
Abstract
The dynamics and phenotypes of intratumoral myeloid cells during tumor progression are poorly understood. Here we define myeloid cellular states in gliomas by longitudinal single-cell profiling and demonstrate their strict control by the tumor genotype: in isocitrate dehydrogenase (IDH)-mutant tumors, differentiation of infiltrating myeloid cells is blocked, resulting in an immature phenotype. In late-stage gliomas, monocyte-derived macrophages drive tolerogenic alignment of the microenvironment, thus preventing T cell response. We define the IDH-dependent tumor education of infiltrating macrophages to be causally related to a complex re-orchestration of tryptophan metabolism, resulting in activation of the aryl hydrocarbon receptor. We further show that the altered metabolism of IDH-mutant gliomas maintains this axis in bystander cells and that pharmacological inhibition of tryptophan metabolism can reverse immunosuppression. In conclusion, we provide evidence of a glioma genotype-dependent intratumoral network of resident and recruited myeloid cells and identify tryptophan metabolism as a target for immunotherapy of IDH-mutant tumors.
Similar content being viewed by others


Main
The glioma microenvironment orchestrates tumor evolution, progression and resistance to therapy1. In high-grade gliomas (HGG), microglia and monocyte-derived macrophages, collectively referred to as glioma-associated myeloid cells (GAM), constitute up to 70% of the tumor mass2. Current concepts propose a recruitment of blood-borne macrophages to the glioma microenvironment, in which phenotypic and functional shaping of invading macrophages and resident microglia is dependent on the tumor genotype, such as disease-defining mutations in the gene encoding IDH type 1 (IDH1) that are causally related to profound tumor cell-intrinsic epigenetic and metabolic alterations associated with a favorable prognosis of patients with glioma2. Functionally altered GAM in turn promote tumor growth by a variety of mechanisms. A striking feature of GAM is a poor antigen-presenting capacity and the acquisition of an immunosuppressive phenotype. While studies to date suggest a continuum rather than a bimodal distribution of microglia-specific versus macrophage-specific genes in myeloid cells3, temporal cell type-specific functional states within the glioma microenvironment have not been defined. Such analyses would reveal important molecular determinants of functional myeloid states as well as therapeutic targets within the myeloid compartment. Here we comprehensively define longitudinal homeostatic and antigen-presenting myeloid cellular states, assess their tumor-genotype dependence and reveal underlying metabolic mechanisms controlling them.
Results
First, we performed RNA-seq on 30,000–600,000 sorted microglia and macrophages from 14 HGG (Fig. 1a and Supplementary Tables 1 and 2). Principal component analysis showed a remarkable separation of samples based on the mutational status of IDH (Fig. 1a). To further dissect these genotype-dependent signatures, we performed single-cell RNA-seq (scRNA-seq) on flow cytometry-purified CD45+CD3−CD19−CD20− hematopoietic cells isolated from IDH-wild-type (WT) (n = 5) and IDH-mutant (n = 5) HGG in comparison to control brain tissues (n = 7) (Fig. 1b). Seurat analysis of 4,460 cells that passed quality control using the data integration workflow revealed ten transcriptionally distinct clusters corresponding to different cell types and states (Fig. 1c)4. Using hypergeometric tests for enrichments that considered different numbers of cells per condition, we found that myeloid cell clusters (C) C0, C3, C4 and C6 were enriched in cells from control tissues, while C1 and C5 were enriched in cells from IDH-WT HGG, and C2 was enriched in cells from IDH-mutant HGG (Fig. 1d and Extended Data Fig. 1a). Differential gene expression analysis of IDH-WT-enriched C1 and C5 showed a downregulation of microglial steady-state genes (TMEM119, P2RY12, CSF1R) with a concomitant upregulation of interferon (IFN) signaling and hypoxia-associated genes, including IFI44L and HIF1A, respectively. Furthermore, C1 and C5 expressed genes that are associated with acutely activated macrophages, including APOE, CD163 and S100A11, suggesting that these clusters contain acutely infiltrating hematopoietic cells3,5. The IDH-mutant-enriched cluster C2 showed upregulation of genes encoding chemokines (Fig. 1e). Control-enriched clusters C0, C3, C4 and C6 showed upregulation of microglia-defining genes such as TMEM119 and OLFML3 (Fig. 1e). Accordingly, we found an increased cumulative expression of genes associated with homeostatic microglia (P2RY12, CX3CR1 and CSF1R) in the control-enriched clusters C0, C3, C4 and C6 (Fig. 1e). Differential gene expression analysis of cells assigned to microglia and macrophage clusters C0–C6 showed upregulation of major histocompatibility complex (MHC) class I and II-coding genes, including HLA-B, CD74 and HLA-DPA1, in IDH-WT HGG-associated GAM with respect to their IDH-mutant HGG-associated counterparts (Fig. 1f and Extended Data Fig. 1b). The latter cells displayed upregulation of steady-state microglia and inflammatory mediator-coding genes, such as P2RY12 and IL1B (Fig. 1f). Next, we validated scRNA-seq findings at the protein level using cytometry by time-of-flight (CyTOF). Unsupervised clustering of n = 65,909 purinergic receptor (P2RY12)+ microglia cells (n = 25,973 controls, n = 17,646 cells from IDH-mutant HGG and n = 22,290 cells from IDH-WT HGG) identified ten clusters with similar protein expression profiles (Extended Data Fig. 1c–g). For ensuring robust clusterwise comparisons of protein expression between conditions, we excluded four clusters (C4, C7, C8 and C10) containing less than 0.05% of all cells per condition (Fig. 1g). While clusters C1 and C5 consisted mostly of cells from control tissues, C3 predominantly contained cells derived from IDH-WT HGG, and C6 and C9 predominantly contained cells from IDH-WT and IDH-mutant HGG (Fig. 1g and Extended Data Fig. 1e). C3 showed an activation gene expression profile with downregulation of homeostatic microglia proteins, including chemokine receptor CX3CR1, transmembrane protein TMEM119 and P2RY12, as well as upregulation of microglial activation proteins, such as apolipoprotein (APOE) and receptor EMR1 (Fig. 1h and Extended Data Fig. 1g). Furthermore, clusters C6 and C9, which were enriched in both IDH-mutant and IDH-WT HGG, showed upregulation of antigen-presentation (AP)-associated proteins including HLA-DR and CD74 (Fig. 1h). Analysis of MHC class II and co-stimulatory protein markers present in the antibody panel showed a consistent upregulation in HGG samples with respect to controls, with the exception of C2 (Fig. 1h). Notably, myeloid cells were previously distinguished in immunogenic and tolerogenic states based on the expression of MHCII and co-stimulatory genes6. In line with transcriptomic profiling, IDH-mutant HGG-derived myeloid cells showed a less pronounced downregulation of the microglia homeostatic signature and a less marked upregulation of the AP signature than IDH-WT HGG (Fig. 1h). Similar differences could be detected using a different CyTOF antibody panel (Supplementary Table 2 and Extended Data Fig. 2). Collectively, these data suggest a striking and differential genotype-dependent shaping of GAM in human HGG toward an immunosuppressive phenotype.

a, Principal component (PC) analysis (PCA) biplots of bulk RNA-seq data of myeloid cells from human IDH-WT (n = 8 patients) and IDH-mutant (n = 6 patients) HGG. N(macrophages) = 13, of which n(IDH-WT) = 8, and n(IDH-mutant) = 5; n(microglia) = 14, of which n(IDH-WT) = 8, and n(IDH-mutant) = 6. MΦ, macrophage. b, Workflow applied to human scRNA-seq samples included in the present study. c, Visualization of scRNA-seq data using uniform manifold approximation and projection (UMAP). The left panel is color coded based on the clustering output of the Seurat algorithm. Cell type assignment was conducted based on the expression of cell type-enriched gene signatures. UMAPs color coded for the cumulative gene expression of microglia- (top right) and monocyte-enriched (bottom right) genes. n = 7 patients for control tissue, n = 5 patients with IDH-WT HGG, n = 5 patients with IDH-mutant HGG; n values per condition are consistent with those in d–f. NKT, natural killer T cell. d, Visualization of the distribution of cells from control, IDH-WT and IDH-mutant HGG tissue samples, respectively, using UMAP (left) and a Marimekko plot (right) (**P < 0.01, ***P < 0.001) for clusterwise enrichment analysis in the one-tailed hypergeometric test. Adjustment for multiple testing was achieved using the Bonferroni method. e, Single-cell heatmap of the top 15 cluster marker genes with representative genes annotated on the left. f, Volcano plot depicting differentially expressed genes in IDH-WT GAM (right half) with respect to IDH-mutant GAM (left half). Highlighted genes represent top differentially expressed genes with P value < 0.05 and average log-transformed fold change > 0.25. g, Left, t-distributed stochastic neighbor embedding (t-SNE) map color coded for proteomically similar clusters of cells. Right, t-SNE plot depicting the distribution of control, IDH-mutant and IDH-WT GAM across the clusters. h, Violin plots depicting the cumulative expression of homeostatic and AP protein signatures in the respective conditions. P values were calculated using the one-way Kruskal–Wallis test followed by Dunn’s post hoc test and adjusted for multiple testing using the Benjamini–Hochberg method. The color scheme is consistent with that in g. NS, not significant.
To investigate the dynamics and underlying molecular mechanisms of this glioma genotype-dependent immunosuppressive phenotype of GAM, we made use of an experimental HGG mouse model, GL261 episomally overexpressing WT and mutant IDH (Fig. 2a and Extended Data Fig. 3a–g). We conducted scRNA-seq of flow cytometry-purified CD45+ cells isolated from IDH-mutant and IDH-WT GL261 gliomas comprising microglia, monocytes, macrophages, monocyte-derived dendritic cells (DCs), mast cells, granulocytes and T and B cells at two time points during glioma progression: early after primary tumor establishment at day 7 (d7) and at a late-stage time point at d28 after tumor injection (Fig. 2a–d). Expectedly, at d7, microglia made up to >75% of the myeloid cells in the tumor (75% ± 17.6% in IDH-WT samples and 92% ± 2.1% in IDH-mutant samples), whereas, at d28, invading cells dominated the myeloid compartment (macrophages, 23% ± 11.6% in IDH-WT samples and 36% ± 7.1% in IDH-mutant samples; DCs, 11% ± 7.7% in IDH-WT samples and 14% ± 3.2% in IDH-mutant samples; Fig. 2e). Interestingly, at d7, invading immune cells were more abundant in IDH-WT compared to IDH-mutant gliomas, while at the late stage, hematopoietic immune cell contents were comparable between both experimental HGG (Fig. 2e). Comparative analysis considering cell type and IDH status of experimental HGG between d7 and d28 revealed a drop in relative microglia content accompanied by a concomitant increase in macrophage and DC numbers in IDH-mutant HGG, suggesting a higher influx of circulating immune cells between early and late time points in these HGG. Importantly, these differences were not a result of differential tumor growth (Fig. 2e,f and Extended Data Fig. 3e). Based on these observations, we hypothesized that, dependent on their IDH status, microglia shaped by the early HGG microenvironment drive differential recruitment of invading immune cells, particularly blood-borne macrophages. Indeed, d7 microglia in IDH-WT tumors, in accordance with human IDH-WT HGG, show increased expression of Ccl12, the gene encoding the murine ligand of CCR2, which is involved in peripheral myeloid cell recruitment to the central nervous system (CNS). Ccl12 was differentially upregulated in microglia from IDH-WT GL261 gliomas (Fig. 2g). Differential expression analysis of microglia in early-stage experimental HGG further showed increased expression of genes encoding MHC and co-stimulatory molecules in microglia from IDH-WT gliomas, while those from IDH-mutant gliomas displayed higher expression of steady-state microglia genes such as P2ry12 in accordance with our findings in human GAM (Fig. 2g). To assess gradual changes between steady-state and activated microglia, we conducted a pseudotime analysis of early microglia using StemID2 (ref. 7) that showed a shift from a homeostatic to a differentiated state. Consistent with our hypothesis, we found enrichment of steady-state microglia in IDH-mutant HGG and enrichment of activated microglia in IDH-WT HGG (Fig. 2h–j and Extended Data Fig. 3h,i). In summary, IDH-mutant experimental HGG showed an attenuated immunogenic microglia transcriptional profile and reduced content of infiltrating myeloid cells at the early time point that increased during tumor progression.

