict a series of graph edits that transform a product into synthons that are further completed into reactants with leaving groups predicted by another GNN (see Fig. 2d)222. A similar strategy is adopted by Shi et al., where synthons are expanded to reactants by a variational graph translation223.
In 2021, Sacha et al. proposed a model that formulates both retrosynthesis and forward synthesis tasks as a sequence of graph edit actions that are predicted by an encoder-decoder structure constructed by stacking graph convolutional layers130.
Crystalline and solid state systems
Compared to molecules, crystal structures and solid-state materials have some additional challenges, such as periodic boundary conditions for crystals and multiple kinds of disorder, either in form of perturbations in the crystal structure itself or in the (lack of) long-range ordering of atoms. We will present different recent applications of GNNs in solid state systems, from predicting global properties of crystal structures with and without disorder, over driving atomistic simulations, to the design of new materials aided by materials synthesis prediction, active learning, and inverse design. Table 4 gives an overview of datasets used in the following applications. We will also discuss approaches in which the trained GNN models have been analyzed to enable further insight into specific scientific questions, which is often equally important as accurate numerical predictions.
Materials property prediction and materials design
GNNs can be used to predict a multitude of materials properties, ranging from formation energies24,224,225,226,227,228 and synthesizability prediction229 over band-gaps230,231,232 and other functional properties to mechanical properties61,233. The most straightforward application of property prediction models is the screening of large (unlabeled) crystal databases, where exhaustive screening using conventional simulation techniques (e.g., DFT) is often not feasible. Screening using GNNs only requires labels from simulation or experiment for the training set while providing fast predictions on the full database - provided the model generalizes well.
Wang et al. use a crystal GNN to predict methane adsorption volumes in metal-organic frameworks (MOFs)234. The pooling function leverages domain knowledge by additionally including structural properties (see Fig. 2f), e.g., the pore limiting diameter, achieving better performance than previous work235. They apply the model to the screening of a hypothetical MOF database by Wilmer et al.236 and find several high-performing candidates234. Gu et al. use an ensemble of attention crystal GNNs to screen alloy catalysts for CO2 reduction237. Only bulk-relaxed structures without the adsorbate (e.g., from the Materials Project (MP)100 database) are needed as input, removing the costly DFT relaxation from the screening process. The performance approaches that of base-line models trained on fully relaxed structures237.
An interesting application of GNNs was presented by Goodall et al., who use GNNs for representation learning of chemical formulas238. The chemical formula is represented as a dense weighted graph, in which each node corresponds to a chemical element weighted by the fraction of this element in the chemical formula. This graph representation was used to train a GNN in a supervised way to obtain a mapping from the chemical formula to an embedding. It was demonstrated that this representation has a better sample efficiency than other structure agnostic approaches.
Schmidt et al. use a crystal graph attention network for stability-screening of non-relaxed hypothetical materials, predicting the convex hull distance24. Only graph distances are included, making the model usable for hypothetical materials where the exact coordinates might be unknown. A vast database is used, combining MP, AFLOW239 and group-internal datapoints. Transfer learning then allows the screening of 15 million tetragonal perovskites of composition ABCD2. Dai et al. use GNNs to predict magnetostriction of polycrystalline systems (see Fig. 2g)240. Instead of using GNNs to model the atoms in the unit cell, individual grains and their interactions to neighboring grains are represented by nodes and edges in the GNN. This shows that GNNs can be used on different scales to predict materials properties.
Typically, screening of materials databases using ML models is only possible if training data is available which covers the target materials distribution, i.e., which adequately allows generalization to the rest of the database. Active learning offers a promising solution when the available data does not fulfill this criterion. Based on uncertainty quantification, the training dataset can then be iteratively extended to include previously uncertain data points and thereby efficiently explore the chemical space. Lu et al. use active learning to search for 2D ferromagnetic materials with a custom crystal graph multilayer descriptor in a small dataset241. Further applications of active learning based GNNs in materials science are promising and can be expected in the future.
Disordered systems and defects
Disordered materials are a wide and particularly interesting field of study, as the effects of disorder can influence or even dominate material properties. Disorder ranges from weak disorder (defects, dislocations, grain boundaries) to strong disorder (e.g., glassy systems and inhomogeneous systems such as porous materials) and includes topological/structural disorder, orientational disorder (spins, dipoles), or substitutional disorder (chemical doping, compositional disorder)242. Due to its inherent multi-scale nature, disorder poses severe challenges not only to materials modeling and simulation, but also to materials synthesis, characterization, and fundamental description. Graph neural networks can be extended to model various forms of disordered systems and predict their local and global properties. In contrast to quantum mechanical methods such as DFT, crystalline systems with substitutional disorder can be modeled using larger, more representative unit cells or by defining atoms/nodes with mixed occupations. Furthermore, amorphous systems can be modeled explicitly by using representative simulation boxes and aggregating over learned atom representations characteristic of the properties of the amorphous system. In the following, we will present seminal work in that direction.
One important challenge for machine learning models is how to represent substitutional disorder such as doping and fractional occupancies in the crystal graph. Chen et al. addressed this question with a MEGNet model with trainable 16-dimensional embeddings for each atom which were pre-trained on a dataset with only ordered structures243. The pre-trained embeddings were used to represent doped crystal sites by doing a weighted average of the elemental embeddings, weighted with (logarithmic or appropriately scaled) occupancies of the elements on the crystal sites. Additionally, it was also demonstrated how to perform multi-fidelity training with band gaps of different levels of DFT by encoding the DFT level in the global state of the MEGNet. Similarly, Wang et al. have trained a crystal GNN model on predicting the phase stability of materials (see Fig. 2f), classifying materials as metal or insulator and predicting the band gap of semiconductors244. They train a crystal GNN on undoped crystal structures and use this trained model to predict the properties of doped crystal structures, which they validate with DFT calculations. They find that the crystal GNN and the DFT usually predict the same trend, despite the crystal GNN not being trained on doped crystal structures244. Frey et al. use a crystal GNN model to predicted the properties of 2D materials with point defects (see Fig. 2f)245. However, the crystal GNN is only used to screen the properties of ordered structures to find promising host structures. The properties of the disordered structures were then partially calculated with DFT to train a random forest model on physics-based features which was used to screen additional structures. Another study predicted the properties of bcc iron structures with point defects246.
A very special application of GNNs is glassy systems. There have been works that predicted the properties of glasses247 and which used inverse design to find glasses with new properties248. Swanson et al. trained a classifier on predicting if a system is in a liquid or a glassy phase only by the positions of the atoms (see Fig. 2h)249. They verified that a GNN was significantly better than a CNN for this task and used self-attention to interpret the reasoning of the trained classifier. Consequently, the trained GNN was interpreted to find previously unknown relationships. Three simple and previously unknown formulas were developed, which describe the reasoning of the classifier and which could be used to differentiate the two phases. Initial work on predicting polymer properties using GNNs was based on learning representations of single monomers250 or of unit cells of crystalline polymers251.
Aiding simulations
Analogous to atomistic dynamics simulations of molecular systems, GNNs can also be used to simulate the dynamic behavior of crystals, i.e., to predict potential energy, forces, and partial charges, in order to drive molecular dynamics simulations. To predict forces for molecular dynamics of crystals, most approaches use conventional machine learning methods252, but there are first examples that use GNNs190,253. Raza et al. use a GNN with node-level readouts to predict partial charges of MOFs for molecular simulations of gas adsorption254. For each atom, a probability distribution of charge is learned and optimized globally using the maximum likelihood principle under a charge neutrality constraint. Park et al. predict forces for molecular dynamics directly using a GNN with edge-level readouts that are used to predict the magnitude of the force between atoms190 (see also Section ‘Molecular systems’). This avoids calculating the derivative of the potential energy and speeds up the simulation, yielding good accuracy in chemical reactions occurring at the surface of a test system of Al2O3 in contact with HF gas (see Fig. 2g). Also, good scaling accuracy transferring a model trained on a 1 × 2 × 1 supercell to a 1 × 2 × 2 supercell (Li7P3S11) is achieved. This approach allows simulations of a larger scale than possible using ab initio methods with similar accuracy and might make it possible to simulate mesoscopic phenomena such as grain boundaries in the future.
Solid state synthesis prediction
While predicting synthesizability overall229 is important in the design process of new materials, predicting the products of solid state synthesis is a challenging task and interesting application area of GNNs. Malik et al. have developed a pipeline to predict the major outcome of solid-state reactions255. In this pipeline, GNNs were used to represent the precursors of a reaction in a set-like fashion as a dense graph while a long short-term memory layer is used to represent the series of processing actions in the reaction. Current limitations in the availability of systematic (experimental) synthesis data, including reaction conditions11, hinder further progress in this area. The use of electronic lab notebooks and repositories specifically designed for chemistry and materials science has a large potential to alleviate that challenge256,257.
Periodic graph generation
A major requirement for the graph-based inverse design of crystal structures is the possibility to generate new periodic graphs based on a vector representation. Recently, this problem has been addressed with a new architecture called PGD-VAE162, a variational autoencoder capable of generating new periodic graph structures. Another work using a VAE focuses on predicting stable crystal structures using GNNs258. New structures are generated in a three-step approach: First, the composition, lattice, and number of atoms of a sampled point in the latent space are decoded using a multilayer perceptron. Afterward, a random structure with these properties is assembled and a GNN trained on moving atoms to their equilibrium positions is used to generate stable structures. Despite these promising efforts, the design of solid state materials and crystal structures using GNNs is only at the beginning. Multiple challenges need to be solved to achieve wide applicability and transferability of methods to multiple classes of materials, ranging from densely packed crystals over porous materials to amorphous systems need to be solved.
