
Abstract
Generative models for molecules based on sequential line notation (for example, the simplified molecular-input line-entry system) or graph representation have attracted an increasing interest in the field of structure-based drug design, but they struggle to capture important three-dimensional (3D) spatial interactions and often produce undesirable molecular structures. To address these challenges, we introduce Lingo3DMol, a pocket-based 3D molecule generation method that combines language models and geometric deep learning technology. A new molecular representation, the fragment-based simplified molecular-input line-entry system with local and global coordinates, was developed to assist the model in learning molecular topologies and atomic spatial positions. Additionally, we trained a separate non-covalent interaction predictor to provide essential binding pattern information for the generative model. Lingo3DMol can efficiently traverse drug-like chemical spaces, preventing the formation of unusual structures. The Directory of Useful Decoys-Enhanced dataset was used for evaluation. Lingo3DMol outperformed state-of-the-art methods in terms of drug likeness, synthetic accessibility, pocket binding mode and molecule generation speed.
Similar content being viewed by others


Main
Structure-based drug design, which involves designing molecules that can specifically bind to a desired target protein, is a fundamental and challenging drug discovery task1. De novo molecule generation using artificial intelligence has recently gained attention as a tool for drug discovery. Earlier molecular generative models relied on either molecular string representations2,3,4,5 or graph representations6,7,8,9. However, both representations disregard three-dimensional (3D) spatial interactions, rendering them suboptimal for target-aware molecule generation. The increase of 3D protein–ligand complex structures data10 and advances in geometric deep learning have paved the way for artificial intelligence algorithms to directly design molecules with 3D binding poses11,12. For example, methods using 3D convolutional neural networks13 are used to capture 3D inductive bias, but they still struggle to convert atomic density grids into discrete molecules.
Some studies14,15,16,17 proposed to represent pocket and molecule as 3D graphs and used graph neural networks (GNNs) for encoding and decoding. These GNN models use an autoregressive generation process that linearizes a molecule graph into a sequence of sampling decisions. Although these methods can generate molecules with 3D conformations, they share some common drawbacks: (1) the generated molecules often contain problematic, non-drug-like or not synthetically available substructures such as very large rings (rings containing seven or more atoms) and honeycomb-like arrays of parallel, juxtaposed rings; (2) problematic topology: the generated molecules often contain an excessive number of rings or none at all. An autoregressive sampling method has its inherent limitations. It can easily get stuck in local optima during the initial stages of molecule generation and may accumulate errors introduced at each step of the sampling process. For example, as mentioned by ref. 18, although the model can accurately position the nth atom to create a benzene ring when the preceding n − 1 carbon atoms are already in the same plane16, accurate placement of the initial atoms is often problematic because of insufficient context information, resulting in unrealistic fragments.
In addition, some methods based on other technical routes have been proposed for 3D molecule generation, such as those based on diffusion models18,19,20. The representative method is TargetDiff18, which uses a graph-based diffusion model for non-autoregressive molecule generation. Despite its efforts to avoid an autoregressive method, it still generates a notable proportion of undesirable structures. This problem is possibly caused by the model’s relatively weak perception of molecular topology, which is associated with its weak ability to directly encode or predict bonds. Consequently, although TargetDiff achieved improved performance compared to earlier models, it still has room for improvement in metrics such as quantitative estimate of drug likeness (QED)21 and synthetic accessibility score (SAS)22, highlighting the urgency of confining the generated molecules to a drug-like chemical space23.