a, Workflow applied to murine HGG samples included in the present study. n = 14 samples from n = 4 IDH-WT tumor-bearing mice (d7), n = 4 IDH-mutant tumor-bearing mice (d7), n = 3 IDH-WT tumor-bearing mice (d28), n = 3 IDH-mutant tumor-bearing mice (d28). n values are identical for b–j. b, UMAP visualization of murine scRNA-seq data, color coded for the clustering output of the Seurat algorithm. c, UMAP color coded for cell types based on the expression of cell type-enriched gene signatures. moMΦ, monocyte-derived macrophages. d, UMAP color coded for the HGG genotype and separated by time point. e, Donut plots depicting composition of tumor myeloid compartments in the HGG with respect to the time point and IDH-mutation status. P values were calculated for enrichment of cell types between IDH-WT and IDH-mutant tumors at each time point by one-tailed hypergeometric test. Adjustment for multiple testing was achieved using the Bonferroni method. Cell types with absolute average counts less than 20 were excluded from the analysis. f, Dot–line plot depicting the relative composition of the myeloid compartment in HGG with respect to the IDH-mutation status and the time point. P values were calculated by fitting a negative binomial function to the percentage of cells as a function of the time point and cell type followed by post hoc comparison between time points using the emmeans method in R with multiple-testing adjustment using the Tukey method. Error bars, s.e.m. Cell types with absolute average counts less than 20 were excluded from the analysis. Monoc., monocytes. g, Volcano plot depicting differentially expressed genes in IDH-WT microglia (right half) with respect to IDH-mutant microglia (left half). Highlighted genes represent top differentially expressed genes with P value < 0.05 and average log-transformed fold change > 0.25. h, GAM trajectory suggested by the StemID2 algorithm. i, Single-cell heatmap of marker genes of clusters along the trajectory from h. Representative genes are highlighted on the left. j, Heatmap depicting z scores of genes expressed by cells along the trajectory from h. Representative genes are highlighted on the right. Below the heatmap (middle), a stacked bar plot depicts the cell composition along the trajectory with respect to the IDH-mutation status. Bottom, a simple linear regression of the abundance of microglia in IDH-mutant experimental HGG in relation to those in IDH-WT experimental HGG along the trajectory, with two-tailed Spearman correlation analysis (F = 30.11, degrees of freedom for numerator (DFn) = 1, degrees of freedom for denominator (DFd) = 28) of the relative abundance of IDH-mutant microglia along the trajectory.
As late-stage experimental tumors were more enriched for recruited myeloid cells, we investigated functional phenotypes of monocyte-derived cells at late time points and found robust expression of the previously defined AP signature in both cell compartments (Fig. 3a)8. While DCs showed only moderate differential signatures in IDH-mutant compared to IDH-WT experimental tumors, macrophages from IDH-mutant gliomas showed upregulation of Il1b with a concomitant downregulation of Arg1 (Fig. 3b,c). To assess mechanisms of attenuating the AP signature in late-stage IDH-mutant HGG, we performed pseudotime analyses of macrophages and monocyte-derived DCs isolated from IDH-mutant compared to IDH-WT tumors (Fig. 3d–f and Extended Data Fig. 3h,i). Strikingly, we observed lower abundance of both cell types in IDH-mutant experimental HGG toward the end of each trajectory, suggesting either delayed functional polarization or a differentiation block of monocyte-derived cells in late-stage IDH-mutant tumors (Fig. 3e,f). Comparing our murine and human datasets, the relatively high intratumoral abundance of DCs was restricted to our HGG model, and current studies observe that DCs account for less than 5% of the human glioma immune cell infiltrate9. Therefore, we aimed to functionally validate the attenuated AP signature inferred by transcriptomic profiling in microglia and macrophages. In ex vivo co-cultures of naive T cells and microglia and macrophages isolated from experimental HGG at the late stage, we observed a consistent and ratio-dependent decrease in IFN-γ production and upregulation of programmed cell death protein (PD)-1 in both cytotoxic and T helper cells. In addition, production of granzyme (Grz)B was reduced in cytotoxic T cells (Fig. 3g,h and Extended Data Fig. 3a). Strikingly, we observed a differential level of T cell suppression by macrophages, but not by microglia, based on IDH-mutation status. Suppression of T cells was significantly increased in co-cultures with macrophages infiltrating IDH-mutant experimental HGG. Based on previous observations in T cells10, we hypothesized that an attenuated AP signature of macrophages in IDH-mutant experimental tumors is dependent on the neomorphic enzymatic activity of mutant IDH. Treatment of mice bearing intracranial IDH-mutant and IDH-WT HGG with the blood–brain-barrier-permeable mutant IDH inhibitor BAY 1436032 (ref. 11) revealed partial reversibility of an IDH-mutant-associated attenuated AP signature in macrophages but not in microglia (Fig. 3i).

a, UMAP visualization of infiltrating hematopoietic myeloid cells from murine scRNA-seq data, color coded for clustering (left) and for cumulative gene expression of selected genes involved in AP (right). n = 6 samples from n = 3 IDH-WT tumor-bearing mice (d28), n = 3 IDH-mutant tumor-bearing mice (d28). b, Volcano plot depicting differentially expressed genes in IDH-WT-associated macrophages (right half) with respect to IDH-mutant-associated macrophages (left half). c, Volcano plot depicting differentially expressed genes in IDH-WT-associated DCs (right half) with respect to IDH-mutant-associated DCs (left half). In b,c, highlighted genes represent top differentially expressed genes with P value < 0.05 and average log-transformed fold change > 0.25. moDC, monocyte-derived DC. d, Infiltrating hematopoietic myeloid cell trajectories suggested by the StemID2 algorithm. e, Heatmap depicting z scores of genes expressed by cells along the macrophage trajectory from d. Representative genes are highlighted on the right. Below the heatmap (middle), a stacked bar plot depicts the cell composition along the trajectory with respect to the IDH-mutation status. Bottom, a simple linear regression with two-tailed Spearman correlation analysis (F = 16.87, DFn = 1, DFd = 28) of the relative abundance of IDH-mutant-associated macrophages along the trajectory. f, Heatmap depicting z scores of genes expressed by cells along the DC trajectory from d. Representative genes are highlighted on the right. Below the heatmap (middle) a stacked bar plot depicts the cell composition along the trajectory with respect to the IDH-mutation status. Bottom, a simple linear regression with two-tailed Spearman correlation analysis (F = 65.98, DFn = 1, DFd = 28) of the abundance of IDH-mutant-associated DCs in relation to IDH-WT-associated DCs along the trajectory. g, Ex vivo glioma-associated microglia T cell-suppression assay. Top, CD4+ T cells after co-culture with microglia. n = 10 individual microglia samples isolated from IDH-WT tumor-bearing mice, n = 9 individual microglia samples isolated from IDH-mutant tumor-bearing mice. Bottom, CD8+ T cells after co-culture with microglia; n values are identical to those on top. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. E:T, efffector-to-target ratio. h, Ex vivo glioma-associated macrophage T cell-suppression assay. Procedure and n values are identical to those in g. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. i. Inhibition of mutant IDH is efficient in reverting the IDH-mutant-induced tumor-associated macrophage (TAM) phenotype. n = 12; n = 4 IDH-WT tumor-bearing mice, n = 4 IDH-mutant tumor-bearing mice treated with vehicle, n = 4 IDH-mutant tumor-bearing mice treated with BAY 1436032. Flow cytometry analysis of glioma-associated microglia and macrophages. Box and whiskers (minimum to maximum, median as center) are shown. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. IDHi, IDH inhibitor; p.o., per os. If not mentioned otherwise, all data are represented as mean ± s.e.m.
To define the molecular mechanism underlying this time-dependent and tumor-genotype-dependent functionality shift, we exposed human monocytes and macrophages to the neomorphic enzymatic product of mutant IDH, R-2-hydroxyglutarate (R-2-HG)12. Co-incubation of R-2-HG-pretreated monocytes or macrophages with T cells revealed a dose-dependent suppression of T cell proliferation (Fig. 4a). To validate that macrophage exposure to R-2-HG leads to an attenuated AP capacity, we assessed CD80, CD86 and HLA-DR expression by flow cytometry. Indeed, a dose-dependent downregulation of these proteins after R-2-HG exposure was observed (Fig. 4b,c). Macrophages and microglia took up exogenous R-2-HG independently of activation status (Fig. 4d and Extended Data Fig. 4a). Overexpression of amino acid transporters known to transport R-2-HG, such as solute carrier (SLC)13A3, but not SLC22A6, SLC16A5 or SLC3A2, resulted in increased uptake of R-2-HG (Fig. 4e and Extended Data Fig. 4b)13. To dissect the mechanism that mediates reprogramming of GAM in IDH-mutant tumors, we performed an in vitro transcriptome screen of R-2-HG-treated primary macrophages isolated from healthy human donors (Fig. 4f and Extended Data Fig. 4c). Pathway analysis revealed that the top regulated pathway after R-2-HG exposure was induced by the synthetic toxin 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, Fig. 4f and Supplementary Table 3). TCDD is one of the strongest dioxin-like compounds to act via a specific ligand–receptor interaction with the aryl hydrocarbon receptor (AHR)14. Strikingly, the expression of AHR target genes was significantly higher in the Cancer Genome Atlas (TCGA) datasets of IDH-mutant gliomas (n = 226) than in those of IDH-WT tumors (n = 55) but only when normalized for intratumoral myeloid cell abundance assessed by ITGAM (CD11b) expression (Extended Data Fig. 4d). AHR target genes, such as AHRR (encoding an AHR repressor), TIPARP (encoding TCDD-inducible poly(ADP-ribose) polymerase) and CYP1A1 were induced in monocytes by R-2-HG to a similar extent as by the known endogenous AHR ligand l-kynurenine (l-Kyn) in vitro (Fig. 4g). AHR target gene activation by R-2-HG in human macrophages was further observed to be dose dependent (Extended Data Fig. 4e). To validate the specific expression of AHR target genes in GAM, we conducted pseudotime trajectory analyses of human glioma infiltrates. We found upregulation of AHR target genes along a trajectory (C3 → C2) enriched in IDH-mutant-derived GAM but not IDH-WT-derived GAM (Fig. 4h). In contrast to this, IDH-WT-derived GAM were predominantly found along another, more immunogenic trajectory with expression of antigen-presenting cell signature genes (C3 → C0 → C1, Fig. 4h). In accordance with the human dataset, all clusters forming the myeloid cell trajectory in experimental HGG showed differential cumulative expression of an AHR activation gene signature between IDH-mutant-derived and IDH-WT-derived cells (Fig. 4i). As AHR transcripts were most abundant in monocytes as compared to other immune cells (Extended Data Fig. 4f), these findings are indicative of an immune cell subtype-specific vulnerability to reprogramming by R-2-HG. Notably, AHR was identified as a critical cofactor for immunosuppressive transforming growth factor (TGF)-β and interleukin (IL)-1β signaling15,16. In addition, AHR directly promotes IL-10 production17,18. We found that, following exposure to R-2-HG, macrophages demonstrated an increased dose-dependent production of IL-10 and TGF-β in an AHR-dependent fashion (Fig. 4j and Extended Data Fig. 4g). Analysis of monocyte-derived macrophages revealed that IL-10 production was in fact induced in an IDH-mutant experimental HGG microenvironment and required functional AHR (Fig. 4k). Consequently, IL-10 levels in freshly isolated tumor lysates were higher in IDH-mutant experimental HGG when grown in an AHR-WT compared to an AHR-deficient microenvironment (Fig. 4l). To verify whether AHR-dependent reprogramming of GAM by R-2-HG is a result of increased AHR translocation, we used an in vitro AHR translocation reporter assay in which GFP expression is driven by a DRE-dependent promoter (DRE-GFP). Incubation with concentrated supernatants of glioma cell lines overexpressing mutant IDH resulted in increased AHR translocation and transcriptional activity (Extended Data Fig. 4h). To determine whether the observed activation of AHR was driven by R-2-HG rather than by other differentially released substances, we tested synthetic R-2-HG and observed increasing nuclear AHR translocation within 60 min of incubation (Fig. 4m and Extended Data Fig. 4i). We verified these results in an independent, luciferase-based endpoint assay (Fig. 4n). Notably, R-2-HG-induced AHR translocation was comparable to that induced by l-Kyn (Fig. 4m–o and Extended Data Fig. 4i). As deprivation of l-Trp reduced AHR translocation by R-2-HG, but not that by l-Kyn, our cumulative data suggested that R-2-HG is not a direct AHR ligand but leads to increased intracellular levels of l-Kyn, presumably preceded by l-Trp catabolism and subsequent AHR activation (Fig. 4o). To assess whether R-2-HG-mediated suppression of T cells by macrophages is indeed dependent on l-Trp, we performed co-culture assays with macrophages in l-Trp-free and control media, respectively. Strikingly, tryptophan deprivation of R-2-HG-exposed macrophages led to increased effector functions of co-incubated T cells (Fig. 4p).