Outlook
GNNs became a very versatile and important tool very quickly. A lot has been achieved already, not only in terms of fundamental method development tailor-made for requirements of materials science and chemistry (see Section ‘Graph neural networks in materials science and chemistry’), but also in terms of applications (see Section ‘Applications’), where GNNs were successfully applied for materials simulation, screening, design, and synthesis. However, there is a wide range of open questions and challenges which need to be solved in order to leverage the full potential of GNNs in materials science and chemistry.
Despite the growing amount of research and publications on GNN model development and application, GNN models remain expert tools, i.e., they are comparably hard to implement, adapt, transfer, train, and apply to given datasets and applications. Libraries such as PyTorch Geometric259, DGL260 or the Keras based KGCNN261 implement a selection of state-of-the-art GNN layers and models. However, the use of such libraries in many cases requires expert knowledge which goes beyond the knowledge needed for the successful application of more established machine learning models. One of the reasons for this is certainly related to the fast development of new GNN variants which are partially hard to compare. The widespread use and further development of common benchmarks (e.g., the QM9 dataset) as well as open communication of models and training workflows, including open-source code, inspired by common practice in the machine learning community, are essential for a reliable quantitative comparison of future developments. However, non-optimal hyperparameters and the (prohibitively) high computational cost of hyperparameter optimization is an open challenge. A further challenge hindering the transfer of state-of-the-art models to applications is the discrepancy in the data distribution between widely used benchmark datasets (e.g., QM9) and actually relevant datasets, which typically contain larger molecules (i.e., larger graphs), which are more diverse, e.g., in terms of the chemical elements used and in the size variance, and which sample the chemical or materials space less densely then QM9262.
Generally, there is more research and model development activity for molecules and chemistry, compared to materials science. As a consequence, the transfer of existing GNNs to new application areas (e.g., porous or amorphous materials) might be challenging and requires further development of GNN models. To some extent, this can be attributed to a lack of generic benchmark datasets for crystalline materials and, even more importantly, (partially) disordered structures and amorphous materials. OC20 is one of the few examples of datasets covering both materials science and chemistry102. Nonetheless, there are many promising application areas of GNNs for solid-state materials. In contrast to quantum mechanical simulation methods such as DFT, GNNs are particularly suited for representing disordered systems and predicting their properties, e.g., systems with compositional disorder such as high-entropy alloys. However, there is a lack of large datasets of disordered systems, particularly labeled datasets with consistent measured or simulated properties. Overall, screening of (hypothetical) materials spaces with predictive ML models promises the rapid discovery of new materials with optimized target properties, especially when combined with transfer and active learning to improve data efficiency and reduce computational cost.
The first steps towards GNN-based generative models exist, but there are many open challenges of reliability and transferability. Despite the high potential of GNN-based algorithms for the inverse design of new molecules and materials, convincing success stories of ML-designed materials are rare. To make generative models more application-relevant, new methods are required that e.g., allow to include constraints in the design process, in the simplest case symmetries of generated molecules and materials, or in more complex scenarios additional (empirical or analytical) objectives such as synthesizability. A large step in that direction is new representations, not only for organic molecules but also for (3D) materials263,264,265.
Finally, more research on the explainability and interpretability of GNNs and machine learning in general will help to better understand underlying correlations and eventually causal relations in large and complex datasets, eventually contributing to scientific understanding and progress266,267,268.
References
von Lilienfeld, O. A. Introducing machine learning: science and technology. Mach. Learn. Sci. Technol. 1, 010201 (2020).
Karamad, M. et al. Orbital graph convolutional neural network for material property prediction. Phys. Rev. Mater. 4, 093801 (2020).
Vamathevan, J. et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 463–477 (2019).
Jorissen, R. N. & Gilson, M. K. Virtual screening of molecular databases using a support vector machine. J. Chem. Inform. Model. 45, 549–561 (2005).
Ekins, S. et al. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 18, 435–441 (2019).
Noh, J., Gu, G. H., Kim, S. & Jung, Y. Machine-enabled inverse design of inorganic solid materials: promises and challenges. Chem. Sci. 11, 4871–4881 (2020).
Zunger, A. Inverse design in search of materials with target functionalities. Nat. Rev. Chem. 2, 1–16 (2018).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Friederich, P., Häse, F., Proppe, J. & Aspuru-Guzik, A. Machine-learned potentials for next-generation matter simulations. Nat. Mater. 20, 750–761 (2021).
Shields, B. J. et al. Bayesian reaction optimization as a tool for chemical synthesis. Nature 590, 89–96 (2021).
Luo, Y. et al. Mof synthesis prediction enabled by automatic data mining and machine learning. Angew. Chem. Int. Ed. 202200242 (2021).
Kalinin, S. V. et al. Machine learning in scanning transmission electron microscopy. Nat. Rev. Methods Primers 2, 1–28 (2022).
Velasco, L. et al. Phase–property diagrams for multicomponent oxide systems toward materials libraries. Adv. Mater. 33, 2102301 (2021).
Häse, F., Roch, L. M. & Aspuru-Guzik, A. Next-generation experimentation with self-driving laboratories. Trends Chem. 1, 282–291 (2019).
LeCun, Y., Kavukcuoglu, K. & Farabet, C. Convolutional networks and applications in vision. In Proc. IEEE International Symposium on Circuits and Systems, 253–256 (2010).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Central Sci. 5, 1572–1583 (2019).
Sperduti, A. & Starita, A. Supervised neural networks for the classification of structures. IEEE Trans. Neural Netw. 8, 714–735 (1997).
Gori, M., Monfardini, G. & Scarselli, F. A new model for learning in graph domains. In Proc. IEEE International Joint Conference on Neural Networks, 2005, vol 2, 729–734 (2005).
Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M. & Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 20, 61–80 (2009).
Duvenaud, D. et al. Convolutional networks on graphs for learning molecular fingerprints. In Cortes, C., Lawrence, N. D., Lee, D. D., Sugiyama, M. & Garnett, R. (eds.) Advances in Neural Information Processing Systems 28: Annual Conference on Neural Information Processing Systems 2015, December 7–12, 2015, Montreal, Quebec, Canada, 2224–2232 (2015). https://proceedings.neurips.cc/paper/2015/hash/f9be311e65d81a9ad8150a60844bb94c-Abstract.html.
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In Proc. 34th International Conference on Machine Learning—Volume 70, ICML’17, 1263–1272 (JMLR.org, 2017).
Schütt, K. T., Sauceda, H. E., Kindermans, P.-J., Tkatchenko, A. & Müller, K.-R. SchNet—a deep learning architecture for molecules and materials. The J. Chem. Phys. 148, 241722 (2018).
Unke, O. T. & Meuwly, M. Physnet: a neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Schmidt, J., Pettersson, L., Verdozzi, C., Botti, S. & Marques, M. A. L. Crystal graph attention networks for the prediction of stable materials. Sci. Adv. 7, eabi7948 (2021).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 1–11 (2022).
Klicpera, J., Becker, F. & Günnemann, S. Gemnet: Universal directional graph neural networks for molecules. In Beygelzimer, A., Dauphin, Y., Liang, P. & Vaughan, J. W. (eds.) Advances in Neural Information Processing Systems (2021). https://openreview.net/forum?id=HS_sOaxS9K-.
von Lilienfeld, O. A. & Burke, K. Retrospective on a decade of machine learning for chemical discovery. Nat. Commun. 11, 1–4 (2020).
Klicpera, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs. In Proc. 8th International Conference on Learning Representations, ICLR 2020, Addis Ababa, Ethiopia, April 26–30, 2020 (OpenReview.net, 2020). https://openreview.net/forum?id=B1eWbxStPH.
Schütt, K., Unke, O. & Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra. In Proc. International Conference on Machine Learning, 9377–9388 (PMLR, 2021).
Cayley, F. R. S. LVII. on the mathematical theory of isomers. London, Edinburgh, Dublin Philos. Mag J. Sci. 47, 444–447 (1874).
Bonchev, D. & Rouvray, D. H. Chemical Graph Theory: Introduction and Fundamentals (Routledge, London, 1991). https://doi.org/10.1201/9781315139104.
Biggs, N., Lloyd, E. K. & Wilson, R. J. Graph Theory. 1736–1936 (Clarendon Press, USA, 1986).
Merkwirth, C. & Lengauer, T. Automatic generation of complementary descriptors with molecular graph networks. J. Chem. Inform. Model. 45, 1159–1168 (2005)..
Kipf, T. N. & Welling, M. Semi-supervised classification with graph convolutional networks. In Proc. 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24–26, 2017, Conference Track Proceedings (OpenReview.net, 2017). https://openreview.net/forum?idS¯JU4ayYgl.
Defferrard, M., Bresson, X. & Vandergheynst, P. Convolutional neural networks on graphs with fast localized spectral filtering. In Lee, D., Sugiyama, M., Luxburg, U., Guyon, I. & Garnett, R. (eds.) Advances in Neural Information Processing Systems, vol. 29 (Curran Associates, Inc., 2016). https://proceedings.neurips.cc/paper/2016/file/04df4d434d481c5bb723be1b6df1ee65-Paper.pdf.
Estrach, J. B., Zaremba, W., Szlam, A. & LeCun, Y. Spectral networks and deep locally connected networks on graphs. In Proc. 2nd International Conference on Learning Representations, ICLR, vol. 2014 (2014).
