
Abstract
In piezocatalysis the polarization field found in piezoelectric materials enables and enhances catalytic redox reactions. Here, we explore piezocatalytic dye degradation through transparent glass–ceramics containing piezoelectric crystals. 30SiO2–35Li2O–35Nb2O5 (in mol%) glass–ceramics containing varying amounts of LiNbO3 crystallites were fabricated by melt-quenching, followed by heat-treatment at a crystallization temperature of 650 °C for 2, 3 and 6 hours. During piezocatalysis, the 2 hour heat-treated sample showed up to 90% degradation of methylene blue dye within 150 min of ultrasonication, with no significant change in performance after three piezocatalysis cycles. This sample showed promising activity for degrading cationic and neutral dyes, and is optically transparent. This work demonstrates that transparent ferroelectric glass–ceramics are promising for water-cleaning applications by piezocatalysis.
Similar content being viewed by others


Introduction
Glasses are commonly used material in our daily life such as in windows, display panels, kitchenwares, and optical fibers, etc.1. Glasses are known for their excellent transparency, tunable chemical composition, easy fabrication, and flexibility of shape and size2. Glass–ceramics consist of two regions: the glassy matrix and the crystals. These are commonly formed from their parent glasses by providing either one step (simultaneous occurrence of crystals nucleation and growth) or two step (nucleation followed by growth of crystals) heat-treatment3. The improved optical, luminescence properties, mechanical strength, physical, chemical stability, bioactivity, corrosion strength, and temperature resistance were noticed due to crystalline phase embedded in glassy matrix4,5,6. Similarly, some unique properties such as photocatalysis7,8,9, antibacterial activity7,8,9 and cancer treatment capability through hypherthermia treatment10, etc., are also induced through selected crystalline phase in glass–ceramics.
Recently, piezocatalysis has been evolved as a potential process for water cleaning applications11,12. Piezocatalysis process makes use of piezoelectric materials to generate polarization field under applied mechanical deformation13. The polarization field (piezoelectricity) forces the free holes (h+)/electrons (e−) to move in opposite direction toward opposite charge polarity of polarization field and thus increases their separation14. Moreover, the piezoelectric polarization induces favorable band bending, which makes it easy for free holes (h+)/electrons (e−) to participate in catalytic redox reactions15. Piezocatalysis phenomenon has been generally observed in piezoelectric materials such as BaTiO316,17, ZnO18, MoS219, LiNbO320, ZnSnO315, etc. The piezocatalytic activity has been generally reported using powder particles21,22 or nanostructures such as nanorods, nanosheets, etc.23,24. However, the powder or nanostructures based piezocatalysts are difficult to recover and reused25. This limitation has been removed by using poled ceramic pellets for piezocatalytic dye degradation26. However, the poling needs high electric field, which is not a safe practical solution. Our recent article pointed out the fact that the unpoled micron-sized powder sample of LiNbO3 could provide the comparative piezocatalytic performance as observed in other reported piezocatalysts20. In this article, it was mentioned that the individual domain can perform its own piezocatalysis20. Thus, an unpoled ceramics can also provide promising piezocatalytic activity. If an unpoled sample can perform piezocatalysis, then the unpoled glass–ceramics containing piezoelectric crystals should also be examined for piezocatalytic dye degradation applications. Glass–ceramics containing piezoelectric crystals are widely reported in dielectric, optical, and electro-optical applications27. However, there is no report on piezocatalysis through glass–ceramics containing piezoelectric crystals. Glass–ceramics provide many advantages over polycrystalline ceramics. Glass–ceramics can be fabricated in large sizes and in variety of shapes. Physical/chemical properties of glass–ceramics can be tuned as per applications, as the crystallization can be controlled using careful selection of heat-treatment time and temperature4,6,7. Due to these unique characteristics, glass–ceramics have large number of applications in diverse scientific fields.
So far, no glass–ceramics have been explored for piezocatalytic applications. Thus, herein, transparent 30SiO2–35Li2O–35Nb2O5 (in mol%) glass–ceramics containing piezoelectric LiNbO3 crystals is explored for dye degradation using piezocatalysis process. The results showed that these glass–ceramics possessed promising piezocatalytic activity for degrading cationic and neutral dyes. This works demonstrates that ferroelectric glass–ceramics are promising for water-cleaning applications by piezocatalysis.