a, Tritiated thymidine (3H-dT) proliferation assay of primary human T cells stimulated in co-culture in vitro with autologous peripheral blood-derived monocytes or differentiated macrophages that were pre-exposed to varying doses of R-2-HG. n = 20 individual monocyte–T cell co-cultures. n = 19 individual macrophage–T cell co-cultures. Statistical significance was assessed by two-tailed Spearman correlation analysis (Pcorr) between 20 and 19 x–y pairs and by two-tailed Student’s t-tests between 0 mM and 20 mM conditions (P0 mM vs. 20 mM). cpm, counts per minute. b,c, Protein abundance of CD86, CD80 and HLA-DR on unstimulated and stimulated peripheral blood-derived monocytes treated with R-2-HG as determined by flow cytometry. b, Representative flow cytometry pseudocolor plot from one healthy donor. LPS, lipopolysaccharide. c, Quantification of CD86+, CD80+ and HLA-DR+ macrophages. Statistical significance was determined by two-tailed Student’s t-tests; n = 6 healthy human donors. d, Intracellular measurements of R-2-HG in primary macrophages after incubation in vitro with R-2-HG and stimulation with IFN-γ and LPS for 24 h. Nonlinear regression is shown. n = 3 experimental repeats. e, Intracellular measurements of R-2-HG in cells overexpressing the indicated SLC isoforms after incubation in vitro with R-2-HG for 24 h. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. n = 3 experimental repeats. EC, extracellular; IC, intracellular. f, DNA-microarray screen of macrophages from human donors (n = 8) treated with exogenous R-2-HG in a matched-pair analysis. Ingenuity pathway analysis of the dataset, indicating a TCDD-induced regulated canonical network. g, Induction of AHR target genes in monocyte-derived macrophages by varying doses of R-2-HG or l-Kyn as determined by a PCR assay. Fold changes relative to vehicle treatment are shown (n = 5–6 independent healthy donors). h, Pseudotime analysis of stepwise changes between control clusters (C3) and clusters enriched for cells from IDH-WT GBMs (C0, C1) and IDH-mutant GBMs (C2), respectively. Analysis was conducted on n = 1,957 cells from control patients, n = 690 cells from IDH-mutant GBMs and n = 809 cells from IDH-WT GBMs. i, Analysis of infiltrating hematopoietic myeloid cells along the trajectories shown in Fig. 3d with UMAP representations color coded for RaceID clusters and cumulative expression of the AHR activation signature. j, ELISA for IL-10 and TGF-β in bone marrow-derived macrophages (BMDMs) from Ahr+/+ versus Ahr−/− mice exposed to increasing concentrations of R-2-HG in vitro. Nonlinear regression is shown. Statistical significance was determined by two-tailed Student’s t-test at a concentration of 10−2 M. n = 3 Ahr+/+ versus n = 3 Ahr−/− mice. k, Flow cytometry analysis of macrophages isolated from GL261 tumors intracranially implanted in Ahr+/+ (n = 10, of which IDH-WT n = 6 and IDH-mutant n = 4) versus Ahr−/− (n = 12, of which IDH-WT n = 6 and IDH-mutant n = 6) mice. Box and whiskers (minimum to maximum, median as center) are shown. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. l, IL-10 ELISA of hemisphere washout from GL261 IDH-mutant tumor-bearing Ahr+/+ (n = 12 matched samples) versus Ahr−/− (n = 6 matched samples) mice. Autologous contralateral hemispheres were used as controls. m, AHR translocation reporter assay. Time-dependent quantification of AHR translocation based on the DRE-GFP reporter. Arbitrary units (AU) are shown. Representative experiment of three independent repeats outlined in Extended Data Fig. 4i. n, Luciferase-based endpoint reporter assay for AHR translocation. Reporter assay after treatment with the indicated compounds for 6 h. CMV-luciferase (CMV_Luc) was used as the positive control (n = 3 independent assay runs, each using two different transduced cell lines for R-2-HG and l-Kyn conditions). RLU, relative luminescence units. o, Left, AHR reporter assay after treatment with increasing doses of R-2-HG or vehicle (PBS) for 6 h. Right, AHR reporter assay after treatment with increasing doses of l-Kyn or vehicle (PBS) for 6 h. Cells were kept in RPMI 1640 medium containing 5 mg l−1 l-Trp with FBS or l-Trp-free medium with dialyzed FBS. Data are represented as means of n = 3 independent assay runs, with s.e.m. projected as error bands. p, Macrophage T cell-suppression assay. BMDMs were differentiated in vitro with R-2-HG or vehicle in medium containing 5 mg l−1 l-Trp with FBS or l-Trp-free medium with dialyzed FBS for 24 h. Cells were then co-cultured with stimulated syngeneic T cells for 72 h. Relative measurements (R-2-HG/vehicle, %/%) of IFN-γ+ or GrzB+ T cells are shown. Statistical significance was determined by two-tailed Student’s t-tests. n = 6 (IFN-γ) or n = 8 (proliferation, GrzB) paired samples from three individual mice as BMDM and T cell donors. If not mentioned otherwise, all data are represented as mean ± s.e.m.
Based on our findings, we hypothesized that immunosuppressive l-Trp catabolism via the kynurenine pathway drives reprogramming of macrophages infiltrating IDH-mutant tumors19. To define dynamics of l-Trp metabolism in immune cells under the influence of R-2-HG, we undertook an LC–MS/MS-based study in macrophages and T cells (Fig. 5a,b and Extended Data Fig. 5a). Here we found that exogenous l-Trp was taken up by T cells in a dose-dependent fashion. While in T cells, a linear increase in l-Trp levels was accompanied by a matching increase in l-Kyn levels; intracellular l-Trp levels remained stable in macrophages with increasing l-Kyn levels (Fig. 5a). Based on this observation, it is reasonable to hypothesize that R-2-HG-exposed macrophages produce l-Kyn from imported l-Trp as a consequence of l-Trp catabolism via the kynurenine pathway. Tryptophan 2,3-dioxygenase (TDO2) and indoleamine 2,3-dioxygenase (IDO)1 and IDO2 catalyze the rate-limiting step of the kynurenine pathway and together account for 90% of dietary l-Trp degradation20,21. The plasma kynurenine-to-tryptophan ([l-Kyn][l-Trp]−1) concentration ratio has frequently been used to express or reflect the activity of these enzymes22. Indeed, when exposed to R-2-HG, macrophages demonstrate a significant increase in the [l-Kyn][l-Trp]−1 ratio with increasing concentrations of extracellular l-Trp. By contrast, T cells do not show an increase in l-Kyn production beyond a linear increase, which is the probable result of a shifted equilibrium reaction due to increased substrate levels (Fig. 5b). To identify the mechanism that underlies l-Trp degradation in macrophages, but not in T cells, we performed cell-free enzymatic assays with described rate-limiting enzymes in the kynurenine pathway. Here, dose-dependent R-2-HG supplementation resulted in increased kynurenine production by TDO2 but not by IDO1 or IDO2, suggesting a TDO2-inductive effect of R-2-HG (Fig. 5c–e). Based on induction kinetics, a new role of R-2-HG as an allosteric activator within the TDO2 tetramer protein complex interface seems likely, as TDO2 is not dependent on α-ketoglutarate, similar to other enzymes that were described to be affected by R-2-HG23,24. TDO2 is generally believed to be constitutively active in hepatocytes to achieve l-Trp homeostasis, while many cell types demonstrate low basal IDO expression that can be rapidly induced by proinflammatory cytokines25. However, TDO2 expression analyses based on publicly available RNA-seq datasets across different immune cell populations revealed a moderate expression level in monocytes and macrophages (Fig. 5f)26. In functional ex vivo assays using cells from Tdo2−/− mice, we demonstrated dependence of R-2-HG-associated reduced T cell proliferation on Tdo2 expression in primary macrophages as well as IL-10 and TGF-β signaling. Interestingly, genetic ablation of Tdo2 in primary macrophages reversed R-2-HG-associated reduced T cell proliferation when PD-ligand (L)1 immune checkpoint blockade was applied. Consequently, direct application of l-Kyn mimicked the phenotypic effects of R-2-HG in TDO2-deficient cells (Fig. 5g). As AHR and TDO2 transcripts are more abundant in human monocytes than in other immune cells (Extended Data Fig. 4f)27, these findings are indicative of an immune cell subtype-specific vulnerability of macrophages to paracrine reprogramming by IDH-mutant tumors.

a,b, l-Trp import and degradation dynamics in immune cell subsets exposed to R-2-HG. Intracellular LC–MS/MS measurements of l-Trp and l-Kyn in T cells and monocyte-derived macrophages exposed to 20 mM R-2-HG and varying doses of l-Trp. Top, linear regression of dose-dependent metabolite accumulation. Bottom, quantification of intracellular l-Trp and l-Kyn levels and the [l-Kyn][l-Trp]−1 ratio. s.e.m is shown as error bands (top) or bars (bottom). n = 3 independent healthy donors. s.e.m. is projected as error bands. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. c–e, Cell-free assay of IDO1, IDO2 and TDO2 enzymatic activities with increasing concentrations of synthetic R-2-HG. A nonlinear regression model (log (R-2-HG) versus response) is shown. Regression curves are representative of y = 0 + (0.16 − 0)(1 + 10(log(EC50)−x))−1 (EC50, half-maximum effective concentration). Produced l-Kyn levels were measured by absorbance at 321 nm (A321). Gray boxes indicate the concentration range of R-2-HG detected in patient glioma tissues12. n = 2 independent measurements for IDO1, n = 4 independent measurements for IDO2, n = 4 independent measurements for TDO2. f, Expression of Tdo2 in different murine immune cell subsets. RNA-seq data were derived from ref. 27. Tn, naive T cell; Teff, effector T cell; Tem, effector memory T cell; Tgd, γδ T cell; NK, natural killer cell (NK); Mo, monocyte; MG, microglia cell. g, Monocyte-derived macrophage–T cell 3H-dT proliferation assay. Monocyte-derived macrophages from n = 3 Tdo2+/+ and n = 3 Tdo2−/− mice were pretreated with 10 mM R-2-HG or vehicle; 10 ng ml−1 anti-IL-10 and anti-TGF-β antibodies (αIL-10/TGF-β) or an isotype (iso) control; 10 ng ml−1 anti-PD-L1 antibody or isotype control; or 10 mM l-Kyn or vehicle as indicated for 24 h and co-cultured with stimulated C57BL6/J T cells for 24 h. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. If not mentioned otherwise, all data are represented as mean ± s.e.m.
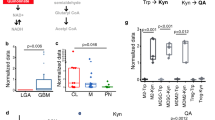
We showed that, in macrophages, TDO2 is directly induced by R-2-HG, leading to an accumulation of the AHR ligand l-Kyn. This pathological activation of the kynurenine pathway can only be sustained if decreasing l-Trp levels are sensed and extracellular l-Trp is imported to a sufficient extent, as its de novo synthesis is impossible for animal cells. Interestingly, exposure of human monocyte-derived macrophages to R-2-HG resulted in a similar amino acid transporter expression pattern as deprivation of extracellular l-Trp, suggesting that R-2-HG drives an amino acid starvation-like response as a result of increased l-Trp degradation in macrophages (Fig. 6a). Remarkably, the expression of SLC3A2 was higher in R-2-HG-exposed cells than in l-Trp-deprived cells. Of note, not Slc13a3, encoding the described transporter of R-2-HG, but Slc3a2 (CD98) contributed to the monocyte-to-macrophage trajectory in murine HGG (Fig. 3e). Slc3a2 and Slc7a5 (LAT1), forming the heterodimer LAT1–CD98 upon translation, were differentially upregulated in IDH-mutant compared to IDH-WT experimental tumors and displayed consistent expression patterns over the monocyte-to-macrophage trajectory (Fig. 6b,c). SLC3A2 (CD98) has multiple binding partners. Expression and in situ hybridization analyses of Slc7a5 across different tissue-resident macrophage populations revealed highest levels of Slc7a5 in CNS macrophages (Fig. 6c and Extended Data Fig. 5b,c)28. It was shown that transmembrane transport of branched-chain amino acids such as l-Trp is preferentially mediated by LAT1–CD9829 and might therefore provide the l-Trp needed to sustain activation of the kynurenine pathway by R-2-HG. Next, we investigated whether increased LAT1–CD98-dependent l-Trp import attenuated the AP capacity of macrophages in experimental HGG via AHR signaling. To this end, we investigated LAT1–CD98 inhibition in vitro and in vivo (Fig. 6d,e). Preconditioning of monocyte-derived macrophages with a small-molecule LAT1–CD98 inhibitor30 rescued the induction of 17 of 22 AHR targets that were induced by R-2-HG (Fig. 6d). Similarly, administration of the LAT1–CD98 inhibitor to glioma-bearing animals led to an increased abundance of MHCII+CD80+CD86+ immune-stimulatory macrophages in IDH-mutant tumors and thus enhanced the AP signature (Fig. 6e). This is consistent with our previous finding that, in the context of macrophage exposure to R-2-HG, tryptophan deprivation led to increased effector functions of co-incubated T cells (Fig. 4p). We aimed to investigate whether the microenvironment of human IDH-mutant gliomas is in fact configured for the maintenance of this l-Trp-dependent axis. Using a matrix-assisted laser desorption–ionization (MALDI)-MS imaging (MSI)-based analysis approach, we discovered the extracellular accumulation of l-Trp in our human HGG tissue cohort in situ (Fig. 6f and Extended Data Fig. 6a,b). As expected, all tumors diagnosed IDH mutant showed accumulation of R-2-HG, whereas there was no R-2-HG detectable in IDH-WT HGG or control cortex samples (Fig. 6f,g). Interestingly, we observed a strong accumulation of extracellular l-Trp in IDH-mutant HGG across all human replicate sets that was significantly higher than the abundance of l-Trp in IDH-WT HGG. l-Trp levels in IDH-WT HGG were moderate, while, in control cortex samples, they were comparable to the background level and thus significantly lower than in tumor samples (Fig. 6f,g). Increased levels of l-Trp in tumor cell lines upon mutant IDH1 and/or IDH2 overexpression or exogenous R-2-HG exposure was previously reported31. We similarly found a moderate-to-high increase in both intracellular and extracellular l-Trp levels, respectively, in IDH-mutant experimental HGG (Fig. 6h). While macrophages showed moderate expression levels of TDO2 as compared to other immune cell subsets, RNA-seq analysis of TCGA glioma datasets showed no increase in TDO2 expression levels in IDH-mutant tumors (Fig. 6i)32. Collectively, these data suggest that an overstimulated, R-2-HG-dependent uptake of l-Trp by myeloid cells via LAT1–CD98 maintains the immunosuppressive reprogramming of GAM in IDH-mutant HGG.