Alon, U. & Yahav, E. On the bottleneck of graph neural networks and its practical implications. In Proc. 9th International Conference on Learning Representations, ICLR 2021, Virtual Event, Austria, May 3–7, 2021 (OpenReview.net, 2021). https://openreview.net/forum?idī80OPhOCVH2.
Li, Q., Han, Z. & Wu, X. Deeper insights into graph convolutional networks for semi-supervised learning. In McIlraith, S. A. & Weinberger, K. Q. (eds.) Proc. Thirty-Second AAAI Conference on Artificial Intelligence, (AAAI-18), the 30th innovative Applications of Artificial Intelligence (IAAI-18), and the 8th AAAI Symposium on Educational Advances in Artificial Intelligence (EAAI-18), New Orleans, Louisiana, USA, February 2–7, 2018, 3538-3545 (AAAI Press, 2018). https://www.aaai.org/ocs/index.php/AAAI/AAAI18/paper/view/16098.
Vinyals, O., Bengio, S. & Kudlur, M. Order matters: sequence to sequence for sets. In: Bengio, Y. & LeCun, Y. (eds.) Proc. 4th International Conference on Learning Representations, ICLR 2016, San Juan, Puerto Rico, May 2–4, 2016, Conference Track Proceedings (2016).
Yang, K. et al. Analyzing learned molecular representations for property prediction. J. Chem. Inform. Model. 59, 3370–3388 (2019).
Schütt, K. T. et al. SchNetPack: a deep learning toolbox for atomistic systems. J. Chem. Theory Comput. 15, 448–455 (2018).
Shui, Z. & Karypis, G. Heterogeneous molecular graph neural networks for predicting molecule properties. In Plant, C., Wang, H., Cuzzocrea, A., Zaniolo, C. & Wu, X. (eds.) Proc. 20th IEEE International Conference on Data Mining, ICDM 2020, Sorrento, Italy, November 17–20, 2020, 492–500 (IEEE, 2020). https://doi.org/10.1109/ICDM50108.2020.00058.
Finzi, M., Stanton, S., Izmailov, P. & Wilson, A. G. Generalizing convolutional neural networks for equivariance to lie groups on arbitrary continuous data. In III, H. D. & Singh, A. (eds.) Proc. 37th International Conference on Machine Learning, vol. 119 of Proceedings of Machine Learning Research, 3165–3176 (PMLR, 2020). https://proceedings.mlr.press/v119/finzi20a.html.
Bodnar, C. et al. Weisfeiler and lehman go topological: Message passing simplicial networks. In Proc. ICLR 2021 Workshop on Geometrical and Topological Representation Learning (2021). https://openreview.net/forum?idR¯ZgbB-O3w6Z.
Maron, H., Ben-Hamu, H., Serviansky, H. & Lipman, Y. Provably powerful graph networks. In Wallach, H. et al. (eds.) Advances in Neural Information Processing Systems, vol. 32 (Curran Associates, Inc., 2019). https://proceedings.neurips.cc/paper/2019/file/bb04af0f7ecaee4aae62035497da1387-Paper.pdf.
Xu, K., Hu, W., Leskovec, J. & Jegelka, S. How powerful are graph neural networks? In Proc. 7th International Conference on Learning Representations, ICLR 2019, New Orleans, LA, USA, May 6-9, 2019 (OpenReview.net, 2019). https://openreview.net/forum?idr¯yGs6iA5Km.
Feng, Y., You, H., Zhang, Z., Ji, R. & Gao, Y. Hypergraph neural networks. Proc. AAAI Conf. Artif. Intell. 33, 3558–3565 (2019).
Jo, J. et al. Edge representation learning with hypergraphs. Adv. Neural Inform. Process. Syst. 34, 7534–7546 (2021).
Dym, N. & Maron, H. On the universality of rotation equivariant point cloud networks. In Proc. International Conference on Learning Representations (2021). https://openreview.net/forum?id6131NFBvWlRXaG.
Morris, C. et al. Weisfeiler and leman go neural: higher-order graph neural networks. Proc. AAAI Conf. Artif. Intell. 33, 4602–4609 (2019).
Maziarka, L. et al. Molecule attention transformer. arXiv preprint arXiv:2002.08264 (2020). https://arxiv.org/abs/2002.08264.
Chen, Y., Tang, X., Qi, X., Li, C.-G. & Xiao, R. Learning graph normalization for graph neural networks. Neurocomputing 493, 613–625 (2022).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Xiong, Z. et al. Pushing the boundaries of molecular representation for drug discovery with the graph attention mechanism. J. Med. Chem. 63, 8749–8760 (2020).
Kruger, F., Stiefl, N. & Landrum, G. A. rdscaffoldnetwork: The scaffold network implementation in rdkit. J. Chem. Inform. Model. 60, 3331–3335 (2020).
RDKit: Open-source cheminformatics. http://www.rdkit.org. [Online; 1 April 2013].
O’Boyle, N. M. et al. Open label: an open chemical toolbox. J. Cheminform. 3, 33 (2011).
Chen, Z.-H. et al. Prediction of drug-target interactions from multi-molecular network based on deep walk embedding model. Front. Bioeng. Biotechnol. 8 (2020). https://www.frontiersin.org/article/10.3389/fbioe.2020.00338.
Perozzi, B., Al-Rfou, R. & Skiena, S. Deepwalk: online learning of social representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’14, 701–710 (Association for Computing Machinery, New York, NY, USA, 2014). https://doi.org/10.1145/2623330.2623732.
Grover, A. & Leskovec, J. Node2vec: Scalable feature learning for networks. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, KDD ’16, 855-864 (Association for Computing Machinery, New York, NY, USA, 2016). https://doi.org/10.1145/2939672.2939754.
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mate. 31, 3564–3572 (2019).
Ramakrishnan, R., Dral, P. O., Rupp, M. & von Lilienfeld, O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 1, 140022 (2014).
von Lilienfeld, O. A., Müller, K.-R. & Tkatchenko, A. Exploring chemical compound space with quantum-based machine learning. Nat. Rev. Chem. 4, 347–358 (2020).
Behler, J. Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys. 134, 074106 (2011).
Seko, A., Takahashi, A. & Tanaka, I. Sparse representation for a potential energy surface. Phys. Rev. B 90, 024101 (2014).
Behler, J. Perspective: Machine learning potentials for atomistic simulations. J. Chem. Phys. 145, 170901 (2016).
Bartók, A. P., Kondor, R. & Csányi, G. On representing chemical environments. Phys. Rev. B 87, 184115 (2013).
Schütt, K. T. et al. How to represent crystal structures for machine learning: Towards fast prediction of electronic properties. Phys. Rev. B 89, 205118 (2014).
Huo, H. & Rupp, M. Unified representation of molecules and crystals for machine learning. Mach. Learn.: Sci. Technol. 3 045017 (2022).
Huang, B. & von Lilienfeld, O. A. Quantum machine learning using atom-in-molecule-based fragments selected on the fly. Nat. Chem. 12, 945–951 (2020).
Christensen, A. S., Bratholm, L. A., Faber, F. A. & Anatole von Lilienfeld, O. Fchl revisited: Faster and more accurate quantum machine learning. J. Chem. Phys. 152, 044107 (2020).
Klicpera, J., Giri, S., Margraf, J. T. & Günnemann, S. Fast and uncertainty-aware directional message passing for non-equilibrium molecules. Machine Learning for Molecules Workshop, NeurIPS (2020).
Flam-Shepherd, D., Wu, T. C., Friederich, P. & Aspuru-Guzik, A. Neural message passing on high order paths. Mach. Learn. Sci. Technol. 2, 045009 (2021).
Pukrittayakamee, A. et al. Simultaneous fitting of a potential-energy surface and its corresponding force fields using feedforward neural networks. J. Chem. Phys. 130, 134101 (2009).
Bronstein, M. M., Bruna, J., LeCun, Y., Szlam, A. & Vandergheynst, P. Geometric deep learning: going beyond euclidean data. IEEE Signal Process Mag. 34, 18–42 (2017).
Atz, K., Grisoni, F. & Schneider, G. Geometric deep learning on molecular representations. Nat. Mach. Intell. 3, 1023–1032 (2021).
Cao, W., Yan, Z., He, Z. & He, Z. A comprehensive survey on geometric deep learning. IEEE Access 8, 35929–35949 (2020).
Monti, F. et al. Geometric deep learning on graphs and manifolds using mixture model cnns. In Proc. IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (2017).
Nguyen, A. & Le, B. 3d point cloud segmentation: A survey. In Proc. 6th IEEE Conference on Robotics, Automation and Mechatronics (RAM), 225–230 (2013).
Qi, C. R., Yi, L., Su, H. & Guibas, L. J. PointNet++: Deep Hierarchical Feature Learning on Point Sets in a Metric Space. In Guyon, I. et al. (eds.) Adv. Neural Inform. Process. Syst., vol. 30 (Curran Associates, Inc., 2017). https://proceedings.neurips.cc/paper/2017/file/d8bf84be3800d12f74d8b05e9b89836f-Paper.pdf.
Lee, J., Lee, I. & Kang, J. Self-attention graph pooling. In Chaudhuri, K. & Salakhutdinov, R. (eds.) Proc. 36th International Conference on Machine Learning, vol. 97 of Proceedings of Machine Learning Research, 3734–3743 (PMLR, 2019). https://proceedings.mlr.press/v97/lee19c.html.
Zhang, Z. et al. Hierarchical Graph Pooling with Structure Learning. arXiv preprint arXiv:1911.05954 (2019).