Results and discussion
XRD and Raman analysis
The obtained XRD patterns of AQ-glass, HT-2h, HT-3h, and HT-6h samples are presented in Fig. 1a. The absence of any sharp crystalline peak indicated the amorphous nature of AQ-sample. The sharp peaks observed in all the heat-treated samples (HT-2h, HT-3h, and HT-6h) were well matched with LiNbO3 reference (JCPDS File No. 01-078-0250). Thus, it is clear from the XRD patterns that all the heat-treated samples possessed LiNbO3 crystalline phase. Figure 1b represents Raman spectra of AQ-glass, HT-2h, HT-3h, and HT-6h samples. In case of the AQ-glass sample, no sharp band was observed, though two weak broad bands were present at ~230 and 650 cm−1, which are generally observed modes in niobate glasses due to vibrations of NbO6 type of octahedron28,29. All the heat-treated samples showed sharp Raman modes at 155, 238, 261, 274, 327, 365, 432, and 630 cm−1. All these Raman modes belongs to LiNbO3 crystalline phase in the heat-treated samples, as these modes were closely matched with Raman modes observed in LiNbO3 crystalline materials reported in various reports30,31. The modes present at 155, 238, 327, 365, and 432 cm−1 are optical transverse E(TO) phonon modes of crystalline LiNbO330,31. The remaining bands at 261, 274, and 630 cm−1 positions are optical transverse A1(TO) phonon modes of crystalline LiNbO330,31. It is to be noted that the weak broad bands were not visible in all the heat-treated samples, which may be due to high intensity of crystalline modes of LiNbO3. Hence, Raman spectroscopy gave another proof of presence of LiNbO3 crystalline phase in the heat-treated (HT-2h, HT-3h, and HT-6h) samples.

a XRD patterns of AQ-glass, HT-2h, HT-3h, and HT-6h samples indicating the amorphous nature of AQ-glass and the presence of crystalline LiNbO3 phase in HT-2h, HT-3h, and HT-6h samples. b Raman spectra of AQ-glass, HT-2h, HT-3h, and HT-6h samples support XRD results evidenced the presence of crystalline LiNbO3 phase in HT-2h, HT-3h, and HT-6h samples.
SEM and TEM results
The observed surface morphology of present investigated samples is shown in Fig. 2. Figure 2a belongs to AQ-glass sample, where only a few and very small sized crystallites of LiNbO3 can be observed. It is worth-mentioning here that the XRD pattern and Raman spectrum of AQ-glass showed amorphous nature (i.e., no crystallite phase embedded in the glass matrix). However, SEM micrographs clearly indicated a presence of very few small sized LiNbO3 particles, which were not detected in XRD and Raman spectrum due to their less counts and small sizes. On the other side, the extensive LiNbO3 crystallites on the surface can be observed in all the heat-treated samples (HT-2h, HT-3h, and HT-6h). HT-2h sample showed uniformly distributed small sized LiNbO3 crystals (Fig. 2b and its inset). HT-3h sample mainly showed leaf type of morphology (Fig. 2c, d). The crystallites size was large, mostly in the range of 10–50 μm. The HT-6h sample showed leaf type and moon type of morphology as depicted in Fig. 2e, f. The moon-type of morphology is basically the aggregates of small sized particles (Fig. 2g, h). Moreover, the number of crystals were also highest in this sample. The crystallites sizes were also in the range of 30–50 μm. Thus, SEM micrographs clearly evidenced the presence of extensive LiNbO3 crystallites on the surface of all heat-treated samples (HT-2h, HT-3h, and HT-6h). Also, these micrographs indicated the growth of particles with increase in heat-treatment time from 2 to 6 h. Figure 3a shows TEM micrographs of the HT-2h sample. It confirmed the existence of nanosized crystals (shown by dark region) in the bulk of HT-2h glass–ceramic matrix (shown by light region) in addition to micron-sized crystals observed on its surface in SEM micrographs. Thus, it can be concluded that the LiNbO3 crystallites were present at both sites, i.e., at surface and in bulk of the heat-treated samples. Figure 3b shows the d-spacing of crystalline phase and the inset present the selective area electron diffraction (SAED) pattern obtained for HT-2h sample. The observed d-spacing value of 3.7 Å was correspond to (012) plane of LiNbO3 crystal. SAED pattern shows the concentric circles of (012) and (104) diffractions planes of crystalline LiNbO3.

SEM micrographs obtained for a AQ-glass, b HT-2h, c, d HT-3h, and e–h HT-6h samples showing the presence of large number of LiNbO3 crystallites on the surface of HT-2h, HT-3h, and HT-6h samples.

a, b TEM micrographs, d-spacing, and SAED pattern of the HT-2h sample showing the existence of large number of LiNbO3 crystallites in the bulk of HT-2h, HT-3h, and HT-6h samples.
Transmittance and absorbance spectra
The transmittance and absorption spectra of all investiagted samples are presented in Fig. 4a, b, respectively. As depicted from the Fig. 4a, the transparency decreased from the AQ-glass to the HT-6h sample. The inset of the Fig. 4b provides the photographs of all the samples. The photographs clealry shows that the HT-6h sample is nearly opaque. The decrease in transparency of the AQ-glass to that of the HT-6h sample was due to increase in number and growth of LiNbO3 crystallites from AQ-glass to HT-6h sample, due to which more scattering of light ocurred. The attenuation in light intensity through the medium is majorly caused by atomic scattering32. If the particle size is less than the typical visible light wavelength, then Rayleigh scattering occurs, which can be quantified by using following Eq. (1)32.