a, Arrayed PCR analysis of human amino acid and drug transporters in monocyte-derived macrophages treated in vitro with R-2-HG or vehicle in medium containing 5 mg l−1 l-Trp with FBS or l-Trp-free medium with dialyzed FBS. ΔΔCt values relative to the vehicle condition are shown. n = 3 pooled healthy human donors. b,c, Local polynomial curve fit of Slc3a2 (b) and Slc7a5 (c) expression along the murine HGG monocyte-to-macrophage trajectory (top); expression analyses of Slc3a2 and Slc7a5 across different tissue-resident macrophage populations by RNA-seq (bottom, dataset derived from ref. 27, mean values shown). Bl., blood; emb., embryonal; Kupff, Kupffer; mo, monocyte; Per, peritoneal; PP, Peyer’s patch; q.d., quaque die, once a day; spl., spleen. d, Induction of AHR target genes in macrophages by 20 mM R-2-HG ± 1 µM LAT1–CD98 inhibitor (LAT1–CD98i) or vehicle as determined by a PCR AHR signaling array. n = 3 pooled healthy human donors. e, Inhibition of LAT1–CD98 is efficient in reverting the IDH-mutant-induced GAM phenotype. n = 4 IDH1-mutant GL261 HGG-bearing mice treated with vehicle, n = 7 IDH1-mutant GL261 HGG-bearing mice treated with the LAT1–CD98 inhibitor. Flow cytometry analysis of glioma-associated macrophages. Box and whiskers (minimum to maximum, median as center) are shown. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. f,g, Spatial distribution and colocalization of R-2-HG and l-Trp assessed by MALDI-MSI in tissue of HGG patient and control sample sets. n = 5 control cortex samples, n = 5 IDH-mutant patient samples, n = 5 IDH-WT patient samples. f, Illustration of corresponding hematoxylin and eosin (H&E)-stained tissues (first columns), two-dimensional (2D) and three-dimensional (3D) ion surface plots of R-2-HG signal intensity (second and third columns, respectively) and 2D and 3D ion surface plots of l-Trp signal intensity (fourth and fifth columns, respectively). g, Left, violin plot of the estimated signal intensity distribution (intraset scaled to (0, 1) AU) for R-2-HG. Right, violin plot of the estimated signal intensity distribution (intraset scaled to (0, 1) AU) for l-Trp. Statistical significance was determined by one-way ANOVA in combination with Tukey’s honest significant difference test (***P < 0.001). h, Quantification of extracellular and intracellular l-Trp metabolite levels along the kynurenine pathway in GL261 IDH-mutant and GL261 IDH-WT tumors by LC–MS/MS. Concentrations were normalized to volume or cell count. A log–log plot is shown. i. Expression of TDO2 in TCGA low-grade glioma (LGG, n = 286) and HGG (n = 402) datasets. Data were derived from ref. 33. Statistical significance was determined by one-way ANOVA in combination with Tukey’s honest significant difference test. If not mentioned otherwise, all data are represented as mean ± s.e.m.
Our previous findings suggested that R-2-HG-induced T cell suppression by GAM is dependent on accumulation and degradation of l-Trp and functional AHR. Inhibition of AHR or LAT1–CD98 by small-molecule inhibitors was effective in reverting R-2-HG-mediated reprogramming of macrophages as revealed by an AHR target screening array (Figs. 6d and 7a and Extended Data Fig. 4h). When co-cultured with AHR-deficient macrophages exposed to R-2-HG, there was furthermore no consistent suppression of T cell proliferation or effector function, irrespective of l-Trp abundance (Fig. 7b). We thus aimed to test whether IDH-mutant experimental HGG harbor a unique immunological vulnerability that could be therapeutically exploited by AHR inhibition. We therefore combined T cell-activating immunotherapy by immune checkpoint blockade (anti-PD-L1 antibody) with a small-molecule AHR inhibitor33 to test the impact of the AHR-mediated dysregulated immune microenvironment found in IDH-mutant HGG (Fig. 7c,d). In total, 40% of mice inoculated with IDH-WT HGG and treated with immune checkpoint blockade were long-term survivors, whereas there were no responders to immune checkpoint blockade in mice bearing IDH-mutant tumors, supporting the observation that IDH-mutant tumors in fact foster a more tolerogenic alignment of their immune microenvironment. While administration of an AHR inhibitor did not improve outcomes in mice bearing IDH-WT tumors (Fig. 7c), we observed statistically significantly prolonged survival of mice bearing IDH-mutant tumors (Fig. 7d). In line with previous results (Figs. 3i and 4k), monocyte-derived macrophages from IDH-mutant gliomas treated with the AHR inhibitor displayed a higher level of AP markers. Production of IL-10 and TGF-β by monocyte-derived cells was reduced by AHR inhibition in IDH-mutant tumors but not in IDH-WT tumors (Fig. 7c,d).

a, Induction of AHR target genes in macrophages by treatment with 20 mM R-2-HG ± 1 µM AHR inhibitor (AHRi) or vehicle as determined by the TaqMan real-time (RT)–PCR AHR signaling array. Fractional differences are shown as indicated. n = 3 pooled healthy human donors. b, Monocyte-derived macrophage T cell-suppression assay. BMDMs of Ahr−/− mice were differentiated in vitro with R-2-HG or vehicle in RPMI 1640 medium containing 5 mg l−1 l-Trp with FBS or l-Trp-free medium with dialyzed FBS for 24 h. Cells were then co-cultured for 72 h with syngeneic T cells stimulated with anti-CD3 and anti-CD28 antibodies. T cells were subsequently analyzed by flow cytometry. Relative measurements (R-2-HG/vehicle, %/%) of T cell proliferation and IFN-γ+ or GrzB+ T cells are shown. Statistical significance was determined by two-tailed Student’s t-tests. n = 8 paired samples from four individual mice were used to assess proliferation and GrzB+ cells, n = 6 paired samples from three individual mice were used to assess IFN-γ+ cells. c,d, Therapeutic benefit of AHR inhibition is restricted to IDH-mutant glioma. C57BL/6J mice were treated with 100 µg anti-PD-L1 antibody every 3 d (q3d), AHR inhibitor (50 mg per kg twice a day (bid) in 40% 2-hydroxypropyl)-β-cyclodextrin (HPBCD) in water, pH 7.2) or vehicle (40% HPBCD in water, pH 7.2, bid) after magnetic resonance imaging (MRI)-based randomization of tumor volume at d8 after intracranial injection of GL261 IDH-WT or GL261 IDH1-R132H tumors (n = 19 IDH-WT tumor-bearing mice versus n = 17 IDH-mutant tumor-bearing mice). Top, Kaplan–Meier survival curves using the log-rank Mantel–Cox test for statistical testing. Bottom, flow cytometry analysis of tumor-infiltrating macrophages (CD45hiCD11b+F4/80+Gr-1−). Statistical significance was determined by the Mann–Whitney test. If not otherwise stated, all data are represented as mean ± s.e.m.
Discussion
Tumor-associated macrophages play a crucial role in a wide array of pathological hallmarks of tumors including gliomas2,34. In line with previous studies35,36,37,38, our study defines biologically relevant functional states of GAM that are controlled by mutant IDH, a disease-defining driver mutation in gliomas39. Moving beyond single-time-point assessments, we now longitudinally describe differential immune cell infiltration and phenotype dynamics during glioma progression that are orchestrated by a fluctuating network of resident microglial cells and educated recruited myeloid cells. In late-stage experimental tumors, monocyte-derived macrophages further drive the tolerogenic alignment of the glioma microenvironment. In IDH-mutant gliomas, we define the molecular mechanism as causally related to dynamic, R-2-HG-dependent tryptophan degradation by myeloid cells via TDO2 and LAT1–CD98, resulting in the activation of the immunity master regulator AHR.
Our study shows that R-2-HG not only profoundly shapes the glioma microenvironment but regulates targetable immunosuppressive tryptophan catabolism in myeloid cells. The robustness of IDH genotype-dependent effects on the innate immune microenvironment in mouse and human datasets presented is underscored by the recent recognition of considerable interspecies variation of microglial programs40. Our study not only supports the notion that HGG-associated microglia lose the homeostatic gene expression signature present in normal brain tissue to enter a functionally altered cellular stage but also proposes a concept whereby the immunosuppressive phenotype in IDH-mutant HGG is a result of an altered differentiation route of tumor-infiltrating monocytes.
Importantly, and in addition to tumor cell-autonomous metabolic vulnerabilities conferred by IDH mutations41,42,43, our study reveals an unexpected function of R-2-HG in regulating amino acid metabolism in immune cells. R-2-HG is taken up by myeloid cells to enzymatically induce TDO2-dependent activation of the kynurenine pathway and, subsequently, the AHR. This pathological tryptophan degradation results in an amino acid starvation-like response that triggers the expression of LAT1–CD98, a key transporter for tryptophan in proliferating cells25, which was previously linked to T cell activation and differentiation44,45. We here provide evidence that LAT1–CD98 is critically involved in the differentiation and activation of GAM and that the previously observed altered amino acid metabolism in IDH-mutant gliomas31 is also responsible for shaping an immunosuppressive tumor microenvironment through maintenance of this complex metabolic axis. We show that this regulatory metabolic network is particularly active in macrophages infiltrating IDH-mutant gliomas through their distinct expression profile, which constitutes a metabolic vulnerability. Our study supports the hypothesis that gliomas with immunogenic driver mutations, such as IDH1-R132H46, in addition to cell-autonomous epigenetic aberrations47, evolve by suppressing immune responses toward this neoantigen through production and secretion of R-2-HG. Importantly, we show that, similar to tumor cell-autonomous metabolic vulnerabilities conferred by IDH mutations, specific R-2-HG-mediated immune vulnerabilities can be therapeutically exploited to sensitize gliomas to immunotherapy.
On a broader level, our data suggest that genetic alterations of driver genes, beyond tumor-intrinsic prognostic implications, result in specific configurations of the immune microenvironment that can be therapeutically exploited by addressing tumor and immune cell metabolism.
Methods
Prospective tissue collection and histopathological validation
Human control (n = 7; mean age, 50 ± 19.07 years (21–74 years); sex, female:male (F:M)(4:3)), IDH1-WT HGG (n = 5; mean age, 65.9 ± 14.9 years (32–81 years); sex, F:M(1)) and IDH1-R132H-mutant HGG (n = 5; mean age, 48.5 ± 6.81 years (39–55 years); sex, F:M(0:5)) samples were prospectively collected from adult patients undergoing brain surgery after informed consent. Patient sample collection at the Freiburg site was regulated under the ethics protocol 472/15. Briefly, after surgical removal, samples were transferred into ice-cold PBS. A representative piece of the sample was snap frozen, cryosectioned, stained with H&E and examined by two board-certified neuropathologists. After inclusion into the study, samples were fixed with formalin (4% paraformaldehyde in PBS) and embedded in paraffin for in situ validation. The remaining sample was maintained on ice at all times. After removal of the meninges, tissue samples were transferred into ice-cold HBSS containing 10 mM glucose and 10 mM HEPES and mechanically dissociated using glass shearing with a 10-ml Potter-Elvehjem pestle and glass-tube homogenizer (Merck). The suspension was passed through a 70-µm cell strainer (BD Biosciences). Myelin was removed using gradient centrifugation with 37% Percoll (Merck) at 800g for 30 min without a brake. Cell pellets were dissolved and cryopreserved in FCS:DMSO (9:1; Merck).
Flow cytometry sorting
Human samples
A MoFlo Astrios (Beckman Coulter) was used for cell sorting. The cell suspension was stained with the following antibodies: anti-CD45 (clone HI30, BD Biosciences), anti-CD11b (clone M1/70, eBioscience), anti-CD3 (clone SP34–2, BD Biosciences), anti-CD19 (clone SJ25C1, BioLegend) and anti-CD20 (clone 2H7, BioLegend). Before surface staining, Fc receptors were blocked (BD Biosciences). DAPI-positive cells were excluded (1:1,000).
Murine samples
Brain tumors were digested with Liberase D (50 µg ml−1) and meshed through a cell strainer to obtain a single-cell suspension. Myelin was removed using a continuous 30% Percoll gradient. The cell suspension was stained with the following antibodies after blocking Fc receptors (anti-mouse CD16/CD32 antibody, BioLegend): anti-CD45.2 (clone 30-F11, BioLegend), anti-CD11b (clone M1/70, BioLegend), anti-NK1.1 (clone REA1162, Miltenyi Biotec), anti-CD3 (clone 17A2, BioLegend) and fixable viability dye eFluor 780 (Invitrogen). Cells were sorted under sterile conditions on a BD FACSAria Fusion equipped with the following lasers: 405 nm, 488 nm, 561 nm and 640 nm, using an 85-µm nozzle and four-way purity mode.