While graph-based 3D molecular generation methods have shown great potential recently, they still face difficulties in reproducing reference molecules on a given pocket without any information leakage, which is an important benchmark for evaluation. To address the abovementioned problems, we propose Lingo3DMol. First, we introduced a new sequence encoding method for molecules, called the fragment-based simplified molecular-input line-entry system24 (FSMILES). FSMILES encodes the size of the ring in all ring tokens, providing additional contextual information for the autoregressive method and adopts ring-first traversal to achieve improved performance. Furthermore, we integrated local spherical12 and global Euclidean coordinate systems into our model. Because bond lengths and bond angles in the ligand are essentially rigid25, directly predicting them is an easier task than predicting the Euclidean coordinates of the atoms. Combining of these two types of coordinate enables the model to consider a larger spatial context while maintaining accurate substructures. Moreover, non-covalent interactions (NCIs)26 and ligand–protein binding patterns were also considered during molecule generation by incorporating a separately trained NCI/anchor predictor. We also used 3D molecule denoising pretraining strategies similar to BART27 and Chemformer28 to improve the generalization ability of the model. Our model was fine-tuned with data from PDBbind2020 (ref. 29). Finally, we evaluated Lingo3DMol on the Directory of Useful Decoys-Enhanced (DUD-E) dataset and compared it with state-of-the-art (SOTA) methods. Lingo3DMol outperformed existing methods on various metrics.
Our main contributions can be summarized as follows:
-
A new FSMILES molecule representation that incorporates both local and global coordinates is introduced, enabling the generation of 3D molecules with reasonable 3D conformations and two-dimensional (2D) topology.
-
A 3D molecule denoising pretraining method and an independent NCI/anchor model are developed to help overcome the problem of limited data and identify potential NCI binding sites.
-
The proposed method outperforms SOTA methods in terms of various metrics, including drug likeness, synthetic accessibility and pocket binding mode.
Results and discussion
Datasets and baselines
The pretraining dataset was derived from an in-house virtual compound library containing structures of more than 20 million commercially accessible compounds that are typically used for the virtual screening of drug hit candidates. Low-energy conformers were generated for each molecule using ConfGen30. To exclude molecules with low drug likeness, we applied a filtering process that removed complex rings and retained molecules with less than three consecutive flexible bonds, resulting in 12 million molecules. In this study, any rings that do not fall into the categories of five- or six-membered rings, as well as fused five- or six-membered rings were considered to be complex rings.
The fine-tuning dataset was sourced from PDBbind (general set, v.2020)29, using the DUD-E dataset31 as homology filters. Specifically, we excluded proteins from the training set that exhibited more than 30% similarity to any target in DUD-E, as determined using MMseqs2 (ref. 32). This process resulted in the selection of 9,024 Protein Data Bank (PDB) IDs, which represented approximately 46% of the protein–ligand PDB IDs in the PDBbind database. Within these selected PDB IDs, non-crystallographic symmetry related protein–ligand complex molecules within an asymmetric unit were considered individual samples. Additionally, samples were excluded from training if no NCIs were recognized between the ligand and the pocket by the Open Drug Discovery Toolkit (ODDT)33. As a result, we obtained a total of 11,800 samples, which encompassed 8,201 PDB IDs (that is, 42% of protein–ligand PDB IDs in PDBbind), in the fine-tuning dataset.
The NCI training dataset had the same samples as the fine-tuning dataset. The NCIs of the hydrogen bond, halogen bond, salt bridge and pi–pi stacking in the PDBbind were labelled using ODDT. The anchors were marked as the atoms in the pocket that are less than 4 Å away from any atom in the ligand. These labelled samples were used for the NCI/anchor prediction model, averaging 4.1 NCI atoms and 32.1 anchor atoms per pocket sample.
Regarding test dataset, the models were evaluated mainly using the DUD-E dataset, which includes more than 100 targets and an average of more than 200 active ligands per target. This dataset spans diverse protein categories such as Kinase, Protease, GPCRs and ion channels. More importantly, the experimentally measured affinity has been reported for the active compounds in DUD-E. Hence, it allows us to compare generated molecules with active ligands for various protein targets. The target with the PDB ID 2H7L in the DUD-E dataset was excluded as it is listed as an obsolete entry in the PDB.
For baselines in this study, two SOTA models, Pocket2Mol (ref. 16) and TargetDiff18, were used. Pocket2Mol is an autoregressive generative GNN model, and TargetDiff is a diffusion-based model. These two models were, respectively, obtained from their official GitHub repository. As mentioned in their original research papers, these two models were trained using the CrossDocked2020 dataset10.