where θ and L represent scattering angle and distance from scattering centers, respectively. I0 and I indicate the initial light intensity and transmitted light intensity, respectively. M represents the ratio between refractive indices of particle to that of surrounding medium and r represents the radius of particle. This equation clearly shows that transmitted light (I) mainly depends upon \(\frac{r}{\lambda }\) ratio. Smaller this \(\frac{r}{\lambda }\) ratio, more the light will be transmitted. In the present case, the particle size majorly less than 0.4 μm range in case of HT-2h sample (see the inset of Fig. 2b, which is less than or equal to the typical wavelength of visible light (400 nm or 0.4 μm). Thus, the sample preserved adequate transparency. However, in case of HT-3h and HT-6h samples, the sizes of majority of the particles lie in 10–50 μm and 30–50 μm range, respectively, which is greater than the typical wavelength of visible light (400 nm or 0.4 μm), due to which these samples showed more scattering and were less transparent. It is to be noted that there is no requirement of transparency for piezocatalysis applications. However, glasses are known for transparency and these have big market due to their unique transparency. For this reason, the control of transparency has given much attention in this work. Figure 4b shows the occurrence of fundamental absorption edge in all samples under study. This absorption edge originates due to transition of a valence electron from an oxygen ion present in the glass network to an excited state33. In absorption spectra, the samples HT-2h, and HT-3h showed no significant change in the absorbance edge as compared to that of AQ-glass sample. However, the sample HT-6h showed a significant blue shift in the absorbance edge than that of other samples. This blue shift was mainly due to decrease in non-bridging oxygens (NBOs) in HT-6h sample as compared to AQ-glass. This statement was verified using XPS O1s scan as discussed later in the paper. The NBOs possessed more basicity than that of BOs34. Thus, the decrease in NBOs decreased the oxygen ions’ ionicity and consequently increase the band gap by lowering the top of valence band. Due to this reason, the heat-treated samples showed blue shift in absorbance edge. Moreoever, the absorbance hump at 500 nm can be observed in all the heat-treated samples, which may be originated due to excitonic behavior. These exciton states generally exist below the conduction band edge, thus the absorption due to excitonic states is mostly observed close to but below the fundamental absorption edge as in the present case35.

a Transmittance spectra of AQ-glass, HT-2h, HT-3h, and HT-6h samples indicating the decrease in transparency from AQ-glass to the HT-6h sample and b absorbance spectra of AQ-glass, HT-2h, HT-3h, and HT-6h samples showing a significant blue shift in the absorbance edge of HT-6h sample than that of AQ-glass, HT-2h, and HT-3h, samples.
XPS analysis
In glass matrix, oxygen (O) element is present in abundant amount36. O element can occupy “bridging” (BO) or “nonbridging” (NBO) sites36. BOs connect two SiO4 tetrahedra through Si–O–Si covalent bond36. NBOs are created when the network modifiers (Li2O, Na2O, K2O, etc.) are added into glass system37. NBOs connect SiO4 framework to network modifying cations as Si–O− Li+, and Si–O− Nb+ ionic bonds38. Increasing content of network modifiers results in increase in NBOs and decrease in BOs37. In order to quantify BOs and NBOs, the XPS O1s scan was performed on AQ-glass and HT-6h sample, which is shown in Fig. 5a, b, resperctively. The O1s curve was fitted using the Guassian fiiting method and Sherley background. Two fitted peaks can be seen in both samples, one at ~532.9 and the other at ~531 eV. The peak at ~532.9 eV was observed due to BO contribution and the peak at ~531 eV was observed due to NBO contribution39,40. In AQ sample, BO peak was very weak as compared to NBOs. This is mainly due to high Li2O network modifier content in the AQ-sample. Moreover, it is also reported in the literature that Nb2O5 when present in more than 15 mol% can act as both; i.e., network former with smallest structural units of NbO6 octahedron and network modifier41. As Nb2O5 content is 35 mol% in the AQ-glass, thus as a network modifier, Nb2O5 helps in increasing more NBOs in the AQ-glass. On the other side, BO peak intensity was increased and NBO peak intensity was decreased in HT-6h sample. This clearly indicated the decrease in NBOs in HT-6h sample. This was caused due to LiNbO3 crystallites formation by consumption of NBOs and network modifier ions, due to which BOs increased and NBOs decreased. The decreased NBO/BO ratio in case of HT-6h sample in comparison to AQ-glass can be easily seen in Table 1, which indicated the superior stability of HT-6h glass–ceramic than that of as quenched AQ-glass.

a XPS O1s scan obtained for AQ-glass, and b XPS O1s scan obtained for HT-6h samples. XPS O1s scan clearly shows the decrease in NBO/BO ratio in case of HT-6h sample in comparison to AQ-glass.