Single-cell RNA sequencing, transcript quantification and analysis
Single-cell transcript amplification and library preparation were performed using mCEL-seq2 as previously described48,49. Paired-end reads were aligned using BWA (version 0.6.2-r126) with default parameters50 to a transcriptome containing all gene models based on human ENCODE release version 24. Isoforms of a given gene were treated as one gene locus. The right mate of each read was mapped to an ensemble of all gene loci and 92 ERCC spike-ins in the sense direction51. The left mate contained the unique molecular identifier (UMI, six bases) and a cell-specific barcode (six bases), followed by a poly-T stretch. The number of distinct UMIs was recorded for each gene locus and assigned to each cell based on the cell barcode. Transcript counts were aggregated from UMI aggregate counts based on binomial statistics52.
Human single-cell RNA sequencing data analysis
Samples were sequenced in batches of eight libraries with 192 cells each per lane on an Illumina HiSeq 3000 sequencing system (paired-end multiplexing run) at a depth of ~130,000–200,000 reads per cell. Fifty-two libraries with 4,460 cells after quality control (control, 2,494; IDH1-WT HGG, 1,000; IDH1-mutant HGG, 966) were integrated and analyzed using Seurat version 3 (ref. 53). Cells expressing >2% of KCNQ1OT1, a previously identified marker of cells with low quality7, were excluded from analysis. Also, transcripts with a correlation coefficient >0.65 with KCNQ1OT1 were excluded. A total of 32,088 gene loci were quantified with a median of 1,296 ± 753.55 (s.d.) genes per cell. Data analysis was performed using the Seurat version 3 pipeline. The counts table was filtered for features expressed by at least three cells and cells with at least 500 detected features, corresponding to the arguments ‘min.cells=3’ and ‘min.features=500’ in the CreateSeuratObject function call. Data were scaled and normalized using the SCTranform function53 with the function set to return 10,000 variable features and regress out the percentage of mitochondrial genes, corresponding to the arguments ‘variable.features.n=10,000’, ‘return.only.var.genes=F’ and ‘vars.to.regress=”percent.mt”’. For dataset integration, 10,000 variable features were used with batch-effect associated features that contained the following patterns in their name filtered out: JUN, FOS, RP, ZFP36, EGR, HSP, MALAT1, XIST, MT- and HIST. These genes were subsequently also removed from the RunPCA function. Subsequently, UMAP and shared nearest-neighbors graph construction were performed on the top 15 principal components. Clusters were identified with resolution set to 0.6.
Analysis of the mouse 10x single-cell RNA sequencing data
First, 10x libraries were prepared from CD45+ cells purified by flow cytometry in a single run. Read alignment and transcript quantification were conducted using Cell Ranger version 3.1.0. The resulting counts files were analyzed using the Seurat version 3 dataset integration workflow. The counts table was filtered for features expressed by at least five cells and cells with at least 500 detected features, corresponding to the arguments ‘min.cells=5’ and ‘min.features=500’ in the CreateSeuratObject function call. Data were scaled and normalized using the SCTranform function, with the function set to return 10,000 variable features and regress out the percentage of mitochondrial genes, corresponding to the arguments ‘variable.features.n=10,000’, ‘return.only.var.genes=F’ and ‘vars.to.regress=”percent.mt”’. For dataset integration, 10,000 variable features were used with batch-effect associated features that contained the following patterns in their name filtered out: Jun, Fos, Gm, Rpl, Rps, Atf3, Zfp36, AY, Egr, Hsp, Malat1, Xist, mt-, Hist and Socs3. These genes were subsequently also removed from the RunPCA function. Subsequently, UMAP and shared nearest-neighbors graph construction were performed on the top 15 principal components. Clusters were identified with resolution set to 2.5.
Differential gene expression analysis of single-cell RNA sequencing data
Differential gene expression analysis was conducted using the FindAllMarkers function of Seurat version 3. For comparisons between two conditions, the FindMarkers function was used. Features with an average log fold change greater than 0.25 and an adjusted P value less than 0.05 were considered significant.
Cluster enrichment analysis
Enrichment analysis for a given condition in a cluster was conducted using a hypergeometric test implemented in R under the phyper function. This test considers the number of cells from condition x in a given cluster with respect to all cells from condition x in the dataset, all cells from condition y in the dataset and the number of cells in a given cluster. We used it to calculate the probability that number n or more cells from condition x could be found in a given cluster by chance. Statistical significance was assumed for probabilities <0.05. Correction for multiple testing was achieved using the Benjamini–Hochberg method.
Clusterwise comparison of gene expression
To compare gene expression between clusters and conditions, we fit a generalized linear model with a negative binomial link function using the ‘glm.nb’ function of the MASS R package. Pairwise testing was achieved by calculating the estimated marginal means for comparisons of diagnoses per cluster using the emmeans function of the emmeans R package with Tukey adjustment.
Pseudotime analysis of single-cell transcriptomes
Pseudotime trajectory analysis of scRNA-seq data was conducted using the StemID2 functionality of RaceID3 and the FateID R package with default settings7,48. First, a lineage tree was computed using the nearest-neighbor mode (nmode=TRUE) with default parameters of StemID2. Next, a list of significant links determined in the previous step was chosen based on the underlying question. A filtered gene expression matrix was obtained through the getfdata function of the FateID R package and used as input for pseudotime gene expression analysis of cells along the given list of links. Genes expressed at less than two normalized transcripts in at least ten cells in mice were filtered out using the filterset FateID function. Genes with similar gene expression profiles were grouped into modules on a self-organizing map using the getsom FateID function with the minimal size of modules set to 3 and the correlation threshold set to 0.85. With the help of the procsom function, modules on self-organizing maps were grouped into larger modules that were used for visualization and downstream gene ontology term and other analyses.
Bulk RNA sequencing of GL261 IDH1-WT and IDH1-R132H cell lines
Bulk RNA-seq data from GL261 IDH1-WT and IDH1-R132H cell lines were analyzed on the usegalaxy.eu platform. Raw FastQ files were mapped to the mm10 genome using STAR aligner version 2.7.5b followed by featureCounts version 1.6.4. For figures, RPKM (reads per million kb) values were compared.
CyTOF sample preparation and measurement
CyTOF was conducted as previously described54 using intracellular barcoding for mass cytometry. Briefly, cells pelleted by Percoll-gradient centrifugation were fixed with a fixation–stabilization buffer and frozen at –80 °C until analysis. Thawed cells were barcoded using premade combinations of six different palladium isotopes: 102Pd, 104Pd, 105Pd, 106Pd, 108Pd and 110Pd (Cell-ID 20-Plex Pd Barcoding kit, Fluidigm) and pooled for further processing. The resulting cell pellet was resuspended in 100 µl antibody cocktail specific for surface markers (Supplementary Tables 1 and 2). Of note, we excluded 169Tm-TGF-β (antibody panel B) from the analysis because of high background in this channel. For intracellular staining, stained cells were subsequently incubated in fixation–permeabilization buffer (Fix/Perm Buffer, eBioscience) for 60 min at 4 °C, washed with permeabilization buffer (eBioscience) and stained with antibody cocktails against intracellular molecules in permeabilization buffer for 1 h at 4 °C. Cells were subsequently washed twice with permeabilization buffer and incubated overnight in 2% methanol-free formaldehyde. The next day, cells were washed and resuspended in iridium intercalator solution (Fluidigm) for 1 h at room temperature. Afterward, samples were washed with cell-staining buffer and ddH2O (Fluidigm). Cells were pelleted and kept at 4 °C until CyTOF measurement. Cells were analyzed using a CyTOF2, upgraded to Helios specifications, with software version 6.5.236. Instrument and acquisition settings were set up as described previously54. Mass cytometry data processing and analysis were performed as described previously using Cytobank and CATALYST54. Clusterwise comparisons of protein expression between conditions was conducted on clusters that contained at least 0.05% of all cells per condition to ensure robust comparisons (>31 cells for antibody panel A and >22 cells for antibody panel B). Clusters that were below this threshold were not considered for visualization or analysis. Pairwise testing was performed using the one-way Kruskal–Wallis test followed by Dunn’s post hoc test with multiple-testing adjustment according to the Benjamini–Hochberg method.
Peripheral blood mononuclear cells
Peripheral blood mononuclear cells were isolated from research-only buffy coat formulations from healthy donors or patients from the Neurology Clinic Heidelberg upon patient consent. EDTA was used as an anticoagulant. Blood formulations were kept at 4 °C before further processing.
Experimental animals
C57BL/6J WT mice were purchased from Charles River. B6.Tdo2tm1Tnak (Tdo2–/–) and B6;129-Ahrtm1Bra/J (Ahr–/–) mice were bred according to local regulatory authorities (breeding approval reference EP-Z124I02). All mice were 7–10 weeks of age at use. Mice were kept under specific pathogen-free conditions at the animal facility in the DKFZ Heidelberg.
Tumor cell inoculation
GL261 tumor cells (1 × 104 cells) were diluted in 2 µl sterile PBS (Sigma-Aldrich) and stereotactically implanted into the right hemisphere of 7–10-week-old female C57BL/6J mice (coordinates, 2 mm right lateral to the bregma and 1 mm anterior to the coronal suture with an injection depth of 3 mm below the dural surface) using a 10-µl Hamilton microsyringe driven by a fine-step stereotactic device (Stoelting).
Cell lines
The murine glioma cell line GL261 was obtained from the Division of Cancer Treatment and Diagnosis at the National Cancer Institute. GL261 cells were cultured in DMEM (Sigma-Aldrich), supplemented with 10% FBS (Sigma-Aldrich), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen).
The embryonic kidney cell line HEK293 was obtained from ATCC and sold by LGC Standards. This cell line was cultured in DMEM (Sigma-Aldrich), supplemented with 10% FBS (Sigma-Aldrich), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen).
Determination of tryptophan metabolites
Frozen cell pellets were processed following an adjusted extraction protocol targeting tryptophan and kynurenine metabolites55,56. Briefly, samples were disrupted in 100 µl acidified mobile phase (0.2% formic acid with 1% acetonitrile in water) and 400 µl ice-cold methanol using a sonication bath (Transsonic 460, Elma) for 15 min at the highest frequency. Afterward, samples were kept at −20 °C for 15 min to precipitate all protein. Subsequently, samples were centrifuged for 15 min at 4 °C and 16,400g, and the resulting supernatant was transferred to a new 1.5-ml microcentrifuge tube (Eppendorf). Finally, the supernatant was dried using the Eppendorf Concentrator Plus, set to no heat, and resuspended in 40 µl acidified mobile phase.
For metabolite separation and detection, an ACQUITY I-class PLUS UPLC system (Waters) coupled to a QTRAP 6500+ (SCIEX) mass spectrometer with an electrospray ionization source was used. In detail, metabolites were separated by reversed-phase chromatography on an ACQUITY HSS T3 column (150 mm × 2.1 mm, 1.7 µm, Waters) kept at 20 °C with a flow rate of 0.4 ml min−1. An overview of multiple-reaction monitoring transitions that were used can be found in Supplementary Table 5. Clear separation of tryptophan and tryptophan-derived compounds was achieved by increasing the concentration of solvent B (acetonitrile with 0.1% formic acid) in solvent A (water with 0.1% formic acid) as follows: 1 min, 5% B; 11 min, 40% B; 13 min, 95% B; 15 min, 95% B; and return to 5% B in 5 min. Data acquisition and processing was performed with the SCIEX OS software suite (SCIEX).
RT–qPCR
For isolation of RNA, cells were lysed without prior washing using TRIzol reagent (Thermo Fisher Scientific) and purified using the RNeasy MinElute Cleanup kit (Qiagen). Next, 1 μg total mRNA was used for cDNA synthesis using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer’s instructions. cDNA was synthesized as described above. RT–qPCR was performed using the primaQuant qPCR SYBR Green Master Mix with ROX (Steinbrenner), and samples were run on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific). All samples were analyzed in quadruplicate, and melting curves were considered to evaluate PCR reactions. Ct values were normalized to both GAPDH and RPL9 (human), or Rpl13 (murine), respectively. Primer-only reactions served as negative controls. All primers were checked for primer efficiency by RT–qPCR and serial dilution of cDNA and were used if efficiency was >90%.
Treatment with R-2-hydroxyglutarate in vitro
d-α-hydroxyglutaric acid disodium salt (≥95.0% purity, determined by gas chromatography) was obtained from Sigma-Aldrich and reconstituted in PBS (Sigma-Aldrich) at a concentration of 2 M. Cells were treated with R-2-HG by diluting the 2 M stock solution in the respective cell medium. R-2-HG-containing medium was then pulse vortexed and added to the cells.