Model evaluation
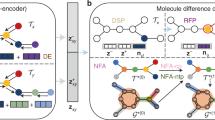
The overall architecture and pretraining strategies are illustrated in Fig. 1a–c. A comprehensive description of the model development process is provided in Methods. In our evaluation, we conducted a comparative analysis of Lingo3DMol with baseline methods. We propose to evaluate the generated molecules from mainly three perspectives: molecular geometry, molecular property distribution and the binding mode within the pocket.

a, Lingo3DMol architecture. Three separate models are included: the pretraining model, the fine-tuning model and the NCI/anchor prediction model. These models share the same architecture with slightly different inputs and outputs. b, Illustration of FSMILES construction. The same colour corresponds to the same fragment. c, Illustration of pretraining perturbation strategy. Step 1, original molecular state; step 2, removal of edge information during pretraining; step 3, perturbation of the molecular structure by randomly deleting 25% of the atoms; step 4, perturbation of the coordinates using a uniform distribution within the range (−0.5 Å, 0.5 Å) and step 5, perturbation of 25% of the carbon element types. These perturbations are applied in no particular order, and the pretraining task aims to restore the molecular structure from step 5 to step 1.
Molecular geometry
The bond length distribution of the generated molecules was assessed using a methodology similar to the one used in the TargetDiff study. Specifically, around 10,000 molecules were generated for each of the three models tested in the study. These molecules were generated for 100 targets in the CrossDocked2020 dataset. Then we compared the bond length distribution of the generated molecules with that of reference molecules, consisting of 100 ligands selected from the CrossDocked2020 dataset, as used in the TargetDiff study. Both our model and benchmark models exhibited favourable performance, as indicated by similar mean bond lengths compared to the reference molecules (Extended Data Table 1).
To assess the dissimilarity between the atom–atom distance distributions of the reference molecules and the model-generated molecules, we used the Jensen–Shannon divergence metric, which was also used in the TargetDiff study. Notably, Lingo3DMol demonstrated the lowest Jensen–Shannon divergence score for all atom distances and ranked second for carbon–carbon bond distances (Extended Data Fig. 1).
Furthermore, we conducted an analysis of ring size, considering that molecules with large rings tend to be challenging to synthesize and may possess poor drug likeness. Our findings, as presented in Extended Data Table 2, revealed that our model exhibited a reduced tendency to generate molecules with a ring size of more than seven compared to the TargetDiff and Pocket2Mol models. This observation suggests that our model shows promise in avoiding the generation of molecules with unfavourable ring size, further enhancing its potential for drug development applications.
Molecular property and binding mode
We proposed to test our model and baseline models using targets from DUD-E, because a notable number of experimentally measured active compounds were documented in this dataset. Specifically, there are over 20,000 active compounds and their affinities against more than 100 targets, an average of over 200 ligands per target. This allows us to analysis the similarity between generated molecules and known active compounds.
Regarding tools for binding pose evaluation, we propose to use Glide34 because it demonstrates superior ability in enriching active compounds and its use is reported as a baseline in research studies investigating scoring functions35,36.
Regarding evaluation metrics for binding pose assessment, three options are available: min-in-place GlideSP score, in-place GlideSP score and GlideSP redocking score. The min-in-place GlideSP score is obtained by using the ‘mininplace’ docking method, where the ligand structure undergoes force-field-based energy minimization within the receptor’s field before scoring. It requires accurate initial placement of the ligand with respect to the receptor. The in-place GlideSP score is generated using the ‘in-place’ docking method, which directly uses the input ligand coordinates for scoring without any docking or energy minimization. The GlideSP redocking score involves docking the generated molecules into the pocket, including the exploration of ligand binding conformations and initial placement.