Piezocatalysis dye degradation results
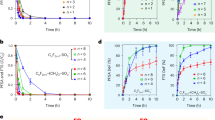
Figure 6a–d shows the results associated with piezocatalytic MB dye degradation experiments using samples under consideration. Figure 6a shows the decrease in characteristic peak intensity of MB dye using the HT-2h sample under ultrasonication for 150 min, which clearly indicated the promising piezocatalytic capability of HT-2h sample. The unknown concentration values corresponding to peak intensity values could be obtained from calibration curve, which is shown in the Supplementary Fig. S1. The MB dye degradation under ultrasonication without and with all samples under study (AQ-glass, HT-2h, HT-3h, and HT-6h) are presented in terms of \(\frac{{\mathrm{C}}}{{{\mathrm{C}}_0}}\) vs. t plots in Fig. 6b. MB dye solution under 150 min ultrasonication without using any sample showed ~15% degradation of MB dye. On the other side, the samples AQ-glass, HT-2h, HT-3h, and HT-6h showed ~33%, 77%, 79%, and 90% degradation of MB dye within 150 min of ultrasonication, respectively. Thus, the degradation performance was greatly improved in case of the heat-treated samples (HT-2h, HT-3h, and HT-6h) as compared to that of AQ-glass under ultrasonication. Generally, the first order kinetics is followed by these catalysis experiments, which is given in the Eq. (2)16,24.

a Decreasing intensity of characteristic peak of MB dye while performing piezocatalysis using HT-2h sample. b, c Piezocatalysis results in terms of \(\frac{{\mathrm{C}}}{{{\mathrm{C}}_0}}\) vs. time plots and the kinetic rate of dye degradation obtained using all samples under study, and d repeatability of piezocatalytic capability of HT-2h sample. All results indicating the promising piezocatalytic dye degradation capability of HT-2h sample.
The rate constant “k” was obtained by calculating the slope of \({\mathrm{ln}}\frac{{\mathrm{C}}}{{{\mathrm{C}}_0}}\) vs. t plots The calculated value of k was 0.00108, 0.00267, 0.00938, 0.00966, and 0.01281 min−1 for MB dye (without sample), AQ-glass, HT-2h, HT-3h, and HT-6h samples, respectively. Thus, the k-value was increased from AQ-glass to HT-6h sample. Though, the piezocatalytic performance of HT-6h was best among all the samples understudy, however this sample is nearly opaque. On the basis of adequate transparency and promising piezocatalytic performance, the sample HT-2h is an optimum choice. To test the repeatability, the piezocatalytic performance of sample HT-2h was observed over the first three cycles. No significant change was found in the piezocatalytic performance even after third cycle indicating its promising repeatable performance.
In order to reveal the mechanism behind the piezocatalysis through the heat-treated samples, the active species were identified by performing radicals trapping experiments. Various radicals such as holes (h+), hydroxyl radicals (˙OH), electrons (e−), and superoxide radicals (˙\({\rm{O}}_{2}^{-}\)) were scavenged/trapped during piezocatalytic experiments by adding ethylenediaminetetraacetic acid (EDTA), isopropanol (IPA), dimethyl sulphoxide (DMSO), and benzoquinone (BQ) scavengers, respectively42,43,44. The arrest of active radical using scavengers leads to decrease of percentage dye degradation. Figure 7a depicts that the percentage dye degradation was reduced after adding scavengers in this order: DMSO (e−) > BQ (˙\({\rm{O}}_{2}^{-}\)) > IPA (˙OH) > EDTA (h+). Thus, the electrons were main active species. The remaining radicals affected the piezocatalytic dye degradation performance in this order: ˙\({\rm{O}}_{2}^{-}\) > ˙OH > h+. In order to check the universal nature of HT-2h sample, the piezocatalysis dye degradation of two more dyes named Rhodamine B (RB dye, neutral dye) and methyl orange (MO dye, negatively charged dye) was performed in addition to MB dye (positive charged dye) and the percentage dye degradation (see Fig. 7b). The percentage dye degradation was found to 77%, 73%, and 32% in case of MB, RB, and MO dyes, respectively. The performance of HT-2h sample was found to be more promising in case of cationic and neutral dyes. Moreover, the power of ultrasonication is an important factor in case of piezocatalysis. Figure 7c clearly showed that the decrease in power from 70 to 50 W decreased the percentage dye degradation from 77 to 35%. The decreasing power decreased the effective compressive stress on the sample, due to which piezocatalysis decreased.

The piezocatalytic dye degradation (%) performance of HT-2h sample using a various scavengers, b various dyes, and c different ultrasonication power.
Piezocatalysis in ceramics is generally showed in terms of piezoelectric coefficient (d33) value. However, in the present case, all the samples (AQ-glass, HT-2h, HT-3h, and HT-6h) showed the overall piezoelectric coefficient (d33) is zero as attempted using d33 meter. Besides zero piezoelectric coefficient, the heat-treated glass–ceramics samples showed promising piezocatalysis. The only possibility exists is the existence of local piezoelectricity in glass–ceramics samples. The presence of local piezoelectricity can be viewed in terms of change of surface potential45,46. The surface potential of AQ-glass and HT-2h samples measured using KPFM is presented in Fig. 8a, b. The high surface potential was clearly observed in the case of HT-2h sample in comparison to that of the AQ-glass sample. This increase in surface potential was mainly due to the existence of large number of piezoelectric LiNbO3 crystallites in the HT-2h sample. It is to be noted that this is a localized surface potential, which is negative over that area. AQ-sample also showed a very small surface potential, which is again due to the presence of a very few crystals of LiNbO3 in it. The large surface potential in case of the heat-treated samples indicated the presence of localized piezoelectricity, which is responsible for piezocatalysis.