[3H]thymidine proliferation measurements
If not mentioned otherwise, T cells or mixed cells from co-cultures were seeded at 500,000 cells per well in a 96-well plate as technical quadruplicates and pulsed with RPMI 1640 (PAN-Biotech) with 10% FBS and human serum AB (both from Sigma-Aldrich), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen), 2 mM l-glutamine (Invitrogen) and 50 μM β-mercaptoethanol (Sigma-Aldrich), supplemented with [methyl-3H]thymidine (PerkinElmer), resulting in a radioactivity concentration of 20 mCi ml−1. Incorporation of radioactively labeled thymidine was allowed for 18 h, after which cells were shock frozen and kept at −20 °C. Scintillation counting was performed to determine radionuclide uptake using a cell harvester (Tomtec) and a scintillation counting device (Wallac MicroBeta TriLux Scintillation Counter, PerkinElmer). Proliferation measurements were given in counts per minute.
Cytokine ELISA
Primary human macrophages and dendritic cells were treated as described above and incubated for 72 h after unspecific stimulation with LPS (Sigma-Aldrich) and recombinant human IFN-γ (PeproTech). Supernatants were transferred to cytokine-specific antibody-coated ultra-low-binding 96-well plates (Corning), and cytokine ELISAs were performed using horseradish peroxidase-conjugated antibodies according to the manufacturer’s instructions (eBioscience). The development process was stopped with 1 M H2SO4, and optical density (OD) was measured at 570 nm and 450 nm. Cytokine concentrations were calculated based on OD450 nm−570 nm according to parallel serial dilutions of cytokine standards included in the respective ELISA kits. ELISA detection was used for the following human cytokines: IL-10, TGF-β (ELISA Ready-SET-Go! kits, eBioscience). Murine cytokines were measured accordingly.
AHR activity assays
Assays were performed as described previously57. Briefly, HEK293T cells were transfected in 96-well plates with a combination of plasmids for expressing AHR and ARNT and reporter plasmids: either the dioxin-responsive element (DRE)-GFP reporter (Qiagen) or the pGudLuc7.1F Cignal xenobiotic response element (XRE) reporter. Cells were transfected using FuGENE HD (Promega) following the manufacturer’s instructions (FuGENE HD:DNA ratio of 3:1). Cells were cultured with the indicated compounds or GL261 cell line supernatant, and the activity of the reporter was measured after 6 h using a Promega luciferase assay kit (E1500) following the manufacturer’s instructions. Luminescence was measured using a PHERAstar FS plate reader (BMG LABTECH). Data represent the mean of three cell lines normalized to the highest intensity result, where, within each cell line replicate, each condition was repeated in triplicate.
MALDI Fourier-transform ion cyclotron resonance mass spectrometry imaging
For MALDI-MSI analyses, frozen tissue sections on ITO slides were dried in a vacuum for 15 min at room temperature and subsequently spray-coated with 1,5-DAN prepared at 10 mg ml−1 with a 50% acetonitrile (vol/vol) matrix using an HTX TM-Sprayer (HTX Technologies). The matrix deposition protocol consisted of ten layers sprayed at a matrix flow rate of 100 µl min−1 and a spray-head velocity of 1,200 mm min–1 with a distance of 3 mm between sprayed lines (HH pattern). The spray nozzle height was set to 40 mm from the ITO slide, and temperature was increased to 60 °C with a pressure of 10 psi and a gas flow rate of 2 l min−1. High-resolution data acquisition was performed using the 7T Fourier-transform ion cyclotron resonance (FTICR) mass spectrometer (SolariX XR, Bruker Daltonics) equipped with an Apollo II dual MALDI/ESI ion source and a 2-kHz Smartbeam II laser. MSI data were recorded in negative-ion mode within a m/z range of 100–12,000 using a raster width of 50 μm and 100 laser shots per pixel at a laser power of 18%. Spectra were recorded using 1 million transient data points (FID 0.4893 s) with an online calibration using an internal lock mass of m/z 157.076025 (deprotonated 1,5-DAN peak). The following parameters were used: ion transfer (funnel 1, 150 V; skimmer 1, 15 V; funnel RF amplitude, 150 Vpp), octopole (frequency, 5 MHz; RF amplitude, 350 Vpp), collision cell (RF frequency, 2 MHz; RF amplitude, 1,900 Vpp), transfer optics (time of flight, 0.5 ms; frequency, 6 MHz; FR amplitude, 350 Vpp); quadrupole (Q1 mass, 140 m/z); excitation mode (sweep excitation, sweep step time, 15 μs) and data reduction and storage (profile spectrum was saved with a data reduction factor of 97%). Data acquisition was performed using ftmsControl 2.2.0 from Bruker Daltonics, and measurement regions were specified using flexImaging 5.0x64 (Bruker Daltonics). MSI data were acquired from each tissue section as well as from matrix control areas to check for matrix interference on analytes.
A mixture of the following amino acids and compounds was used as an external quadratic calibration for the MALDI-FTICR-MS instrument: the deprotonated 1,5-DAN peak (m/z 157.076025), kynurenic acid (m/z 188.034219), tryptophan (m/z 203.081504), kynurenine (m/z 207.076419), sunitinib (m/z 397.203431), olanzapine (m/z 311.132494), sorafinib (m/z 463.077929) and CZC54252 (m/z 503.126278).
Flow cytometry
For intracellular cytokine staining, cells were incubated with 5 μg ml−1 brefeldin A (Sigma-Aldrich) for 5 h at 37 °C with 5% CO2 to allow for intracellular enrichment of cytokines. Brain tumor and spleen cell suspensions were blocked with anti-CD16/CD32 (eBioscience, 93, 14-0161), and extracellular targets were stained for 30 min at 4 °C (Supplementary Table 4). For detection of intracellular antigens, cells were fixed, permeabilized and stained using the FOXP3 Transcription Factor Staining buffer set (eBioscience, 00-5523) and the antibodies listed in Supplementary Table 4. Staining of intracellular targets was performed for 45 min at 4 °C. Stained lymphocytes were analyzed on the FACSCanto II or the LSRFortessa (BD Biosciences) or on the Attune NxT (Thermo Fisher). BD FACSDiva software version 9 and FlowJo version 9 or 10 were used for data analysis.
Statistics and reproducibility
Single-cell profiling
For Figs. 1–3 and Supplementary Fig. 1, no statistical method was used to predetermine sample size. Experiments were not randomized. Investigators were not blinded to allocation during experiments or outcome assessment.
For Fig. 1d, hypergeometric testing was used to test for enrichment. Genes expressed by less than three cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 1e, differentially expressed genes were determined based on the Wilcoxon rank-sum test using the FindAllMarkers Seurat function. The expression of the top 15 marker genes per cluster is shown. Genes expressed by less than three cells and cells with less than 500 detected genes were excluded from the analyses. Furthermore, mitochondrial genes and genes associated with the dissection response were excluded. The pattern these gene names contained are the following: HTRA, LIN, EEF, CTC-, MIR, CTD-, AC0, RP, FOS, JUN, MTRNR, MT-, XIST, DUSP, ZFP36, RGS, PMAIP1, HSP, NEAT1, HIST and MALAT1.
For Fig. 1f, genes differentially expressed between the two indicated clusters were determined based on the Wilcoxon rank-sum test using the FindMarkers Seurat function. Up to the five top marker genes per cluster are indicated. Genes expressed by less than three cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 1h, a Kruskal–Wallis test was conducted followed by Dunn’s test for multiple comparisons. Multiple-testing adjustment was performed using Holm’s method. Clusters with less than 1‰ cells per condition were excluded from the analyses.
For Fig. 2e, hypergeometric testing was used to test for enrichment. Genes expressed by less than ten cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 2f, mean cell type percentages within each glioma genotype were compared based on the negative binomial function. Pairwise comparisons were performed by calculating the estimated marginal means for comparisons of diagnoses per cluster with Tukey adjustment. Genes expressed by less than ten cells, cells with less than 500 detected genes and cells with absolute average counts less than 20 were excluded from the analysis.
For Fig. 2g, genes differentially expressed between microglia from the two glioma genotypes were determined based on the Wilcoxon rank-sum test using the FindMarkers Seurat function. Up to the five top marker genes per cluster are indicated. Genes expressed by less than ten cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 2h, statistically significant intercluster links were determined using the StemID2 algorithm. Genes expressed by less than ten cells with less than two detected transcripts were excluded from the analyses.
For Fig. 2i, differentially expressed genes in the clusters along the trajectory from Fig. 2h were determined based on the Wilcoxon rank-sum test using the FindAllMarkers Seurat function. The expression of the top 15 marker genes per cluster is shown. Genes expressed by less than ten cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 2j, gradual gene changes along the indicated cell trajectory were determined using the StemID2 algorithm. Genes with a correlation >0.85 were arranged into modules with similar expression patterns. Genes expressed by less than ten cells with less than two detected transcripts and modules with less than three genes were excluded from the analyses. The Spearman correlation in the bottom panel was calculated in the bottom panel.
For Fig. 3b,c, genes differentially expressed between the indicated clusters were determined based on the Wilcoxon rank-sum test using the FindMarkers Seurat function. Representative top differentially expressed genes per cluster are indicated. Genes expressed by less than ten cells and cells with less than 500 detected genes were excluded from the analyses.
For Fig. 3d, statistically significant intercluster links were determined using the StemID2 algorithm. Genes expressed by less than ten cells with less than two detected transcripts were excluded from the analyses.
For Fig. 3e,f, gradual gene changes along the indicated cell trajectory were determined using the StemID2 algorithm. Genes with a correlation >0.85 were arranged into modules with similar expression patterns. Genes expressed by less than five cells with less than two detected transcripts and modules with less than three genes were excluded from the analyses. The Spearman correlation in the bottom panel was calculated in the bottom panel.
For Fig. 4i, left, the analysis corresponds to that in Fig. 3d.
For Fig. 4i, right, statistical significance of the interglioma genotype for the respective clusters was determined by fitting a generalized linear model with a negative binomial link function. Pairwise comparisons were performed by calculating the estimated marginal means for comparisons of diagnoses per cluster with Tukey adjustment. Genes from the indicated expression signature were included.
For Supplementary Fig. 1b, hypergeometric testing was used to test for enrichment. No statistical method was used to predetermine sample size. Genes expressed by less than three cells and cells with less than 500 detected genes were excluded from the analyses.
For Supplementary Fig. 1c, the statistical significance of each diagnosis for the respective clusters was determined by fitting a generalized linear model with a negative binomial link function. Pairwise comparisons were performed by calculating the estimated marginal means for comparisons of diagnoses per cluster with Tukey adjustment. Genes from the indicated expression signature were included.
For Supplementary Fig. 1h,j, a Kruskal–Wallis test was conducted, followed by Dunn’s test for multiple comparisons. Multiple-testing adjustment was performed using Holm’s method. Clusters with less than one promille cells per condition were excluded from the analyses.
For Supplementary Fig. 2h,i, the indicated lineage graphs were computed using StemID2 in neighbor mode with a P value cutoff of 0.01, and the scthresh parameter for links shown in the graph was set to 0.9
In vitro and in vivo experiments
For Figs. 2–7 and Extended Data Figs. 2–4, no statistical method was used to predetermine sample size. Experiments were not randomized. Investigators were not blinded to allocation during experiments or outcome assessment.
For in vitro experiments, unless stated otherwise in figure legends, data are represented as individual values or as mean ± s.e.m. Group sizes (n) and applied statistical tests are indicated in figure legends. Significance was assessed by either unpaired t-test analysis, paired t-test analysis or one-way ANOVA analysis with Tukey post hoc testing as indicated in figure legends. Spearman correlation was applied for all correlation analyses, and the Kaplan–Meier method was used to examine survival differences. Statistics were calculated using GraphPad Prism 7.0. Key experiments (TAM phenotyping, AHR reporter assays) were all performed at least three times or with biologically independent healthy human donors or mice. All other experiments were performed as specified in figure legends.
For in vivo experiments, unless stated otherwise in figure legends, data are represented as individual values or as mean ± s.e.m. Group sizes (n) and applied statistical tests are indicated in figure legends. Significance was assessed by either unpaired t-test analysis, paired t-test analysis or one-way ANOVA analysis with Tukey post hoc testing as indicated in figure legends. Survival was analyzed by the log-rank Mantel–Cox test. Sample size was calculated with the help of a biostatistician using R version 3.4.0. Assumptions for power analysis were as follows: α error, 5%; β error, 20%. Values for standard deviations and differences between experimental groups were based on previous experiments (whenever a similar data type was available). In all other cases, a pilot group size was used without using a statistical method to predetermine sample size. Mice were randomized into treatment groups stratified for tumor size (measured by MRI) at the time when treatment started. Intracranial tumor experiments were performed in a blinded manner (MRI, treatment, flow cytometric analyses). In case animals had to be killed before the pre-defined endpoint (due to weight loss or other termination criteria), they were excluded from any downstream analyses. All investigators were blinded to allocation during experiments and outcome assessment.