Among the three docking-related metrics for binding pose evaluation, we advocate using the min-in-place GlideSP score for the following reasons. The in-place GlideSP score is excessively sensitive to atomic distances between the ligand and pocket, making it unsuitable for evaluating the quality of generated poses. In Extended Data Table 3, most molecules generated by all three methods (Lingo3DMol, Pocket2Mol and TargetDiff) exhibit positive scores, indicating steric clashes between the pockets and ligands. However, such clashes do not necessarily denote a poor molecule unless they cannot be rectified by force-field-based minimization. On the other hand, the min-in-place GlideSP score respects the initial binding pose generated by the model and optimizes it through force-field-based adjustments to achieve a robust score. The use of only the GlideSP redocking score, without considering the min-in-place GlideSP score, would contradict the objective of 3D generation as it disregards the original pose. In this study, we recommend considering the min-in-place GlideSP score as the primary metric for binding pose evaluation, while also providing GlideSP redocking scores for contextual information.
Before conducting the binding mode evaluation, we emphasize the importance of examining molecular property distributions. Extended Data Fig. 2 shows three molecules that exhibit notably good docking scores (min-in-place GlideSP scores). However, despite their favourable scores, none of these molecules can be considered as potential drug candidates. Their exclusion stems from their poor drug likeness, as evidenced by low QED values, and low synthetic accessibility, reflected by high SAS values. This intriguing finding underscores the possibility that a superior performance on the binding pose evaluation metric may result from the presence of non-ideal molecules. Consequently, it becomes crucial to eliminate molecules with inadequate drug likeness or limited synthetic accessibility before calculating GlideSP scores. To further investigate the importance of this filtering criterion on a larger scale, we conducted an in-depth analysis of the distributions of various key properties of the generated molecules using heatmaps (Fig. 2a–e).

The drug-like region with QED ≥ 0.3 and SAS ≤ 5 is indicated with red boxes. a–e, Heatmaps show the distribution of key properties for generated molecules, including count (a), molecular weight (b), minimum in-place GlideSP score (c), GlideSP redocking score (d) and r.m.s.d. versus low-energy conformer (e). These distributions are depicted along SAS and QED.
It is important to notice that, as shown in the Fig. 2c, the molecules that possess good min-in-place Glide scores (lower scores) are mostly found outside the drug-like region indicated by the red box for Pocket2mol and TargetDiff. To define the drug-like region, we considered a QED value of 0.3 or higher and a SAS value of 5 or lower, which encompass more than 80% of the molecules in DrugBank37. Unlike benchmark models, Lingo3DMol demonstrates a different pattern. Specifically, Lingo3DMol tends to generate drug-like molecules with relatively good min-in-place GlideSP scores.
For binding mode evaluation, building on the above analysis, we conducted a DUD-E-based evaluation of our model and the baseline models and the results are presented in Table 1. Although the average QED and SAS values do not notably differentiate our model from the baselines, the percentage of drug-like molecules determined by combining QED and SAS indicates the superiority of our model.
Furthermore, in addition to generating drug-like molecules, an effective molecule generative model should be capable of generating active compounds. Since it is impractical to synthesize all generated molecules for real-world testing, an alternative approach is to evaluate whether the model can reproduce known active compounds or generate molecules that are highly similar to known active compounds. To assess this, we introduced the metric ‘ECFP_TS > 0.5’, representing the percentage of targets with generated compounds that demonstrate more than 0.5 Tanimoto similarity in terms of ECFP to active compounds. Among the drug-like molecules generated by the three models, our model yields similar-to-active compounds for 33% of the targets, surpassing Pocket2Mol’s 8% and TargetDiff’s 3%.
Additionally, for a 3D molecule generative model, it is crucial to generate molecules with favourable bindings in the target pockets. This assessment can be approached from two perspectives: binding mode with the pocket (interactions with the pocket) and the ligand’s strain energy, both of which are typically associated with good binding affinity38,39,40. We used the min-in-place GlideSP score to evaluate pocket interactions and root-mean-square deviation (r.m.s.d.) versus low-energy conformers to reflect ligand strain energy. Although the r.m.s.d. versus low-energy conformers metric does not directly quantify strain energy in the unit of kcal mol−1, it provides valuable information on how closely the generated conformers resemble the low-energy conformers in terms of their overall geometry. This metric serves as a proxy for evaluating the ligand’s strain energy. The generated conformations were compared against the top 20 lowest-energy conformers from ConfGen30. Lingo3DMol outperforms the baselines in terms of the min-in-place GlideSP score and r.m.s.d. versus low-energy conformers, indicating the high quality of our generated conformations relative to the baselines. Moreover, our molecular generation speed is faster than benchmark methods (Extended Data Table 4).