Li, C. et al. 3dmol-net: learn 3d molecular representation using adaptive graph convolutional network based on rotation invariance. IEEE J. Biomed. Health Inform. 1–1 (2021).
Montavon, G. et al. Learning invariant representations of molecules for atomization energy prediction. In Pereira, F., Burges, C. J. C., Bottou, L. & Weinberger, K. Q. (eds.) Adv. Neural Inform. Process. Syst., vol. 25 (Curran Associates, Inc., 2012). https://proceedings.neurips.cc/paper/2012/file/115f89503138416a242f40fb7d7f338e-Paper.pdf.
Satorras, V. G., Hoogeboom, E. & Welling, M. E(n) equivariant graph neural networks. In Meila, M. & Zhang, T. (eds.) Proc. 38th International Conference on Machine Learning, vol. 139 of Proceedings of Machine Learning Research, 9323-9332 (PMLR, 2021). https://proceedings.mlr.press/v139/satorras21a.html.
Nigam, J., Willatt, M. J. & Ceriotti, M. Equivariant representations for molecular hamiltonians and n-center atomic-scale properties. J. Chem. Phys. 156, 014115 (2022).
Thomas, N. et al. Tensor field networks: Rotation- and translation-equivariant neural networks for 3d point clouds. arXiv preprint arXiv:1802.08219 (2018).
Cheng, J., Zhang, C. & Dong, L. A geometric-information-enhanced crystal graph network for predicting properties of materials. Commun. Mater. 2, 92 (2021).
Blum, L. C. & Reymond, J.-L. 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13. J. Am. Chem. Soc. 131, 8732 (2009).
Montavon, G. et al. Machine learning of molecular electronic properties in chemical compound space. N. J. Phys. 15, 095003 (2013).
Wang, R., Fang, X., Lu, Y., Yang, C.-Y. & Wang, S. The PDBbind database: methodologies and updates. J. Med. Chem. 48, 4111–4119 (2005).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 3, e1603015 (2017).
Chmiela, S., Sauceda, H. E., Müller, K.-R. & Tkatchenko, A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 9, 3887 (2018).
Mobley, D. L. & Guthrie, J. P. FreeSolv: a database of experimental and calculated hydration free energies, with input files. J. Comput.-Aided Mol. Des. 28, 711–720 (2014).
Wu, Z. et al. Moleculenet: a benchmark for molecular machine learning. Chem. Sci. 9, 513–530 (2018).
Richard, A. M. et al. Toxcast chemical landscape: paving the road to 21st century toxicology. Chem. Res. Toxicol. 29, 1225–1251 (2016).
Martins, I. F., Teixeira, A. L., Pinheiro, L. & Falcao, A. O. A bayesian approach to in silico blood-brain barrier penetration modeling. J. Chem. Inform. Model. 52, 1686–1697 (2012).
Kuhn, M., Letunic, I., Jensen, L. J. & Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 44, D1075–D1079 (2015).
Altae-Tran, H., Ramsundar, B., Pappu, A. S. & Pande, V. Low data drug discovery with one-shot learning. ACS Cent. Sci. 3, 283–293 (2017).
Jain, A. et al. Commentary: The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Kirklin, S. et al. The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies. npj Comput. Mater. 1, 15010 (2015).
Chanussot, L. et al. Open catalyst 2020 (oc20) dataset and community challenges. ACS Catal. 11, 6059–6072 (2021).
Morris, C. et al. Tudataset: A collection of benchmark datasets for learning with graphs. arXiv preprint arXiv:2007.08663 (2020). https://arxiv.org/abs/2007.08663.
Cho, K., van Merrienboer, B., Bahdanau, D. & Bengio, Y. On the properties of neural machine translation: Encoder-decoder approaches. In Wu, D., Carpuat, M., Carreras, X. & Vecchi, E. M. (eds.) Proc. SSST@EMNLP 2014, Eighth Workshop on Syntax, Semantics and Structure in Statistical Translation, Doha, Qatar, 25 October 2014, 103–111 (Association for Computational Linguistics, 2014). https://aclanthology.org/W14-4012/.
Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput. 9, 1735–1780 (1997).
Hamilton, W., Ying, Z. & Leskovec, J. Inductive representation learning on large graphs. Adv. Neural Inform. Process. Syst. 30 (2017).
Vaswani, A. et al. Attention is all you need. Adv. Neural Inform. Process. Syst. 30 (2017).
Velickovic, P. et al. Graph attention networks. In Proc. 6th International Conference on Learning Representations, ICLR 2018, Vancouver, BC, Canada, April 30 - May 3, 2018, Conference Track Proceedings (OpenReview.net, 2018). https://openreview.net/forum?idr¯JXMpikCZ.
Jørgensen, P. B., Jacobsen, K. W. & Schmidt, M. N. Neural Message Passing with Edge Updates for Predicting Properties of Molecules and Materials. Paper presented at 32nd Conference on Neural Information Processing Systems, Montreal, Canada (2018).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. npj Comput. Mater. 7 (2021). https://doi.org/10.1038/s41524-021-00650-1.
Zhang, S., Liu, Y., & Xie, L. Molecular Mechanics-Driven Graph Neural Network with Multiplex Graph for Molecular Structures. NeurIPS-W. (2020).
Hsu, T. et al. Efficient and interpretable graph network representation for angle-dependent properties applied to optical spectroscopy. npj Comput Mater 8, 151 (2022).
Ganea, O. et al. Geomol: Torsional geometric generation of molecular 3d conformer ensembles. Adv. Neural Inform. Process. Syst. 34, 13757–13769 (2021).
Anderson, B., Hy, T. S. & Kondor, R. Cormorant: covariant molecular neural networks. Adv. Neural Inform. Process. Syst. 32 (2019).
Qiao, Z. et al. Unite: unitary n-body tensor equivariant network with applications to quantum chemistry. arXiv preprint arXiv:2105.14655 (2021).
Schütt, K. T., Gastegger, M., Tkatchenko, A., Müller, K.-R. & Maurer, R. J. Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat. Commun. 10, 5024 (2019).
Qiao, Z., Welborn, M., Anandkumar, A., Manby, F. R. & Miller, T. F. Orbnet: deep learning for quantum chemistry using symmetry-adapted atomic-orbital features. J. Chem. Phys. 153, 124111 (2020).
Unke, O. et al. Se (3)-equivariant prediction of molecular wavefunctions and electronic densities. Adv. Neural Inform. Process. Syst. 34, 14434–14447 (2021).
Liu, Q., Nickel, M. & Kiela, D. Hyperbolic graph neural networks. Adv. Neural Inform. Process. Syst. 32 (2019).
Wieder, O. et al. A compact review of molecular property prediction with graph neural networks. Drug Discov. Today: Technol. 37 (2020). https://www.sciencedirect.com/science/article/pii/S1740674920300305.
Zhou, J. et al. Graph neural networks: a review of methods and applications. AI Open 1, 57–81 (2020).
Sun, M. et al. Graph convolutional networks for computational drug development and discovery. Brief. Bioinform. 21, 919–935 (2019).
Zhang, S., Tong, H., Xu, J. & Maciejewski, R. Graph convolutional networks: a comprehensive review. Comput. Soc. Netw. 6, 1–23 (2019).
Wu, Z. et al. A comprehensive survey on graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. 32, 4–24 (2021).
Yuan, Y., Wang, W. & Pang, W. A systematic comparison study on hyperparameter optimisation of graph neural networks for molecular property prediction. In Proc. Genetic and Evolutionary Computation Conference (2021). https://doi.org/10.1145/3449639.3459370.
Jiang, D. et al. Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminform. 13, 1–23 (2021).
Banitalebi-Dehkordi, A. & Zhang, Y. ML4CO: is GCNN all you need? graph convolutional neural networks produce strong baselines for combinatorial optimization problems, if tuned and trained properly, on appropriate data. arXiv preprint arXiv:2112.12251 (2021). https://arxiv.org/abs/2112.12251.
Sanchez-Lengeling, B. & Aspuru-Guzik, A. Inverse molecular design using machine learning: generative models for matter engineering. Science 361, 360–365 (2018).
Zang, C. & Wang, F. Moflow: An invertible flow model for generating molecular graphs. In Proc. 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining (2020). https://doi.org/10.1145/3394486.3403104.
Sacha, M. et al. Molecule edit graph attention network: modeling chemical reactions as sequences of graph edits. J. Chem. Inform. Model. 61, 3273–3284 (2021).
Gilbert, E. N. Random graphs. Ann Math. Stat. 30, 1141–1144 (1959).
Watts, D. J. & Strogatz, S. H. Collective dynamics of ‘small-world’ networks. Nature 393, 440–442 (1998).
Leskovec, J., Chakrabarti, D., Kleinberg, J., Faloutsos, C. & Ghahramani, Z. Kronecker graphs: an approach to modeling networks. J. Mach. Learn. Res. 11, 985–1042 (2010).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. In Bengio, Y. & LeCun, Y. (eds.) Proc. 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, April 14–16, 2014, Conference Track Proceedings (2014).
Goodfellow, I. et al. Generative adversarial nets. In Ghahramani, Z., Welling, M., Cortes, C., Lawrence, N. & Weinberger, K. Q. (eds.) Adv. Neural Inform. Process. Syst., vol. 27 (Curran Associates, Inc., 2014). https://proceedings.neurips.cc/paper/2014/file/5ca3e9b122f61f8f06494c97b1afccf3-Paper.pdf.
Sutton, R. S. & Barto, A. G. Reinforcement learning: An Introduction (MIT press, 2018).