MALDI Fourier-transform ion cyclotron resonance mass spectrometry imaging
After being acquired, all MALDI-FTICR-MSI datasets were imported directly into R 3.6.0 (R Foundation for Statistical Computing) and processed using the MALDIquant package58. Datasets were acquired with on-the-fly centroid detection natively run on the measurement equipment, and, therefore, the imported datasets represented already centroided mass spectra peak-picked with a signal-to-noise ratio of 3. Resulting mass spectra were normalized to their total ion count. All subsequent analyses and visualization were performed in R. Mass resolution, which was calculated based on the full width at half maximum of analytes’ peaks, was approximately 200,000 for R-2-HG and 145,000 for both l-Trp and l-Kyn. After acquisition, the mass accuracy for the amino acid mixture mentioned in the previous section was recalculated on a pixel-wise basis. The maximum mass shift observed was <2.5 ppm with a median mass shift <1 ppm for all amino acid mixture analytes. To avoid picking spurious signals originating from tissue-cutting artifacts and tears, background matrix pixels were computationally dropped based on their total ion count and corresponding optical image pixel gray level intensity. To estimate the level of the detected intensities of the analytes of interest, a search window of 2 ppm (of the theoretical mass) was used to search for the presence of the corresponding analyte. The corresponding signal intensity was then collected and rescaled to an intensity range of (0, 1) to simplify visualization.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Bulk and scRNA-seq data that support the findings of this study were deposited in the Gene Expression Omnibus under the SuperSeries accession code GSE166420. This consists of the following SubSeries: GSE166218 (mouse 10x data), GSE166418 (human CEL-seq2 data) and GSE166521 (GL261 bulk RNA-seq data). Mass cytometry data were deposited in the FlowRepository at https://flowrepository.org/id/FR-FCM-Z3G7. TCGA dataset was downloaded from http://gliovis.bioinfo.cnio.es/. Imaging source data for this manuscript can be found at https://doi.org/10.6084/m9.figshare.14166983. All other data supporting the findings of this study are available from the corresponding author on reasonable request (m.platten@dkfz.de). Source data are provided with this paper.
Code availability
Custom code for transcriptomic and proteomic analyses can be found at https://github.com/rsankowski/friedrich-et-al-IDH1wt-mut-micr.git.
References
Broekman, M. L. et al. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 14, 482–495 (2018).
Gutmann, D. H. & Kettenmann, H. Microglia/brain macrophages as central drivers of brain tumor pathobiology. Neuron 104, 442–449 (2019).
Venteicher, A. S. et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902 (2019).
Müller, S. et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 18, 23 (2017).
Schmidt, S. V., Nino-Castro, A. C. & Schultze, J. L. Regulatory dendritic cells: there is more than just immune activation. Front. Immunol. 3, 274 (2012).
Grün, D. et al. De novo prediction of stem cell identity using single-cell transcriptome data. Cell Stem Cell 19, 266–277 (2016).
Jordão, M. J. C. et al. Neuroimmunology: single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019).
Darmanis, S. et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. 21, 1399–1410 (2017).
Bunse, L. et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 24, 1192–1203 (2018).
Pusch, S. et al. Pan-mutant IDH1 inhibitor BAY 1436032 for effective treatment of IDH1 mutant astrocytoma in vivo. Acta Neuropathol. 133, 629–644 (2017).
Dang, L. et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744 (2009).
Hagos, Y. et al. Organic anion transporters OAT1 and OAT4 mediate the high affinity transport of glutarate derivatives accumulating in patients with glutaric acidurias. Pflugers Arch. Eur. J. Physiol. 457, 223–231 (2008).
Mimura, J. & Fujii-Kuriyama, Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochim. Biophys. Acta 1619, 263–268 (2003).
Gramatzki, D. et al. Aryl hydrocarbon receptor inhibition downregulates the TGF-β/Smad pathway in human glioblastoma cells. Oncogene 28, 2593–2605 (2009).
Silginer, M. et al. The aryl hydrocarbon receptor links integrin signaling to the TGF-β pathway. Oncogene 35, 3260–3271 (2016).
Gandhi, R. et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3+ regulatory T cells. Nat. Immunol. 11, 846–853 (2010).
Zhu, J. et al. Aryl hydrocarbon receptor promotes IL-10 expression in inflammatory macrophages through Src–STAT3 signaling pathway. Front. Immunol. 9, 2033 (2018).
Platten, M., Nollen, E. A. A., Röhrig, U. F., Fallarino, F. & Opitz, C. A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 18, 379–401 (2019).
Pfefferkorn, E. R., Rebhun, S. & Eckel, M. Characterization of an indoleamine 2,3-dioxygenase induced by gamma-interferon in cultured human fibroblasts. J. Interferon Res. 6, 267–279 (1986).
Pfefferkorn, E. R. Interferon γ blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan (tryptophan/kynurenine/N-formylkynurenine). Proc. Natl Acad. Sci. USA 81, 908–912 (1984).
Badawy, A. A. B. & Guillemin, G. The plasma [kynurenine]/[tryptophan] ratio and indoleamine 2,3-dioxygenase: time for appraisal. Int. J. Tryptophan Res. 12, 1178646919868978 (2019).
Gagné, M. L., Boulay, K., Topisirovic, I., Huot, M.-É. & Mallette, F. A. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol. 27, 738–752 (2017).
Friedrich, M., Bunse, L., Wick, W. & Platten, M. Perspectives of immunotherapy in isocitrate dehydrogenase-mutant gliomas. Curr. Opin. Oncol. 30, 368–374 (2018).
Kaper, T. et al. Nanosensor detection of an immunoregulatory tryptophan influx/kynurenine efflux cycle. PLoS Biol. 5, e257 (2007).
Shay, T. & Kang, J. Immunological genome project and systems immunology. Trends Immunol. 34, 602–609 (2013).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 (2019).
Schmiedel, B. J. et al. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715 (2018).
Scalise, M., Galluccio, M., Console, L., Pochini, L. & Indiveri, C. The human SLC7A5 (LAT1): the intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front. Chem. 6, 243 (2018).
Kim, C. S. et al. BCH, an inhibitor of system l amino acid transporters, induces apoptosis in cancer cells. Biol. Pharm. Bull. 31, 1096–1100 (2008).
Reitman, Z. J. et al. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl Acad. Sci. USA 108, 3270–3275 (2011).
Bowman, R. L., Wang, Q., Carro, A., Verhaak, R. G. W. & Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 19, 139–141 (2017).
Schmees, N. et al. Abstract 4454: identification of BAY-218, a potent and selective small-molecule AhR inhibitor, as a new modality to counteract tumor immunosuppression. Cancer Res. https://doi.org/10.1158/1538-7445.AM2019-4454 (2019).
Kierdorf, K., Masuda, T., Jordão, M. J. C. & Prinz, M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat. Rev. Neurosci. 20, 547–562 (2019).
Friebel, E. et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181, 1626–1642 (2020).
Klemm, F. et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–1660 (2020).
Poon, C. C. et al. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 10, 3129–3143 (2019).
Sankowski, R. et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22, 2098–2110 (2019).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166 (2018).
Geirsdottir, L. et al. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179, 1609–1622 (2019).
Tateishi, K. et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell 28, 773–784 (2015).
Badur, M. G. et al. Oncogenic R132 IDH1 mutations limit NADPH for de novo lipogenesis through (d)2-hydroxyglutarate production in fibrosarcoma cells. Cell Rep. 25, 1018–1026 (2018).
McBrayer, S. K. et al. Transaminase inhibition by 2-hydroxyglutarate impairs glutamate biosynthesis and redox homeostasis in glioma. Cell 175, 101–116 (2018).
Sinclair, L. V. et al. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14, 500–508 (2013).
Cibrian, D. et al. CD69 controls the uptake of l-tryptophan through LAT1–CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat. Immunol. 17, 985–996 (2016).
Schumacher, T. et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 512, 324–327 (2014).
Núñez, F. J. et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 11, eaaq1427 (2019).
Herman, J. S., Sagar & Grün, D. FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nat. Methods 15, 379–386 (2018).
Masuda, T. et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392 (2019).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1756 (2009).
Baker, S. C. et al. The external RNA Controls Consortium: a progress report. Nat. Methods 2, 731–734 (2005).
Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat. Methods 11, 637–640 (2014).
Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20, 296 (2019).
Bottcher, C. et al. Human microglia regional heterogeneity and phenotypes determined by multiplexed single-cell mass cytometry. Nat. Neurosci. 22, 78–90 (2019).
Fuertig, R. et al. LC–MS/MS-based quantification of kynurenine metabolites, tryptophan, monoamines and neopterin in plasma, cerebrospinal fluid and brain. Bioanalysis 8, 1903–1917 (2016).
Sadok, I., Rachwał, K. & Staniszewska, M. Application of the optimized and validated LC–MS method for simultaneous quantification of tryptophan metabolites in culture medium from cancer cells. J. Pharm. Biomed. Anal. 176, 112805 (2019).
Schulte, K. W., Green, E., Wilz, A., Platten, M. & Daumke, O. Structural basis for aryl hydrocarbon receptor-mediated gene activation. Structure 25, 1025–1033 (2017).
Gibb, S. & Strimmer, K. MALDIquant: a versatile R package for the analysis of mass spectrometry data. Bioinformatics 28, 2270–2271 (2012).
Acknowledgements
We are grateful to all patients who have donated tissue for this study. We acknowledge the support of the DKFZ Light Microscopy Facility, the DKFZ Genomics and Proteomics Core Facility, the Transgenic Service of the Center for Preclinical Research, the DKFZ and the DKFZ–Bayer Alliance. We also acknowledge support from the Flow Cytometry Core Facility at the German Cancer Research Center and the Flow Cytometry Core Facility at the Medical Faculty Mannheim of Heidelberg University. We thank the Metabolomics Core Technology Platform at the Excellence Cluster ‘CellNetworks’ (University of Heidelberg) and the German Research Foundation (DFG, grant ZUK 40/2010-3009262) for support with UPLC-based metabolite quantification. We acknowledge the data storage service SDS@hd supported by the Ministry of Science, Research and the Arts Baden-Württemberg (MWK) and the German Research Foundation (DFG) through grants INST 35/1314-1 FUGG and INST 35/1503-1 FUGG. We thank D. Grün and Sagar, MPI-IE, Freiburg, for their excellent support with processing human samples for scRNA-seq. We acknowledge the assistance of the Charité | BIH Cytometry Core (Charité, Universitätsmedizin Berlin, Germany). We thank M. Fischer and J. Meyer for technical support. We acknowledge the support of the Freiburg Galaxy Team: R. Backofen, Bioinformatics, University of Freiburg (Germany), funded by the Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and the German Federal Ministry of Education and Research BMBF grant 031 A538A de.NBI-RBC. M.F. and L.B. are members of the MD–PhD program at Heidelberg University. M.F. received fellowships from the Heidelberg Biosciences International Graduate School, the Konrad-Adenauer Foundation, the German Academic Exchange Service (DAAD), the German Academic Scholarship Foundation (SDV) and the Excellence Initiative of the German Council of Science and Humanities and the German Research Foundation (DFG). L.B. was funded by Heidelberg Medical Faculty and the Else Kröner Fresenius Foundation. R.S. was funded by the Berta-Ottenstein-Programme for Clinician Scientists, Faculty of Medicine, University of Freiburg. M.K. and K.S. are supported by the Helmholtz International Graduate School. K.S. is supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 404521405, SFB 1389–UNITE Glioblastoma, Work Package B01. T.B. is supported by the Medical Faculty Mannheim and the University Hospital Mannheim. J.K.S. was funded by German Cancer Aid (project 70112399). K.A. was supported by the German-Israeli Helmholtz Research School in Cancer Biology (2536). F.S. was supported by the Else Kröner-Fresenius Excellence Program of the EKFS. C.B. was supported by the German Research Foundation (SFB TRR167 B05). J.P. was supported by the German Research Foundation (SFB TRR167 B05 and B07, TRR265 B04) and the UK DRI Momentum Award. T.T. is supported by a fellowship from the Germany Cancer Aid non-profit organization (Deutsche Krebshilfe). L.S. is supported by the Hertie Foundation (medMS-MyLab program; P1180016), the National Multiple Sclerosis Society (FG-1607-25111) and the Medical Faculty Mannheim, University of Heidelberg. M. Prinz was supported by the Sobek Foundation, the Ernst Jung Foundation, the DFG (SFB 992, SFB1160, SFB1479, SFB/TRR167, a Reinhart Koselleck grant and the Gottfried Wilhelm Leibniz prize) and the Ministry of Science, Research and Arts, Baden-Wüerttemberg (Sonderlinie ‘Neuroinflammation’). This study was supported by the DFG under Germany’s Excellence Strategy (CIBSS, EXC-2189, project 668 ID390939984) and by the Helmholtz Gemeinschaft, Zukunftsthema ‘Immunology and Infection’ (ZT0027), the Dr. Rolf M. Schwiete Foundation and the Sonderförderlinie ‘Neuroinflammation’ of the Ministry of Science of Baden-Württemberg, the German Ministry of Education and Science (the National Center for Tumor Diseases Heidelberg NCT 3.0 program ‘Precision immunotherapy of brain tumors’ and the DKTK program), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 404521405, SFB 1389–UNITE Glioblastoma, Work Package B01, and Project-ID-406052676; PL-315/5-1–Impact of dietary Tryptophan on the gut microbiome and autoimmune neuroinflammation, and the German Cancer Aid (projects 70112399 and 70113515) to M. Platten. This study was supported by grants from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project-ID 404521405, SFB 1389–UNITE Glioblastoma, Work Package B01 or B03 to T.B. or L.B. and S.P., respectively, and the Medical Faculty Mannheim at Heidelberg University (“Anerkennung von Spitzenleistung” and SEED program) and the Swiss Cancer Foundation to L.B.