While our model exhibited slightly lower diversity in generating drug-like molecules compared to the baselines, it is important to note that the baselines’ higher diversity does not translate into a satisfactory ability to generate similar-to-active compounds. This observation suggests that the baselines may explore the chemical space in a direction that deviates from the region where known active compounds are typically found.
Last, we used Dice score to access the 3D shape similarity between the reference compound observed in the crystal structure and the generated 3D molecules. By voxelizing the molecules and comparing their intersected and union points, we quantified the ‘intersection over union’ ratio as a Dice score that ranges from 0 to 1, with 0 indicating no similarity. Although all the models demonstrated similar performance according to this metric (Table 1), the Dice score contributed in improving our model throughout the model development process (further details are provided in the ‘Ablation analysis’ section).
Another important point to consider is the issue of information leakage in the baselines during above DUD-E test. It is essential to note that Pocket2Mol was trained on the CrossDocked2020 dataset, which did not exclude targets with high homology to DUD-E targets. As a result, the performance of Pocket2Mol in this test may be overestimated due to the information leakage problem. On the other hand, our model was trained on a low-homology (less than 30% sequency identity) dataset to mitigate this issue.
To ensure fair comparisons, we categorized DUD-E targets into three groups on the basis of their sequency identity with the Pocket2Mol training targets: severe (more than 90%), moderate (30–90%) and limited (less than 30%) information leakage. Across all categories, Lingo3DMol consistently outperformed Pocket2Mol, particularly in terms of the min-in-place GlideSP score, Glide redocking score and r.m.s.d. versus low-energy conformers (Extended Data Fig. 3). It is intriguing to observe that the performance gap between the two models widens as the impact of information leakage on the baselines becomes negligible.
Case analysis
For the case study, we selected the generated molecules from two perspectives: ECFP fingerprint similarity with known active compounds and the docking score. In particular, a high similarity of ECFP fingerprints with known active compounds indicated the model’s capability of generating similar substructures or topology features with positive molecules, and a good docking score indicated a stronger fit with the pocket.
As shown in Fig. 3a,b, the molecules in ‘high similarity, good docking score’ group resembled positive molecules in terms of structure and binding mode, demonstrating the ability of our model to reproduce active compounds. However, this is insufficient for this study, because drug design researchers in the real world are more interested in retrieving the following two types of molecule: (1) active compounds that the docking program or other virtual screen tools fail to detect, and (2) molecules with new scaffolds and good pocket binding affinity. The first issue, ‘active compounds missed by the docking program’, is often caused by insufficient sampling of the binding pose, which is closely related to the docking score. Our 3D molecule generation model provides a potential solution to this issue. As shown in Fig. 3c,d, the generated molecules in the ‘high similarity, poor docking score’ group are highly similar to the positive molecules but had poor docking scores (that is, −6.5 and −6.4, respectively) when the docking program was used for binding pose sampling (that is, Glide redocking). Conversely, when a binding pose generated by our model was evaluated using the Glide scorer without conformation sampling, we obtained good scores of −8.7 and −8.8 (that is, min-in-place Glide score), respectively, for the two compounds. This demonstrates the effectiveness of our generated 3D conformation for retrieving good molecules with poor docking scores. For the second issue of obtaining molecules with a new scaffold and good binding mode, we present two cases, shown in Fig. 3e,f, to demonstrate that our model can potentially generate molecules with these characteristics.

Six cases are selected based on the docking scores of generated molecules and their similarities to reference active compounds. a,b, Cases with high similarity and good docking scores. c,d, Cases with high similarity but poor docking scores. e,f, Cases with low similarity but good docking scores. The reference binding mode is based on the crystal structure from the PDB. The Lingo3DMol conformation represents the generated conformation, while the GlideSP redocking conformation was obtained by docking the generated compound into the pocket using Glide with specific parameters (that is, docking_method:confgen and precision:sp). The most similar known active compound to the generated molecule is also displayed, noting that it may not necessarily be the reference compound observed in the crystal structure. The information provided includes the PDB ID for the reference binding mode, the Tanimoto similarity between the Bemis–Murcko scaffolds of generated molecule and its most similar active compound, and the GlideSP redocking score.