You, J., Ying, R., Ren, X., Hamilton, W. & Leskovec, J. GraphRNN: Generating realistic graphs with deep auto-regressive models. In Dy, J. & Krause, A. (eds.) Proc. 35th International Conference on Machine Learning, vol. 80 of Proceedings of Machine Learning Research, 5708–5717 (PMLR, 2018). https://proceedings.mlr.press/v80/you18a.html.
Dinh, L., Krueger, D. & Bengio, Y. NICE: non-linear independent components estimation. In Bengio, Y. & LeCun, Y. (eds.) Proc. 3rd International Conference on Learning Representations, ICLR 2015, San Diego, CA, USA, May 7–9, 2015, Workshop Track Proceedings (2015).
Dinh, L., Sohl-Dickstein, J. & Bengio, S. Density estimation using real NVP. In Proc. 5th International Conference on Learning Representations, ICLR 2017, Toulon, France, April 24-26, 2017, Conference Track Proceedings (OpenReview.net, 2017). https://openreview.net/forum?idH¯kpbnH9lx.
Kingma, D. P. & Dhariwal, P. Glow: generative flow with invertible 1x1 convolutions. Adv. Neural Inform. Process. Syst. 31 (2018).
Pu, Y. et al. Variational autoencoder for deep learning of images, labels and captions. In Lee, D., Sugiyama, M., Luxburg, U., Guyon, I. & Garnett, R. (eds.) Adv. Neural Inform. Process. Syst., vol. 29 (Curran Associates, Inc., 2016). https://proceedings.neurips.cc/paper/2016/file/eb86d510361fc23b59f18c1bc9802cc6-Paper.pdf.
Kusner, M. J., Paige, B. & Hernández-Lobato, J. M. Grammar variational autoencoder. In Precup, D. & Teh, Y. W. (eds.) Proc. 34th International Conference on Machine Learning, ICML 2017, Sydney, NSW, Australia, 6-11 August 2017, vol. 70 of Proceedings of Machine Learning Research, 1945–1954 (PMLR, 2017). http://proceedings.mlr.press/v70/kusner17a.html.
Dai, H., Tian, Y., Dai, B., Skiena, S. & Song, L. Syntax-directed variational autoencoder for structured data. In Proc. 6th International Conference on Learning Representations, ICLR 2018, Vancouver, BC, Canada, April 30–May 3, 2018, Conference Track Proceedings (OpenReview.net, 2018). https://openreview.net/forum?idS¯yqShMZRb.
Kipf, T. N. & Welling, M. Variational graph auto-encoders. arXiv preprint arXiv:1611.07308 (2016).
Simonovsky, M. & Komodakis, N. Graphvae: Towards generation of small graphs using variational autoencoders. In Kurková, V., Manolopoulos, Y., Hammer, B., Iliadis, L. S. & Maglogiannis, I. (eds.) Artificial Neural Networks and Machine Learning - ICANN 2018 - 27th International Conference on Artificial Neural Networks, Rhodes, Greece, October 4–7, 2018, Proceedings, Part I, vol. 11139 of Lecture Notes in Computer Science, 412–422 (Springer, 2018). https://doi.org/10.1007/978-3-030-01418-6_41.
Liu, Q., Allamanis, M., Brockschmidt, M. & Gaunt, A. Constrained graph variational autoencoders for molecule design. Adv. Neural Inform. Process. Syst. 31 (2018).
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Dy, J. & Krause, A. (eds.) Proc. 35th International Conference on Machine Learning, vol. 80 of Proceedings of Machine Learning Research, 2323-2332 (PMLR, 2018). https://proceedings.mlr.press/v80/jin18a.html.
Denton, E. L., Chintala, S., Fergus, R. et al. Deep generative image models using a laplacian pyramid of adversarial networks. Adv. Neural Inform. Process. Syst. 28 (2015).
Yu, L., Zhang, W., Wang, J. & Yu, Y. Seqgan: Sequence generative adversarial nets with policy gradient. In Proc. 31st AAAI Conference on Artificial Intelligence (2017). https://ojs.aaai.org/index.php/AAAI/article/view/10804.
Long, T. et al. Constrained crystals deep convolutional generative adversarial network for the inverse design of crystal structures. npj Comput. Mater. 7, 1–7 (2021).
Wang, H. et al. Graphgan: Graph representation learning with generative adversarial nets. In McIlraith, S. A. & Weinberger, K. Q. (eds.) Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, (AAAI-18), the 30th innovative Applications of Artificial Intelligence (IAAI-18), and the 8th AAAI Symposium on Educational Advances in Artificial Intelligence (EAAI-18), New Orleans, Louisiana, USA, February 2–7, 2018, 2508–2515 (AAAI Press, 2018). https://www.aaai.org/ocs/index.php/AAAI/AAAI18/paper/view/16611.
Cao, N. D. & Kipf, T. Molgan: An implicit generative model for small molecular graphs. ICML18 Workshop on Theoretical Foundations and Applicationsof Deep Generative Models, PMLR 80 (2018).
Li, Y., Vinyals, O., Dyer, C., Pascanu, R. & Battaglia, P. Learning deep generative models of graphs. arXiv preprint arXiv:1803.03324 (2018). https://arxiv.org/abs/1803.03324.
You, J., Liu, B., Ying, Z., Pande, V. & Leskovec, J. Graph convolutional policy network for goal-directed molecular graph generation. Adv. Neural Inform. Process. Syst. 31 (2018).
Romeo Atance, S., Viguera Diez, J., Engkvist, O., Olsson, S. & Mercado, R. De novo drug design using reinforcement learning with graph-based deep generative models. ChemRxiv (2021).
Mercado, R. et al. Graph Networks for Molecular Design. Machine Learning: Science and Technology 2 (2021).
Li, Y., Zhang, L. R. & ming Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 10 (2018).
Papamakarios, G., Nalisnick, E. T., Rezende, D. J., Mohamed, S. & Lakshminarayanan, B. Normalizing flows for probabilistic modeling and inference. J. Mach. Learn. Res. 22, 1–64 (2021).
Madhawa, K., Ishiguro, K., Nakago, K. & Abe, M. Graphnvp: An invertible flow model for generating molecular graphs. arXiv preprint arXiv:1905.11600 (2019).
Bengio, E., Jain, M., Korablyov, M., Precup, D. & Bengio, Y. Flow network based generative models for non-iterative diverse candidate generation. In Ranzato, M., Beygelzimer, A., Dauphin, Y. N., Liang, P. & Vaughan, J. W. (eds.) Advances in Neural Information Processing Systems 34: Annual Conference on Neural Information Processing Systems 2021, NeurIPS 2021, December 6–14, 2021, virtual, 27381–27394 (2021). https://proceedings.neurips.cc/paper/2021/hash/e614f646836aaed9f89ce58e837e2310-Abstract.html.
Frey, N. C., Gadepally, V. & Ramsundar, B. Fastflows: Flow-based models for molecular graph generation. arXiv preprint arXiv:2201.12419 (2022).
Wang, S., Guo, X. & Zhao, L. Deep Generative Model for Periodic Graphs. arXiv preprint arXiv:2201.11932 [cs] (2022).
Pham, T.-H., Qiu, Y., Zeng, J., Xie, L. & Zhang, P. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to covid-19 drug repurposing. Nat. Mach. Intell. 3, 247–257 (2021).
St. John, P. C. et al. Message-passing neural networks for high-throughput polymer screening. J. Chem. Phys. 150, 234111 (2019).
Feinberg, E. N., Joshi, E., Pande, V. S. & Cheng, A. C. Improvement in admet prediction with multitask deep featurization. J. Med. Chem. 63, 8835–8848 (2020).
Peng, Y. et al. Enhanced graph isomorphism network for molecular admet properties prediction. IEEE Access 8, 168344–168360 (2020).
Montanari, F., Kuhnke, L., Ter Laak, A. & Clevert, D.-A. Modeling physico-chemical admet endpoints with multitask graph convolutional networks. Molecules 25 (2020). https://www.mdpi.com/1420-3049/25/1/44.
Yaowen, G., Bowen, Z., Si, Z., Fengchun, Y. & Jiao, L. Predicting drug admet properties based on graph attention network. Data Anal. Knowl. Discov. 5, 76–85 (2021).
Xiong, J., Xiong, Z., Chen, K., Jiang, H. & Zheng, M. Graph neural networks for automated de novo drug design. Drug Discov. Today 26, 1382–1393 (2021).
Cheung, M. & Moura, J. M. Graph neural networks for covid-19 drug discovery. In Proc. IEEE International Conference on Big Data (Big Data), 5646–5648 (IEEE, 2020).
Yu, X., Lu, S., Guo, L., Wang, S.-H. & Zhang, Y.-D. Resgnet-c: a graph convolutional neural network for detection of covid-19. Neurocomputing 452, 592–605 (2021).
Kumar, A., Tripathi, A. R., Satapathy, S. C. & Zhang, Y.-D. Sars-net: Covid-19 detection from chest x-rays by combining graph convolutional network and convolutional neural network. Pattern Recogn. 122, 108255 (2022).
Nakata, M. & Shimazaki, T. PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry. J. Chem. Inform. Model. 57, 1300–1308 (2017). Publisher: American Chemical Society.
Lee, C.-K. et al. Transfer learning with graph neural networks for optoelectronic properties of conjugated oligomers. J. Chem. Phys. 154, 024906 (2021).
Lu, C. et al. Deep learning for optoelectronic properties of organic semiconductors. J. Phys. Chem. C 124, 7048–7060 (2020).