KPFM surface potential micrographs of a AQ-glass, and b HT-2h samples indicating higher surface potential in the case of HT-2h sample in comparison to that of the AQ-glass sample.
Mechanism of piezocatalytic dye degradation
During ultrasonication of water, the cavity bubbles firstly form, then grow, and ultimately collapse47. These cavity bubbles generally contain entrapped gases and surrounding water vapors47. This continuous phenomena of the formation/growth/collapsation of bubbles lead to the formation of local hot spots with temperature of 4000–5000 K and in addition to this, a shock wave having ~108 Pa peak pressure releases19,47. These local hot spots decompose the contents of bubbles into free ˙OH radical species using thermolysis (sonolysis) process47,48. The free ˙OH radicals finally degraded MB dye and the products of degradation are formed as indicated in Eqs. (3) and (4)47,48.
During thermolysis (also known as sonolysis):
Thus, the thermolysis was the main reason behind the ~15% degradation of MB dye under 150 min ultrasonication without using any sample.
Moreover, the pressure exerted by shock waves produces local piezoelectricity in the glass–ceramics containing piezoelectric LiNbO3 crystals, due to which piezocatalysis initiates. This local piezoelectricity forced hole (h+)/electron (e−) pairs to move in opposite direction towards opposite polarity of polarization field. During this, some of these reacts with OH− ions and adsorbed O2 and then generate ˙OH and ˙\({\rm{O}}_{2}^{-}\) as shown in Eqs. (5)–(8). These radicals further decompose MB dye into degradation products.
Also, increasing piezoelectric LiNbO3 crystallites in glass–ceramic from HT-2h to HT-6h samples, an increase in piezocatalytic dye degradation performance was observed in this order HT-2h < HT-3h < HT-6h. AQ-sample also showed nearly 33% MB dye degradation within 150 min under ultrasonication, which was possibly comes from a few LiNbO3 crystals present in it as shown in SEM micrograph.
For confirming stability of HT-2h sample, the XRD was performed again after performing three cycles of piezocatalysis. However, no shift/change in peak position and orientation were observed in XRD pattern before and after third cycle of piezocatalysis as shown in Supplementary Fig. S2. Also, no physical change was seen on the sample. This clearly indicated that the present glass–ceramic was stable after catalytic reactions. The piezocatalytic dye degradation performance of present glass–ceramic (HT-6h) was found to be comparable with reported piezocatalysts in the literature as mentioned in Table 2.
Conclusions
Piezocatalysis can be induced in glass by embedded piezoelectric LiNbO3 crystals inside the matrix of glass using heat-treatment at 650 °C. The increase in crystallization time from 2 to 6 h decreased the transparency, however increase the piezocatalytic dye degradation capability due to increase in LiNbO3 crystallites counts and size. The sample heat-treated for 2 h provided sufficient transparency and promising piezocatalytic dye degradation capability. This study provides a promising alternative to powder and pellet ceramics in water-cleaning applications. Since, this is a first report on piezocatalysis glass–ceramics, there is an untapped potential to further explore and optimize the performance of ferroelectric glass–ceramics for catalytic applications.
Methods
Glass–ceramic fabrication
The parent glass (30SiO2–35Li2O–35Nb2O5 (all oxides are mentioned in mol%)) was obtained through melt-quenching process. During this process, various oxide powders (Li2O, Nb2O5, and SiO2) were considered according to stoichiometric calculation. These oxide powders were mixed followed by manually grounded for 30 min. The obtained homogenous fine powder was then transferred to platinum crucible. After this, the powder was melted at 1400 °C for 10 min in furnace (Nabertherm, Germany). The melt was poured on polished stainless-steel plate (having temperature of 200 °C) and then pressed with the help of an electric press (also at 200 °C). At last, as-quenched glass (AQ-glass) was obtained, which was transparent and light brown in color. In order to crystallize LiNbO3 crystals in AQ-glass, the glass was provided with additional heat-treatment at crystallization temperature of 650 °C for 2, 3, and 6 h. The crystallization temperature and time were decided from the reference27. These heat-treated samples were denoted as HT-2h, HT-3h, and HT-6h according to their crystallization time.