Author information
Authors and Affiliations
Contributions
M.F. conceptualized the study, designed and performed experiments, analyzed and interpreted data and wrote the paper. L.B. was involved in study design and wrote the paper. R.S. analyzed scRNA-seq data and wrote the paper. M.F., M.K., J.K.S. and K.A. performed in vivo experiments. E.G. established AHR translocation assays. G.P. performed measurements of l-Trp metabolites and TCA cycle intermediates. S.P. performed R-2-HG measurements. T.B. was involved in study design. M.F., K.S., M.H., P.M., A.v.L. and F.C. performed in vitro experiments. T.T. and L.S. performed histological staining including RNA-ISH. C.R.G., D.A.-S. and C.H. performed MALDI-MSI. D.H.H., O.S. and J.B. obtained informed consent from patients and dissected brain tissues. C.B., C.F.-Z. and J.P. established and performed the CyTOF workflow for microglia as well as data processing and the clustering analysis of the CyTOF data. R.S. performed comparative analyses of CyTOF clusters. F.J.Q., W.W. and A.v.D. were involved in study design and data interpretation. S.H. performed MRI. I.G. provided BAY-218. M. Prinz and M. Platten conceptualized the study, interpreted data and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
M. Platten, W.W. and T.B. are inventors and patent holders on ‘Peptides for use in treating or diagnosing IDH1-R132H positive cancers’ (EP2800580B1). S.P. and A.v.D. are eligible to royalties as co-inventors of BAY 1436032 and are patent holders of ‘Means and methods for the determination of (D)-2-hydroxyglutarate (D2HG)’ (WO2013127997A1). The other authors declare no conflict of interest.
Additional information
Peer review information Nature Cancer thanks Tracy McGaha and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Integrated single-cell profiling of control and HGG-derived GAM.
a, Donut plots showing the cluster-wise cell compositions for the respective conditions. Clusters are arranged in a descending order from largest to smallest counterclockwise starting at 12 o’clock. Numbers indicate clusters with significant enrichment in a given condition based on hypergeometric testing shown in Fig. 1d. N = 7 patients for control tissue, n = 5 IDHwt GBM patients, n = 5 IDHmut GBM patients; N per condition is consistent with panel b. b, Violin plots represent probability density smoothed by a kernel density estimator with a line showing the median of corresponding overlaid boxplots that indicate the cumulative gene expression of indicated genes as part of the antigen presentation gene expression signature in scRNA-Seq analysis. Only clusters with significant differences in expression modelled based on a negative binomial distribution followed by pairwise testing using estimated marginal means with a significance cutoff of adjusted p-value < 0.05 are shown. P-values were calculated using the one-way Kruskal-Wallis test followed by the Dunn post-hoc test and adjusted for multiple testing using the Benjamini-Hochberg method. c, t-SNE map color-coded for similar clusters (obtained from the antibody panel A). d-e, t-SNE and Marimekko plot depicting the distribution of control, IDHmut and IDHwt microglia across the clusters. f, Violin plots showing the clusterwise expression of the proteins assessed. g, Violin plots depicting the cumulative expression of ApoE and EMR1 in the respective conditions. The color scheme is consistent with panel d.
Extended Data Fig. 2 Integrated single-cell profiling of control and HGG-derived GAM.
a, t-SNE map color-coded for similar clusters (obtained from the antibody panel B) of n = 46,384 cells. b-c, t-SNE and Marimekko plot depicting the distribution of control (n = 16,687), IDHmut (n = 12,172) and IDHwt (n = 17,525) microglia across the clusters. d, Violin plots showing the clusterwise expression of the proteins assessed in the experiment by antibody panel B. e, top: t-SNE map color-coded for the log-transformed cumulative expression of the homeostatic microglia signature proteins present in antibody panel B (CD115/CSF1R, P2RY12). The color scale indicates the color values associated with the respective log-transformed cumulative protein expression levels. bottom: t-SNE map color-coded for the log-transformed cumulative expression of the AP signature the protein present in antibody panel B (HLA-DR). The color scale indicates the color values associated with the respective log-transformed cumulative protein expression levels. f, Violin plots depicting the cumulative expression of the homeostatic microglia signature proteins in the respective conditions. N = 7 control cortex tissues, n = 4 IDHmut HGG patients, n = 9 IDHwt HGG patients. P-values were calculated using the one-way Kruskal-Wallis test followed by the Dunn post-hoc test and adjusted for multiple testing using the Benjamini-Hochberg method. The color scheme is consistent with panel b. g, Violin plots depicting HLA-DR expression in the respective conditions. P-values were calculated using the one-way Kruskal-Wallis test followed by the Dunn post-hoc test. Multiple testing adjustment was achieved using the Benjamini-Hochberg method. The color scheme is consistent with panels b and f.
Extended Data Fig. 3 Establishment of an IDH1-R132H-expressing syngeneic murine glioma model.
a, Vector map of scaffold/matrix attachment region (S/MAR) episomal DNA vectors encoding IDH1-R132H and IDHwt. b, R-2-HG measurement in S/MAR-IDHwt (control), IRES-IDH1-R132H, or S/MAR-IDH1-R132H-transfected Gl261 syngeneic murine glioma cell lines. Statistical significance was determined by two-tailed student’s t test. Cell lines were sampled and examined over 4 independent measurements. Data are represented as mean ± SEM. c, Immunofluorescence staining of IDH1-R132H (green) in S/MAR-IDHwt (control) or S/MAR-IDH1-R132H-transfected Gl261 syngeneic murine glioma cell line. Staining was performed once for each cell line shown in b. d, R-2-HG measurement in explanted intracranial tumors of GL261-S/MAR-IDH1-R132H-bearing mice. Median concentration indicated as broken line. N = 5 tumor-bearing mice. e, Inhibition of mutant IDH is efficient in a syngeneic murine glioma model by reverting R-2-HG-induced TAM phenotype. N = 7 IDHwt tumor-bearing mice, n = 7 IDHmut tumor-bearing mice + vehicle, n = 4 IDHmut tumor-bearing mice + BAY 1436032. Tumor volumes measured by T2-weighted MRI. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. f, Bulk RNA-Sequencing of GL261-S/MAR-IDH1-wildtype (IDHwt) and GL261-S/MAR-IDH1-R132H (IDHmut) cell lines indicating genes that have been previously highlighted to indicate mesenchymal and proneural glioma phenotypes, respectively42,53. The color scale indicates z-scores. g, MALDI-MS imaging of R-2-HG in snap-frozen tissue of explanted intracranial GL261 tumors expressing S/MAR-IDH1-R132H or S/MAR-IDHwt. Left: Exemplary heatmap of measured R-2-HG intensity, right: normalized R-2-HG in arbitrary units (AU) shown. Data points aggregated from n = 6 independent explanted tumors. Box and whiskers (10–90 percentile, median as center) shown. h-i, Pseudotime analysis of the stepwise changes between clusters enriched for cells of early stage tumors (h) and late stage tumors (i) respectively. Data is suggested by the StemID2 algorithm generating a GAM lineage tree. N = 14 samples from n = 4 IDHwt tumor-bearing mice (d7), n = 4 IDHmut tumor-bearing mice (d7), n = 3 IDHwt tumor-bearing mice (d28), n = 3 IDHmut tumor-bearing mice (d28).
Extended Data Fig. 4 R-2-HG-driven AHR signaling of GAM.
a, Intracellular measurements of R-2-HG in primary microglia after in vitro incubation with R-2-HG for 24 h. Nonlinear regression is shown. N = 1 healthy donor. b, Intracellular measurements of R-2-HG in human MΦ. Cells were primed for 2 h with 100 μM Candesartan, 100μM N-(p-Amylcinnamoyl)anthranilic acid (NAA) or vehicle only and treated with 20mM R-2-HG for 48 h. N = 4 healthy donors. Data are represented as mean ± SEM. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. c, DNA-microarray screen of MΦ from n = 8 healthy donors, treated with exogenous R-2-HG in a matched-pair analysis: procedural overview. d, Quantification of AHR signature expression in IDHwt (grey) and IDHmut (blue) LGG (absolute (left) and normalized to CD11b- (ITGAM-) expression (right)). TCGA dataset, N = 286, n = 68 IDHwt LGG, n = 218 IDHmut LGG. e, AHR target gene expression in human monocyte-derived MΦ treated with increasing concentrations of R-2-HG in vitro as determined by RT-PCR. N = 4 healthy donors. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test. f, Expression levels of AHR in transcripts per million (TPM) in human immune cell subsets as analyzed in the Database of Immune Cell Expression (DICE). g, Cytokine ELISA measurements of IL-10 and TGF-β in human monocyte-derived MΦ following incubation with increasing doses of R-2-HG. Statistical significance was determined by two-tailed student’s t test. N = 4 independent healthy donors evaluated over 6 experimental conditions h. Induction of AHR translocation by supernatant of IDH1-R132H-expressing glioma cells (blue). Supernatant of IDH1-wt-overexpressing GL261 glioma cell line as control (grey). N = 3 IDHwt cell lines, n = 3 IDHmut cell lines. Statistical significance was determined by two-tailed student’s t test. i, AHR translocation reporter assay. Titration of R-2-HG and L-Kyn in DRE-GFP-reporter, n = 3 assay runs. Statistical significance was determined by one-way ANOVA in combination with Tukey’s test.
Extended Data Fig. 5 LAT1-CD98-dependent L-Trp uptake of GAM.
a, Intracellular L-Trp measurement of R-2-HG-treated MΦ from n = 5 healthy human donors by UPLC-FLR/UV. Fold change R-2-HG/vehicle given on the right. Statistical significance was determined by two-tailed student’s t test. b-c, Multiplex fluorescence in situ hybridization of IDHwt, IDHmut glioma as well as control cortical tissue, showing the expression of CD163 (green), SLC7A5 (red), CD44 (white) and nuclear marker DAPI (blue). White arrowheads mark SLC7A5-expressing macrophages, grey arrowheads mark reactive astrocytes expressing SLC7A5. Staining was repeated with 5 independent patient samples and 1 control cortex tissue.
Extended Data Fig. 6 Establishing a MALDI-MSI approach for detection of R-2-HG-induced microenvironmental changes in primary human tissue.
a, 1, Cryosectioning and mounting of fresh frozen tissue specimens. Cutting scheme: (a) the first 10 µm tissue section of each specimen was mounted onto a conductive ITO slide for MALDI-MSI while (b) the adjacent tissue sections (10 µm) were then mounted on a Superfrost© microscope glass slide for standard histological analysis. 2, MALDI matrix deposition using an automated Sprayer. Frozen tissue sections on ITO slides were spray-coated with 1,5-diaminonaphtalene (1,5-DAN) prepared at 10 mg/mL with 50% ACN (v/v) matrix for analyte extraction and ionization. 3, Pixel-wise MALDI MS analysis using a high-resolution mass spectrometer. Molecular ions generated after laser ablation are separated by their mass to charge ratio. At each pixel a mass spectrum is recorded in a grid like manner for multiple (x,y)-coordinates for data acquisition. 4, Data processing was performed with an in-house developed pre-processing pipeline using the R programming environment. 5, Image reconstruction & histological verification. For each selected m/z (z = 1) value the intensity map is shown as a color-coded distribution map. Merely one ion image per mass can be reconstructed. H&E stained image confirms the tumor sites within the tissue section. 6, Data analysis to estimate the levels of detected intensities of the analytes of interest (m/z) in different tissue specimens. b, Local polynomial regression analysis plot for L-Trp as a function of increasing signal intensity of R-2-HG when both are co-localized. A locally estimated scatterplot smoothing (LOESS) curve was fitted which indicates that L-Trp exhibits a general tendency of increasing signal intensity when R-2-HG is increasing. SEM is projected as error bands. N = 5 control cortex samples, n = 5 IDHmut patient samples, n = 5 IDHwt patient samples c. AHR reporter assay after treatment with increasing doses of R-2-HG + vehicle, R-2-HG after AHRi pretreatment (1 h) or PBS for 6 h. N = 3 independent experiments. SEM is projected as error bands. If not mentioned otherwise, all data are represented as mean ± SEM.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2.
Supplementary Tables
Supplementary Tables 1–8
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Friedrich, M., Sankowski, R., Bunse, L. et al. Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer 2, 723–740 (2021). https://doi.org/10.1038/s43018-021-00201-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43018-021-00201-z
This article is cited by
-
Cancer immunometabolism: advent, challenges, and perspective
Molecular Cancer (2024)
-
Prediction of metabolites associated with somatic mutations in cancers by using genome-scale metabolic models and mutation data
Genome Biology (2024)
-
Lactate dehydrogenase A regulates tumor-macrophage symbiosis to promote glioblastoma progression
Nature Communications (2024)
-
Glioblastoma-instructed microglia transition to heterogeneous phenotypic states with phagocytic and dendritic cell-like features in patient tumors and patient-derived orthotopic xenografts
Genome Medicine (2024)
-
Functional polarization of tumor-associated macrophages dictated by metabolic reprogramming
Journal of Experimental & Clinical Cancer Research (2023)