Pronobis, W., Schütt, K. T., Tkatchenko, A. & Müller, K.-R. Capturing intensive and extensive dft/tddft molecular properties with machine learning. Eur. Phys. J. B 91, 178 (2018).
Atz, K., Isert, C., Böcker, M. N., Jiménez-Luna, J. & Schneider, G. δ-quantum machine learning for medicinal chemistry. ChemRxiv (2021). https://chemrxiv.org/engage/chemrxiv/article-details/61c02f7e7f367e306759a0fd.
Nakata, M., Shimazaki, T., Hashimoto, M. & Maeda, T. PubChemQC PM6: data sets of 221 million molecules with optimized molecular geometries and electronic properties. J. Chem. Inform. Model. 60, 5891–5899 (2020). Publisher: American Chemical Society.
Gastegger, M., Schütt, K. T. & Müller, K.-R. Machine learning of solvent effects on molecular spectra and reactions. Chem. Sci. 12, 11473–11483 (2021).
Gastegger, M., McSloy, A., Luya, M., Schütt, K. T. & Maurer, R. J. A deep neural network for molecular wave functions in quasi-atomic minimal basis representation. J. Chem. Phys. 153, 044123 (2020).
Burkholz, R., Quackenbush, J. & Bojar, D. Using graph convolutional neural networks to learn a representation for glycans. Cell Rep. 35, 109251 (2021).
Li, S. et al. MutagenPred-GCNNs: a graph convolutional neural network-based classification model for mutagenicity prediction with data-driven molecular fingerprints. Interdiscip. Sci. Comput. Life Sci.13, 25–33 (2021).
Deng, D. et al. Xgraphboost: Extracting graph neural network-based features for a better prediction of molecular properties. J. Chem. Inform. Model. 61, 2697–2705 (2021).
Wang, F. et al. Graph attention convolutional neural network model for chemical poisoning of honey bees’ prediction. Sci. Bull. 65, 1184–1191 (2020).
Schweidtmann, A. M. et al. Graph neural networks for prediction of fuel ignition quality. Energy Fuels 34, 11395–11407 (2020).
Kim, Q., Ko, J.-H., Kim, S. & Jhe, W. Gcicenet: a graph convolutional network for accurate classification of water phases. Phys. Chem. Chem. Phys. 22, 26340–26350 (2020).
Ying, Z., Bourgeois, D., You, J., Zitnik, M. & Leskovec, J. Gnnexplainer: Generating explanations for graph neural networks. Adv. Neural Inform. Process. Syst. 32 (2019).
Sanchez-Lengeling, B. et al. Machine learning for scent: Learning generalizable perceptual representations of small molecules. arXiv preprint arXiv:1910.10685 (2019).
Yuan, Q., Szczypiński, F. T. & Jelfs, K. E. Explainable graph neural networks for organic cages. Digital Discov. 1, 127–138 (2022).
Park, C. et al. Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture. npj Comput. Mater. 7 (2021).
Hu, W. et al. Forcenet: A graph neural network for large-scale quantum calculations. arXiv preprint arXiv:2103.01436 (2021).
Haghighatlari, M. et al. Newtonnet: A newtonian message passing network for deep learning of interatomic potentials and forces. Digital Discov. (2022).
Unke, O. T. et al. Spookynet: Learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12 (2021). https://doi.org/10.1038/s41467-021-27504-0.
Li, Z. et al. Graph neural network based coarse-grained mapping prediction. Chem. Sci. 11, 9524–9531 (2020).
Wang, J. et al. Machine learning of coarse-grained molecular dynamics force fields. ACS Cent. Sci. 5, 755–767 (2019).
Husic, B. E. et al. Coarse graining molecular dynamics with graph neural networks. J. Chem. Phys. 153, 194101 (2020).
Westermayr, J. & Marquetand, P. Deep learning for uv absorption spectra with schnarc: first steps toward transferability in chemical compound space. J. Chem. Phys. 153, 154112 (2020).
Westermayr, J., Gastegger, M. & Marquetand, P. Combining schnet and sharc: the schnarc machine learning approach for excited-state dynamics. J. Phys. Chem. Lett. 11, 3828–3834 (2020).
Axelrod, S., Shakhnovich, E. & Gómez-Bombarelli, R. Excited state non-adiabatic dynamics of large photoswitchable molecules using a chemically transferable machine learning potential. Nat. Commun. 13, 1–11 (2022).
Tabor, D. P. et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 3, 5–20 (2018).
Liu, B. et al. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 3, 1103–1113 (2017).
Zheng, S., Rao, J., Zhang, Z., Xu, J. & Yang, Y. Predicting retrosynthetic reactions using self-corrected transformer neural networks. J. Chem. Inform. Model. 60, 47–55 (2020).
Lin, K., Xu, Y., Pei, J. & Lai, L. Automatic retrosynthetic route planning using template-free models. Chem. Sci. 11, 3355–3364 (2020).
Schwaller, P. et al. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 11, 3316–3325 (2020).
Segler, M. H. S. & Waller, M. P. Neural-symbolic machine learning for retrosynthesis and reaction prediction. Chem. Eur. J. 23, 5966–5971 (2017).
Segler, M. H. S. & Waller, M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 555, 604–610 (2018).
Jin, W., Coley, C. W., Barzilay, R. & Jaakkola, T. S. Predicting organic reaction outcomes with weisfeiler-lehman network. In Guyon, I. et al. (eds.) Adv. Neural Inform. Process. Syst. 30: Annual Conference on Neural Information Processing Systems 2017, December 4–9, 2017, Long Beach, CA, USA, 2607–2616 (2017). https://proceedings.neurips.cc/paper/2017/hash/ced556cd9f9c0c8315cfbe0744a3baf0-Abstract.html.
Coley, C. W. et al. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 10, 370–377 (2019).
Struble, T. J., Coley, C. W. & Jensen, K. F. Multitask prediction of site selectivity in aromatic c-h functionalization reactions. React. Chem. Eng. 5, 896–902 (2020).
Guan, Y. et al. Regio-selectivity prediction with a machine-learned reaction representation and on-the-fly quantum mechanical descriptors. Chem. Sci. 12, 2198–2208 (2021).
Nikitin, F., Isayev, O. & Strijov, V. Dracon: disconnected graph neural network for atom mapping in chemical reactions. Phys. Chem. Chem. Phys. 22, 26478–26486 (2020).
Do, K., Tran, T. & Venkatesh, S. Graph transformation policy network for chemical reaction prediction. In Teredesai, A. et al. (eds.) Proc. 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, KDD 2019, Anchorage, AK, USA, August 4–8, 2019, 750-760 (ACM, 2019). https://doi.org/10.1145/3292500.3330958.
Bradshaw, J., Kusner, M. J., Paige, B., Segler, M. H. S. & Hernández-Lobato, J. M. A generative model for electron paths. In Proc. 7th International Conference on Learning Representations, ICLR 2019, New Orleans, LA, USA, May 6-9, 2019 (OpenReview.net, 2019). https://openreview.net/forum?idr¯1x4BnCqKX.
Wen, M., Blau, S. M., Spotte-Smith, E. W. C., Dwaraknath, S. & Persson, K. A. Bondnet: a graph neural network for the prediction of bond dissociation energies for charged molecules. Chem. Sci. 12, 1858–1868 (2021).
St John, P. C., Guan, Y., Kim, Y., Kim, S. & Paton, R. S. Prediction of organic homolytic bond dissociation enthalpies at near chemical accuracy with sub-second computational cost. Nat. Commun. 11, 1–12 (2020).
Pattanaik, L., Ingraham, J. B., Grambow, C. A. & Green, W. H. Generating transition states of isomerization reactions with deep learning. Phys. Chem. Chem. Phys. 22, 23618–23626 (2020).
Grambow, C. A., Pattanaik, L. & Green, W. H. Deep learning of activation energies. J. Phys. Chem. Lett. 11, 2992–2997 (2020).
Dai, H., Li, C., Coley, C. W., Dai, B. & Song, L. Retrosynthesis prediction with conditional graph logic network. In Wallach, H. M. et al. (eds.) Advances in Neural Information Processing Systems 32: Annual Conference on Neural Information Processing Systems 2019, NeurIPS 2019, December 8–14, 2019, Vancouver, BC, Canada, 8870–8880 (2019). https://proceedings.neurips.cc/paper/2019/hash/0d2b2061826a5df3221116a5085a6052-Abstract.html.
Ishida, S., Terayama, K., Kojima, R., Takasu, K. & Okuno, Y. Prediction and interpretable visualization of retrosynthetic reactions using graph convolutional networks. J. Chem. Inform. Model. 59, 5026–5033 (2019).
Chen, S. & Jung, Y. Deep retrosynthetic reaction prediction using local reactivity and global attention. JACS Au 1, 1612–1620 (2021).
Yan, C. et al. Retroxpert: Decompose retrosynthesis prediction like A chemist. In Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M. & Lin, H. (eds.) Advances in Neural Information Processing Systems 33: Annual Conference on Neural Information Processing Systems 2020, NeurIPS 2020, December 6–12, 2020, virtual (2020). https://proceedings.neurips.cc/paper/2020/hash/819f46e52c25763a55cc642422644317-Abstract.html.