Characterization
The crystalline phase induced after heat-treatment was identified with the help of X-ray diffraction (XRD). Rigaku diffractometer, Japan possessing rotating 9 kW Cu anode was used to get XRD patterns of AQ-glass, HT-2h, HT-3h, and HT-6h samples. The scanning of samples was performed over 10–90° 2θ range using the scanning speed of 3°/min. After identification, Raman spectroscopy was performed to confirm phases and to get the knowledge of various vibrational modes of all investigated samples. Horiba (Lab RAM HR evolution) Raman spectrometer was utilized to obtain Raman spectra over 100–800 cm−1 wavenumber range using 25% power of 633 nm laser (1800 grating) for 15 s acquisition time. Scanning electron microscopy (FE-SEM, Nova Nano SEM-450, JFEI, USA) was utilized for visualizing the surface morphology. High resolution-transmission electron microscope (HR-TEM, FP 5022/22-Tecnai G2 20 S-TWIN, FEI, USA) was utilized for visualizing bulk crystals in the heat-treated samples. The absorbance and transparency were quantified using UV–visible spectrophotometer (Shimadzu UV-2600) over the 200–800 nm wavelength range. The NBO/bridging oxygens (BO) present in AQ-glass and the HT-6h samples were quantified using O1s scan of X-ray photoemission spectroscopy (XPS). XPS spectra was recorded using Nexsa X-ray photoelectron spectrophotometer having source of Al-Kα. The surface potential of AQ and HT-2h samples was measured using Kelvin probe force microscopy.
Piezocatalysis experiments
The piezocatalytic dye degradation experiment was performed using a model dye named as Methylene blue (MB dye). The initial concentration of MB dye solution was 5.5 mgL−1. The 5 mL volume of MB dye solution was filled in four different glass vials (15 mL). Samples of ~185 mm2 surface area were dipped in these vials. These vials were subjected to ultrasonic vibrations using an ultrasonicator of 70 W power and 40 kHz frequency. Water was used as an ultrasonication medium. The piezocatalysis experiments were executed under dark so as to exclude the possibility of photocatalysis. The test samples of 1000 μL were taken out from vials after every 30 min of ultrasonication and measured for absorbance under UV–visible spectrophotometer. The percentage degradation (D%) in dye concentration during piezocatalysis was obtained by following Eq. (9).
where “A0” and “A”; the values of dye solution absorbance recorded at times “t” = 0 and t min, respectively. Similarly, “Co” and “C”; the values of dye solution concentration at times “t” = 0 and t min, respectively. The test sample was recovered every time into the same glass vial so that dye volume remains constant. Water was used as medium of ultrasonication, which was regularly replaced after an interval of 15 min to prevent heating.
Data availability
All data are available within the manuscript and supporting information file.
References
Masai, H., Toda, T., Ueno, T., Takahashi, Y. & Fujiwara, T. ZnO glass–ceramics: An alternative way to produce semiconductor materials. Appl. Phys. Lett. 94, 151908 (2009).
Masai, H., Toda, T., Takahashi, Y. & Fujiwara, T. Fabrication of anatase precipitated glass–ceramics possessing high transparency. Appl. Phys. Lett. 94, 151910 (2009).
Bertrand, A. et al. New transparent glass–ceramics based on the crystallization of ‘anti-glass’ spherulites in the Bi2O3-Nb2O5-TeO2 system. Cryst. Growth Des. 15, 5086–5096 (2015).
Xue, J., Wang, X., Jeong, J. H. & Yan, X. Fabrication, photoluminescence and applications of quantum dots embedded glass ceramics. Chem. Eng. J. 383, 123082 (2020).
Chen, D. et al. Advances in transparent glass–ceramic phosphors for white light-emitting diodes—a review. J. Eur. Ceram. Soc. 35, 859–869 (2015).
Zanotto, E. D. A bright future for glass–ceramics. Am. Ceram. Soc. Bull. 89, 19–27 (2010).
Singh, G. et al. Antibacterial and photocatalytic active transparent TiO2 crystallized CaO–BaO–B2O3–Al2O3–TiO2–ZnO glass nanocomposites. J. Am. Ceram. Soc. 102, 3378–3390 (2019).
Singh, G., Kumar, S., Sharma, M. & Vaish, R. Transparent CaF2 surface crystallized CaO–2B2O3 glass possessing efficient photocatalytic and antibacterial properties. J. Am. Ceram. Soc. 102, 5127–5137 (2019).
Singh, G., Kumar, S., Singh, V. P. & Vaish, R. Transparent ZnO crystallized glass ceramics for photocatalytic and antibacterial applications. J. Appl. Phys. 125, 175102 (2019).
Miola, M. et al. Glass–ceramics for cancer treatment: So close, or yet so far? Acta Biomater. 83, 55–70 (2019).
Feng, Y. et al. Engineering spherical lead zirconate titanate to explore the essence of piezo-catalysis. Nano Energy 40, 481–486 (2017).
Lin, E., Wu, J., Qin, N., Yuan, B. & Bao, D. Silver modified barium titanate as a highly efficient piezocatalyst. Catal. Sci. Technol. 8, 4788–4796 (2018).