Ablation analysis
Effective pretraining and fine-tuning analysis
Specifically, for DUD-E targets, the molecules generated by the models with and without pretraining were respectively compared with the molecules in the pretraining set. We demonstrated that the molecules generated by the pretraining model exhibited a higher degree of similarity to the molecules in the pretraining set compared to those generated by the model without pretraining. This indicates that the model retained the effect of pretraining after the fine-tuning. The comparison of these methods is described in Supplementary Information Part 1. As shown in Table 2, pretraining notably improves the percentage of drug-like molecules, mean QED, the percentage of ECFP_TS > 0.5, mean min-in-place GlideSP score and diversity. We attribute this improvement to the effectiveness of pretraining, especially in scenarios with limited fine-tuning data. In deep learning models, pretraining plays a crucial role in capturing relevant chemical patterns and features, allowing the model to generalize and generate molecules that align with desired properties even when fine-tuning data is limited.
NCI prediction model ablation studies
During this ablation study, we compared Lingo3DMol using randomly selected NCI sites to the standard Lingo3DMol that uses a well-trained NCI site predictor. It is important to note that both approaches share the same molecule generation model. As shown in Table 2, standard Lingo3DMol demonstrated superior performance in most of the metrics, especially in drug likeness and ECFP_TS > 0.5. This can be attributed to several factors. One factor is that randomly selected NCI sites may result in the selection of solvent-exposed regions of the pocket where polar groups are more likely to be located. This may offer more accessible space compared to the cavity where the reference molecule is located. Additionally, the random selection of NCI sites tends to result in NCIs that are spaced farther apart from each other. The combination of these factors, including the preference for solvent-exposed regions and the spacing of selected NCIs, may contribute to the generation of larger molecules and subsequently affect the QED score and the percentage of drug-like molecules.
Last, it is worth noting that for over 95% of DUD-E targets, both our training set (PDBbind, general set, v.2020) and the benchmark models’ training set (CrossDocked2020) include at least one molecule with Tanimoto similarity greater than 0.5 to the DUD-E actives in terms of ECFP4 fingerprints. However, the notable improvement in ECFP_TS > 0.5 for standard Lingo3DMol compared to Lingo3DMol with random NCI and baseline models suggests that this improvement cannot be solely attributed to the model reproducing what it has seen during training.
Conclusion
In this study, we proposed a new molecule representation called FSMILES and developed Lingo3DMol, a model based on language modelling and geometric deep learning techniques. Compared with baselines, our model exhibited superior performance in generating drug-like 3D molecules with better binding mode. This indicates the potential of our model for further exploration in drug discovery and design.
Nevertheless, challenges remain. Capturing all NCIs within a single molecule is not straightforward due to the autoregressive generation process, and we plan to investigate this issue further. Representing molecules and intermolecular interactions with electron densities perhaps offers a promising direction, and some related researches may serve as a good starting point26,41,42. Moreover, the equivariance property is a critical aspect of 3D molecule generation19,20,43. There are many studies on rotational and translational equivariant models, such as GVP44 and Vector Neurons45. Currently, we use rotation and translation augmentation to enhance the model, and we use SE(3) invariant features such as distance matrices and local coordinates46 to alleviate the problem. Last, we have assessed drug-like properties through case analysis and used cheminformatics tools such as QED21 and SAS22 from RDKit47. However, a comprehensive and systematic evaluation of these properties is an essential next step for further research.
Methods
In this section, we present an overview of the Lingo3DMol architecture and its key attributes. The methodology comprises two models: the generation model, which is the central component, and the NCI/anchor prediction model, an essential auxiliary module. These models share the same transformer-based architecture. In the following text, unless specifically mentioned, model refers to the generation model.