Somnath, V. R., Bunne, C., Coley, C. W., Krause, A. & Barzilay, R. Learning graph models for retrosynthesis prediction. In Ranzato, M., Beygelzimer, A., Dauphin, Y. N., Liang, P. & Vaughan, J. W. (eds.) Advances in Neural Information Processing Systems 34: Annual Conference on Neural Information Processing Systems 2021, NeurIPS 2021, December 6-14, 2021, virtual, 9405–9415 (2021). https://proceedings.neurips.cc/paper/2021/hash/4e2a6330465c8ffcaa696a5a16639176-Abstract.html.
Shi, C., Xu, M., Guo, H., Zhang, M. & Tang, J. A graph to graphs framework for retrosynthesis prediction. In Proc. 37th International Conference on Machine Learning, ICML 2020, 13–18 July 2020, Virtual Event, vol. 119 of Proceedings of Machine Learning Research, 8818–8827 (PMLR, 2020). http://proceedings.mlr.press/v119/shi20d.html.
Bartel, C. J. et al. A critical examination of compound stability predictions from machine-learned formation energies. npj Comput. Mater. 6, 1–11 (2020).
Park, C. W. & Wolverton, C. Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery. Phys. Rev. Mater. 4, 063801 (2020).
Jørgensen, P., Garijo Del Río, E., Schmidt, M. & Jacobsen, K. Materials property prediction using symmetry-labeled graphs as atomic position independent descriptors. Phys. Rev. B 100 (2019).
Noh, J., Gu, G., Kim, S. & Jung, Y. Uncertainty-quantified hybrid machine learning/density functional theory high throughput screening method for crystals. J. Chem. Inform. Model. 60, 1996–2003 (2020).
Pandey, S., Qu, J., Stevanović, V., St. John, P. & Gorai, P. Predicting energy and stability of known and hypothetical crystals using graph neural network. Patterns 2 (2021).
Jang, J., Gu, G. H., Noh, J., Kim, J. & Jung, Y. Structure-based synthesizability prediction of crystals using partially supervised learning. J. Am. Chem. Soc. 142, 18836–18843 (2020).
Li, X.-G. et al. Graph network based deep learning of bandgaps. J. Chem. Phys. 155, 154702 (2021).
Omprakash, P. et al. Graph representational learning for bandgap prediction in varied perovskite crystals. Comput. Mater. Sci. 196 (2021).
Na, G. S., Jang, S., Lee, Y.-L. & Chang, H. Tuplewise material representation based machine learning for accurate band gap prediction. J. Phys. Chem. A 124, 10616–10623 (2020).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Wang, R. et al. Combining crystal graphs and domain knowledge in machine learning to predict metal-organic frameworks performance in methane adsorption. Microporous Mesoporous Mater. 331 (2022).
Wang, R., Zhong, Y., Bi, L., Yang, M. & Xu, D. Accelerating discovery of metal-organic frameworks for methane adsorption with hierarchical screening and deep learning. ACS Appl. Mater. Interfaces 12, 52797–52807 (2020).
Wilmer, C. E. et al. Large-scale screening of hypothetical metal–organic frameworks. Nat. Chem. 4, 83–89 (2012).
Gu, G. et al. Practical deep-learning representation for fast heterogeneous catalyst screening. J. Phys. Chem. Lett. 11, 3185–3191 (2020).
Goodall, R. E. A. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat. Commun. 11, 6280 (2020).
Curtarolo, S. et al. AFLOW: an automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 58, 218–226 (2012).
Dai, M., Demirel, M., Liang, Y. & Hu, J.-M. Graph neural networks for an accurate and interpretable prediction of the properties of polycrystalline materials. npj Comput. Mater. 7 (2021).
Lu, S. et al. Coupling a crystal graph multilayer descriptor to active learning for rapid discovery of 2D ferromagnetic semiconductors/half-metals/metals. Adv. Mater. 32, 2002658 (2020).
Catlow, R. Defects and disorder, In Crystalline and Amorphous Solids, vol. 418 (Springer Science & Business Media, 2012).
Chen, C., Zuo, Y., Ye, W., Li, X. & Ong, S. P. Learning properties of ordered and disordered materials from multi-fidelity data. Nat. Comput. Sci. 1, 46–53 (2021).
Wang, Z., Han, Y., Cai, J., Wu, S. & Li, J. DeepTMC: A deep learning platform to targeted design doped transition metal compounds. Energy Storage Mater. 45, 1201–1211 (2022).
Frey, N. C., Akinwande, D., Jariwala, D. & Shenoy, V. B. Machine learning-enabled design of point defects in 2D materials for quantum and neuromorphic information processing. ACS Nano 14, 13406–13417 (2020).
Cian, L. et al. Atomistic Graph Neural Networks for metals: application to bcc iron. arXiv preprint arXiv:2109.14012 [cond-mat] (2021).
Bapst, V. et al. Unveiling the predictive power of static structure in glassy systems. Nat. Phys. 16, 448–454 (2020).
Wang, Q. & Zhang, L. Inverse design of glass structure with deep graph neural networks. Nat. Commun. 12, 1–11 (2021).
Swanson, K., Trivedi, S., Lequieu, J., Swanson, K. & Kondor, R. Deep learning for automated classification and characterization of amorphous materials. Soft Matter 16, 435–446 (2020).
Park, J. et al. Prediction and interpretation of polymer properties using the graph convolutional network. ACS Polym. Au (2022).
Zeng, M. et al. Graph convolutional neural networks for polymers property prediction. arXiv preprint arXiv:1811.06231 (2018).
Deringer, V. L., Caro, M. A. & Csányi, G. A general-purpose machine-learning force field for bulk and nanostructured phosphorus. Nat. Commun. 11, 5461 (2020).
Wang, Z. et al. Symmetry-adapted graph neural networks for constructing molecular dynamics force fields. Science China: Phys., Mech. Astron. 64 (2021).
Raza, A., Sturluson, A., Simon, C. M. & Fern, X. Message passing neural networks for partial charge assignment to metal–organic frameworks. J. Phys. Chem. C 124, 19070–19082 (2020).
Malik, S., Goodall, R. & Lee, A. Predicting the outcomes of material syntheses with deep learning. Chem. Mater. 33, 616–624 (2021).
Tremouilhac, P. et al. Chemotion eln: an open source electronic lab notebook for chemists in academia. J. Cheminform. 9, 1–13 (2017).
Brandt, N. et al. Kadi4mat: a research data infrastructure for materials science. Data Sci. J. 20 (2021).
Xie, T., Fu, X., Ganea, O.-E., Barzilay, R. & Jaakkola, T. Crystal diffusion variational autoencoder for periodic material generation. International Conference on Learning Representations (2022).
Fey, M. & Lenssen, J. E. Fast graph representation learning with pytorch geometric. arXiv preprint arXiv:1903.02428 (2019).
Wang, M. et al. Deep graph library: a graph-centric, highly-performant package for graph neural networks. arXiv preprint arXiv:1909.01315 (2019). https://arxiv.org/abs/1909.01315.
Reiser, P., Eberhard, A. & Friederich, P. Graph neural networks in tensorflow-keras with raggedtensor representation (kgcnn). Softw. Impacts 9, 100095 (2021).
Gasteiger, J. et al. How do graph networks generalize to large and diverse molecular systems? arXiv preprint arXiv:2204.02782 (2022).
Krenn, M. et al. Selfies and the future of molecular string representations. Patterns 3, 10:100588 (2022).
Brammer, J. C. et al. Tucan: A molecular identifier and descriptor applicable to the whole periodic table from hydrogen to oganesson. Research Square preprint (2022).
Yao, Z. et al. Inverse design of nanoporous crystalline reticular materials with deep generative models. Nat. Mach. Intell. 3, 76–86 (2021).
Friederich, P., Krenn, M., Tamblyn, I. & Aspuru-Guzik, A. Scientific intuition inspired by machine learning-generated hypotheses. Mach. Learn. Sci. Technol. 2, 025027 (2021).
Krenn, M. et al. On scientific understanding with artificial intelligence. Nat Rev Phys (2022). https://doi.org/10.1038/s42254-022-00518-3.
Lavin, A. et al. Simulation intelligence: towards a new generation of scientific methods. arXiv preprint arXiv:2112.03235 (2021).
Pocha, A., Danel, T., Podlewska, S., Tabor, J. & Maziarka, L. Comparison of atom representations in graph neural networks for molecular property prediction. In Proc. International Joint Conference on Neural Networks, IJCNN 2021, Shenzhen, China, July 18–22, 2021, 1–8 (IEEE, 2021). https://doi.org/10.1109/IJCNN52387.2021.9533698.
Liao, R., Zhao, Z., Urtasun, R. & Zemel, R. Lanczosnet: Multi-scale deep graph convolutional networks. In Proc. International Conference on Learning Representations (2019). https://openreview.net/forum?idB¯kedznAqKQ.
Bruna, J., Zaremba, W., Szlam, A. & LeCun, Y. Spectral networks and locally connected networks on graphs. In International Conference on Learning Representations (ICLR2014), CBLS (2013).
Henaff, M., Bruna, J. & LeCun, Y. Deep convolutional networks on graph-structured data. arXiv:1506.05163 (2015). http://arxiv.org/abs/1506.05163.
Levie, R., Monti, F., Bresson, X. & Bronstein, M. M. Cayleynets: graph convolutional neural networks with complex rational spectral filters. IEEE Trans. Signal Process. 67, 97–109 (2019).
Schlichtkrull, M. et al. Modeling relational data with graph convolutional networks. In Gangemi, A. et al. (eds.) The Semantic Web, 593–607 (Springer International Publishing, Cham, 2018).