Ling, J., Wang, K., Wang, Z., Huang, H. & Zhang, G. Enhanced piezoelectric-induced catalysis of SrTiO3 nanocrystal with well-defined facets under ultrasonic vibration. Ultrason. Sonochem. 61, 104819 (2020).
Yuan, B., Wu, J., Qin, N., Lin, E. & Bao, D. Enhanced piezocatalytic performance of (Ba,Sr)TiO3 nanowires to degrade organic pollutants. ACS Appl. Nano Mater. 1, 5119–5127 (2018).
Biswas, A., Saha, S. & Jana, N. R. ZnSnO3 nanoparticle-based piezocatalysts for ultrasound-assisted degradation of organic pollutants. ACS Appl. Nano Mater. 2, 1120–1128 (2019).
Wu, J., Qin, N. & Bao, D. Effective enhancement of piezocatalytic activity of BaTiO3 nanowires under ultrasonic vibration. Nano Energy 45, 44–51 (2018).
Liu, X., Xiao, L., Zhang, Y. & Sun, H. Significantly enhanced piezo-photocatalytic capability in BaTiO3 nanowires for degrading organic dye. J. Mater. 6, 256–262 (2020).
Ma, J. et al. High efficiency bi-harvesting light/vibration energy using piezoelectric zinc oxide nanorods for dye decomposition. Nano Energy 62, 376–383 (2019).
Pan, M., Liu, S. & Chew, J. W. Unlocking the high redox activity of MoS2 on dual-doped graphene as a superior piezocatalyst. Nano Energy 68, 104366 (2020).
Singh, G., Sharma, M. & Vaish, R. Exploring the piezocatalytic dye degradation capability of lithium niobate. Adv. Powder Technol. 31, 1771–1775 (2020).
Qifeng, L., Jingjun, M., Sharma, M. & Vaish, R. Photocatalytic, piezocatalytic, and piezo-photocatalytic effects in ferroelectric (Ba0.875Ca0.125)(Ti0.95Sn0.05)O3 ceramics. J. Am. Ceram. Soc. 102, 5807–5817 (2019).
Zhang, A. et al. Ultrasonic vibration driven piezocatalytic activity of lead-free K0.5Na0.5NbO3 materials. Ceram. Int. 45, 22486–22492 (2019).
Wang, S. et al. Lead-free sodium niobate nanowires with strong piezo-catalysis for dye wastewater degradation. Ceram. Int. 45, 11703–11708 (2019).
Ma, W. et al. A novel multi-flaw MoS2 nanosheet piezocatalyst with superhigh degradation efficiency for ciprofloxacin. Environ. Sci. Nano 5, 2876–2887 (2018).
Qian, W. et al. Piezoelectric material-polymer composite porous foam for efficient dye degradation via the piezo-catalytic effect. ACS Appl. Mater. Interfaces 11, 27862–27869 (2019).
Kumar, S., Sharma, M., Kumar, A., Powar, S. & Vaish, R. Rapid bacterial disinfection using low frequency piezocatalysis effect. J. Ind. Eng. Chem. 77, 355–364 (2019).
Singh, G., Sharma, M. & Vaish, R. Influence of LiNbO3 crystallization on the optical, dielectric and nanoindentation properties of the 30SiO2-35Li2O-35Nb2O5 glass. J. Appl. Phys. 126, 214101 (2019).
Lipovskii, A. A., Kaganovskii, Y., Melehin, V. G., Tagantsev, D. K. & Yanush, O. V. Electrooptical Kerr phenomenon and Raman spectroscopy of one lithium-niobium-silicate glass-forming system. J. Non. Cryst. Solids 354, 1245–1249 (2008).
Graça, M. P. F., Ferreira Da Silva, M. G. & Valente, M. A. Preparation, structure, morphology, and dc and ac conductivity of the 88SiO2-6Li2O-6Nb2O5 (% mole) sol-gel derived glass–ceramics. J. Sol Gel Sci. Technol. 42, 1–8 (2007).
Chen, Z. et al. Piezoelectric properties of rhombic LiNbO3 nanowires. RSC Adv. 2, 7380–7383 (2012).
Repelin, Y., Husson, E., Bennani, F. & Proust, C. Raman spectroscopy of lithium niobate and lithium tantalate. Force field calculations. J. Phys. Chem. Solids 60, 819–825 (1999).
Hao, X. et al. Preparation and properties of transparent cordierite-based glass–ceramics with high crystallinity. Ceram. Int. 41, 14130–14136 (2015).
Rukang, L. I. The interpretation of UV absorption of borate glasses and crystals. J. Non. Cryst. Solids 111, 199–204 (1989).
Abdel-Baki, M., El-Diasty, F. & Wahab, F. A. A. Optical characterization of xTiO2-(60-x)SiO2-40Na2O glasses: II. Absorption edge, Fermi level, electronic polarizability and optical basicity. Opt. Commun. 261, 65–70 (2006).
Mistrik, J., Kasap, S., Ruda, H. E., Koughia, C. & Singh, J. Optical properties of electronic materials: fundamentals and characterization. In Springer Handbook of Electronic and Photonic Materials (eds Kasap, S. & Capper, P.) (1–1). (Springer, Cham, 2017).
Stebbins, J. F. & Xu, Z. NMR evidence for excess non-bridging oxygen in an aluminosilicate glass. Nature 390, 60–62 (1997).
Singh, S. & Singh, K. Effect of in-situ reduction of Fe3+ on physical, structural and optical properties of calcium sodium silicate glasses and glass ceramics. J. Non Cryst. Solids 386, 100–104 (2014).
Lee, S. K., Mysen, B. O. & Cody, G. D. Chemical order in mixed-cation silicate glasses and melts. Phys. Rev. B Condens. Matter Mater. Phys. 68, 214206 (2003).
Nesbitt, H. W. et al. Bridging, non-bridging and free (O2−) oxygen in Na2O-SiO2 glasses: an X-ray photoelectron spectroscopic (XPS) and nuclear magnetic resonance (NMR) study. J. Non Cryst. Solids 357, 170–180 (2011).
Kang, J. et al. Effects of alkali metal oxides on crystallization behavior and acid corrosion resistance of cordierite-based glass–ceramics. J. Non Cryst. Solids 481, 184–190 (2018).
Kim, Y. M., Reardon, D. E. & Bray, P. J. ESR studies of radiation-induced niobium centers in Nb2O5-Na2O-SiO2 glasses. J. Chem. Phys. 48, 3396–3402 (1968).
Acharya, S., Mansingh, S. & Parida, K. M. The enhanced photocatalytic activity of g-C3N4-LaFeO3 for the water reduction reaction through a mediator free Z-scheme mechanism. Inorg. Chem. Front. 4, 1022–1032 (2017).
Etaiw, S., Din, E. & Marie, H. Sonochemical nanostructure of Mn(II) supramolecular complex: X-ray structure, sensing and photocatalytic properties. Sensors Actuators B Chem 290, 631–639 (2019).
Mansingh, S., Acharya, R., Martha, S. & Parida, K. M. Pyrochlore Ce2Zr2O7 decorated over rGO: A photocatalyst that proves to be efficient towards the reduction of 4-nitrophenol and degradation of ciprofloxacin under visible light. Phys. Chem. Chem. Phys. 20, 9872–9885 (2018).
Cho, A., Kim, D. Bin & Cho, Y. S. Electric-field-dependent surface potentials and vibrational energy-harvesting characteristics of Bi(Na0.5Ti0.5)O3-based Pb-free piezoelectric thin films. ACS Appl. Mater. Interfaces 11, 13244–13250 (2019).
Rastogi, M., Chauhan, A., Kushwaha, H. S., Kumar, R. V. & Vaish, R. Reaping the benefits of ferroelectricity in selectively precipitated lithium niobate microcrystals in silica matrix for photocatalysis. Appl. Phys. Lett. 109, 223901 (2016).
Tezcanli-Güyer, G. & Ince, N. H. Individual and combined effects of ultrasound, ozone and UV irradiation: a case study with textile dyes. in Ultrasonics. 42, 603–609 (2004).
Ghows, N. & Entezari, M. H. Kinetic investigation on sono-degradation of Reactive Black 5 with core-shell nanocrystal. Ultrason. Sonochem. 20, 386–394 (2013).
Wu, J. et al. Synthesis of Bi4Ti3O12 decussated nanoplates with enhanced piezocatalytic activity. Nanoscale 44, 21128–21136 (2019).
Liu, Y. L. & Wu, J. M. Synergistically catalytic activities of BiFeO3/TiO2 core-shell nanocomposites for degradation of organic dye molecule through piezophototronic effect. Nano Energy 56, 74–81 (2019).
Wu, J. et al. Insights into the role of ferroelectric polarization in piezocatalysis of nanocrystalline BaTiO3. ACS Appl. Mater. Interfaces 10, 17842–17849 (2018).
Acknowledgements
R.V. thanks to Biotechnology Industry Research Assistance Council (BIRAC), New Delhi for the project (BT/AMR0235/05/18).
Author information
Authors and Affiliations
Contributions
G.S. and R.V. generated the idea and planned the experiments. G.S and M.S. performed all the experiments. G.S. wrote the paper. R.V. reviewed and corrected the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Primary handling editor: John Plummer.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Singh, G., Sharma, M. & Vaish, R. Transparent ferroelectric glass–ceramics for wastewater treatment by piezocatalysis. Commun Mater 1, 100 (2020). https://doi.org/10.1038/s43246-020-00101-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-020-00101-2
This article is cited by
-
2D MXenes polar catalysts for multi-renewable energy harvesting applications
Nature Communications (2023)
-
Effect of poling and porosity on BaTiO3 for piezocatalytic dye degradation
Journal of Materials Science: Materials in Electronics (2023)
-
Poling direction-driven tuning of piezoelectric properties of ferroelectric materials: an experimental study
Journal of the Australian Ceramic Society (2022)