Niepert, M., Ahmed, M. & Kutzkov, K. Learning convolutional neural networks for graphs. In Balcan, M. F. & Weinberger, K. Q. (eds.) Proceedings of The 33rd International Conference on Machine Learning, vol. 48 of Proceedings of Machine Learning Research, 2014–2023 (PMLR, New York, New York, USA, 2016). https://proceedings.mlr.press/v48/niepert16.html.
Wang, X. et al. Molecule property prediction based on spatial graph embedding. J. Chem. Inform. Model. 59, 3817–3828 (2019).
Yadati, N. Neural message passing for multi-relational ordered and recursive hypergraphs. In Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M. & Lin, H. (eds.) Advances in Neural Information Processing Systems, vol. 33, 3275–3289 (Curran Associates, Inc., 2020). https://proceedings.neurips.cc/paper/2020/file/217eedd1ba8c592db97d0dbe54c7adfc-Paper.pdf.
Strathmann, H., Barekatain, M., Blundell, C. & Veličković, P. Persistent message passing. In Proc. ICLR 2021 Workshop on Geometrical and Topological Representation Learning (2021). https://openreview.net/forum?id=HhOJZT--N23.
Kim, Y. et al. Molnet: A chemically intuitive graph neural network for prediction of molecular properties. Chem. Asian J. (2022).
Coors, B., Condurache, A. P. & Geiger, A. Spherenet: Learning spherical representations for detection and classification in omnidirectional images. In Ferrari, V., Hebert, M., Sminchisescu, C. & Weiss, Y. (eds.) Computer Vision – ECCV 2018, 525–541 (Springer International Publishing, Cham, 2018).
Brody, S., Alon, U. & Yahav, E. How attentive are graph attention networks? International Conference on Learning Representations (2021).
Thekumparampil, K. K., Oh, S., Wang, C. & Li, L.-J. Attention-based graph neural network for semi-supervised learning (2018). https://openreview.net/forum?idr¯Jg4YGWRb.
Withnall, M., Lindelöf, E., Engkvist, O. & Chen, H. Building attention and edge message passing neural networks for bioactivity and physical–chemical property prediction. J. Cheminform. 12, 1–18 (2020).
Xinyi, Z. & Chen, L. Capsule graph neural network. In International Conference on Learning Representations (2019). https://openreview.net/forum?idB¯yl8BnRcYm.
Busbridge, D., Sherburn, D., Cavallo, P. & Hammerla, N. Y. Relational graph attention networks. arXiv preprint arXiv:1904.05811 (2019). http://arxiv.org/abs/1904.05811.
Tang, B. et al. A self-attention based message passing neural network for predicting molecular lipophilicity and aqueous solubility. J. Cheminform. 12, 1–9 (2020).
Li, Z., Yang, S., Song, G. & Cai, L. Conformation-guided molecular representation with hamiltonian neural networks. In Proc. International Conference on Learning Representations (2021). https://openreview.net/forum?idq¯-cnWaaoUTH.
Kondor, R., Lin, Z. & Trivedi, S. Clebsch-gordan nets: a fully fourier space spherical convolutional neural network. In Bengio, S. et al. (eds.) Advances in Neural Information Processing Systems 31: Annual Conference on Neural Information Processing Systems 2018, NeurIPS 2018, December 3–8, 2018, Montréal, Canada, 10138-10147 (2018). https://proceedings.neurips.cc/paper/2018/hash/a3fc981af450752046be179185ebc8b5-Abstract.html.
Brandstetter, J., Hesselink, R., van der Pol, E., Bekkers, E. & Welling, M. Geometric and physical quantities improve E(3) equivariant message passing. International Conference on Learning Representations (2022).
Fuchs, F., Worrall, D., Fischer, V. & Welling, M. Se(3)-transformers: 3d roto-translation equivariant attention networks. In Larochelle, H., Ranzato, M., Hadsell, R., Balcan, M. & Lin, H. (eds.) Advances in Neural Information Processing Systems, vol. 33, 1970–1981 (Curran Associates, Inc., 2020). https://proceedings.neurips.cc/paper/2020/file/15231a7ce4ba789d13b722cc5c955834-Paper.pdf.
Cohen, T. & Welling, M. Group equivariant convolutional networks. In Balcan, M. F. & Weinberger, K. Q. (eds.) Proc. 33rd International Conference on Machine Learning, vol. 48 of Proceedings of Machine Learning Research, 2990–2999 (PMLR, New York, New York, USA, 2016). https://proceedings.mlr.press/v48/cohenc16.html.
Lu, Y. et al. Cnn-g: Convolutional neural network combined with graph for image segmentation with theoretical analysis. IEEE Trans. Cognit. Dev. Syst. 13, 631–644 (2021).
Bekkers, E. J. B-spline cnns on lie groups. In Proc. International Conference on Learning Representations (2020). https://openreview.net/forum?idH¯1gBhkBFDH.
Ying, R. et al. Hierarchical graph representation learning with differentiable pooling. In Proc. 32nd International Conference on Neural Information Processing Systems, NIPS’18, 4805–4815 (Curran Associates Inc., Red Hook, NY, USA, 2018).
Diehl, F. Edge contraction pooling for graph neural networks. arXiv preprint arXiv:1905.10990 (2019). https://arxiv.org/abs/1905.10990.
Gao, H. & Ji, S. Graph u-nets. In Chaudhuri, K. & Salakhutdinov, R. (eds.) Proc. 36th International Conference on Machine Learning, vol. 97 of Proceedings of Machine Learning Research, 2083–2092 (PMLR, 2019). https://proceedings.mlr.press/v97/gao19a.html.
Gao, X., Xiong, H. & Frossard, P. ipool—information-based pooling in hierarchical graph neural networks. IEEE Trans. Neural Netw. Learn. Syst. PP (2021).
Ma, Y., Wang, S., Aggarwal, C. C. & Tang, J. Graph convolutional networks with eigenpooling. In Proc. 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, KDD’19, 723-731 (Association for Computing Machinery, New York, NY, USA, 2019). https://doi.org/10.1145/3292500.3330982.
Bergerhoff, G., Hundt, R., Sievers, R. & Brown, I. D. The inorganic crystal structure data base. J. Chem. Inform. Comput. Sci. 23, 66–69 (1983).
Pickard, C. J. AIRSS data for carbon at 10GPa and the C+N+H+O system at 1GPa (2020).
NREL Materials Database (NRELMatDB). https://materials.nrel.gov/.
Tran, K. & Ulissi, Z. W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 1, 696–703 (2018).
Kononova, O. et al. Text-mined dataset of inorganic materials synthesis recipes. Sci. Data 6, 203 (2019).
Castelli, I. E. et al. New cubic perovskites for one- and two-photon water splitting using the computational materials repository. Energy Environ. Sci. 5, 9034–9043 (2012).
Chung, Y. G. et al. Advances, updates, and analytics for the computation-ready, experimental metal–organic framework database: CoRE MOF 2019. J. Chem. Eng. Data 64, 5985–5998 (2019).
Dragoni, D., Daff, T. D., Csányi, G. & Marzari, N. Achieving DFT accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron. Phys. Rev. Mater. 2, 013808 (2018).
Deringer, V. L., Pickard, C. J. & Csányi, G. Data-driven learning of total and local energies in elemental boron. Phys. Rev. Lett. 120, 156001 (2018).
Haastrup, S. et al. The computational 2D materials database: high-throughput modeling and discovery of atomically thin crystals. 2D Materials 5, 042002 (2018).
Rasmussen, F. A. & Thygesen, K. S. Computational 2D materials database: electronic structure of transition-metal dichalcogenides and oxides. The J. Phys. Chem. C 119, 13169–13183 (2015).
Nazarian, D., Camp, J. S. & Sholl, D. S. A comprehensive set of high-quality point charges for simulations of metal–organic frameworks. Chem. Mater. 28, 785–793 (2016).
Gao, Y. et al. Gnes: learning to explain graph neural networks. In Proc. IEEE International Conference on Data Mining (ICDM), 131–140 (IEEE, 2021).
Acknowledgements
We acknowledge support by the Federal Ministry of Education and Research (BMBF) Grant No. 01DM21001B (German-Canadian Materials Acceleration Center) and Grant No. 01DM21002A (FLAIM). We would like to thank the Federal Ministry of Economics and Energy under Grant No. KK5139001AP0. We acknowledge funding from the Klaus Tschira Stiftung gGmbH (SIMPLAIX project 1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.E., L.T., C.S., H.M., C.v.H. worked on GNN principles and generative models, M.N. and C.Z. worked on applications of GNNs in chemistry, H.S. and T.S. worked on GNN applications in materials science. P.F. and P.R. designed the main structure of the manuscript. All authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Communications Materials thanks Linjiang Chen, Johannes Gasteiger and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Reinhard Maurer and Aldo Isidori.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reiser, P., Neubert, M., Eberhard, A. et al. Graph neural networks for materials science and chemistry. Commun Mater 3, 93 (2022). https://doi.org/10.1038/s43246-022-00315-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-022-00315-6
This article is cited by
-
Advancing material property prediction: using physics-informed machine learning models for viscosity
Journal of Cheminformatics (2024)
-
A general model for predicting enzyme functions based on enzymatic reactions
Journal of Cheminformatics (2024)
-
Cell Painting-based bioactivity prediction boosts high-throughput screening hit-rates and compound diversity
Nature Communications (2024)
-
Automatic feature engineering for catalyst design using small data without prior knowledge of target catalysis
Communications Chemistry (2024)
-
A network analysis-based framework to understand the representation dynamics of graph neural networks
Neural Computing and Applications (2024)