Definitions and notations
The Lingo3DMol learns M ≅ P(M∣Pocket; μ), where μ is the parameter of the model, Pocket = (p1, p2…pn) is the set of atoms in the pocket and pi = (typei, main/sidei, residuei, coordsi, hbd/hbai, NCI/anchori) indicates the information of the ith atom in the pocket, where ‘type’ denotes the element type of the atom, ‘main/side’ denotes an atom on the main or the side chain, ‘residue’ denotes the residue type of the atom, ‘coords’ is the coordinates of the atom, ‘hbd/hba’ denotes whether an element is a hydrogen bond donor or acceptor and ‘NCI/anchor’ records whether it is a possible NCI site or anchor point where a potential ligand atom exists within a 4 Å range. Further details are provided in the section ‘NCI/anchor prediction model’ below. \(M=({{{\rm{FSMILES}}}},{\{({r}_{i})\}}_{i = 1}^{K})\) is the representation of the ligand, ri is the coordinates of the ith atom of the ligand and K is the number of atoms in the ligand.
FSMILES is a modified representation of SMILES24 that reorganizes the molecule into fragments, using the normal SMILES syntax for each fragment. The entire molecule is then constructed by combining these fragments using a specific syntax in a fragment-first then depth-first manner, as illustrated in Extended Data Fig. 4. This approach offers two key advantages: enhanced 2D pattern learning through the use of symbols to represent fragments and local structures, and the prioritization of ring closure, enabling the generation of molecules with accurate ring structures and bond angles. In FSMILES, the size of a ring is indicated from the first atom of the ring. For example, ‘C_6’ represents a carbon atom in a six-membered ring. By providing both the atom type and the ring size in advance, the model can more accurately predict the correct bond angles.
The molecule fragment cutting process in FSMILES involves selecting individual bonds that meet specific criteria, such as not being part of a ring, not connecting hydrogen atoms and having at least one end attached to a ring. This cutting process helps divide the ligand into fragments. The FSMILES construction process occurs in two steps (Fig. 1b). First, the ligand is divided into fragments according to the cutting rule. Second, ring information is embedded in each FSMILES token, with the number following the element type’s underscore indicating the ring size. The symbol ‘*’ denotes the connection points of a fragment, while the preceding atom indicates the connection position. In the depth-first growth model, each subsequent fragment connects to the preceding asterisk. To facilitate this, asterisks are stored in a stack and when encountering a new fragment the topmost asterisk is used to establish a connection.
Pretraining and fine-tuning
Pretraining strategy
In the pretraining phase, as illustrated in Fig. 1c, we introduced perturbations into the 3D molecular structure and fed the perturbed molecule into the encoder. This model, which uses an autoregressive approach, aims to reconstruct the perturbed molecule back to its original state in both 2D and 3D representations.
Fine-tuning
For the fine-tuning phase, we used the pretrained model and further fine-tuned it on the protein–ligand complex data. The primary task during this phase continued to be autoregressive molecule generation. To circumvent overfitting of the fine-tuning dataset, the initial three encoder layers were fixed during fine-tuning.
The pseudocode of the above training process is shown in Supplementary Algorithm 1.
Model architecture
The generation model and NCI/anchor prediction model were built on the transformer-based structure with additional graph structural information encoded into the model similar to the previous study48. The generation model was trained by pretraining and fine-tuning. The NCI/anchor prediction model was trained on the basis of the generation model’s pretrained parameters and fine-tuned additionally by its specific prediction task. The overall architecture is shown in Fig. 1a. In the following sections, we first discuss the encoder and decoder components of the generation model, followed by an introduction to the NCI/anchor prediction model.
Encoder
During the pretraining stage, the input of the encoder is a perturbed 3D molecule, which includes the element type and Euclidean coordinates. We can define the molecule as \({M}^{{{{\rm{enc}}}}}=({m}_{1}^{{{{\rm{enc}}}}},{m}_{2}^{{{{\rm{enc}}}}}\ldots {m}_{n}^{{{{\rm{enc}}}}})\), mi = (typei, coordsi), where coordsi = (xi, yi, zi) and n is the number of atoms. Input feature \({f}_{{{{\rm{pre}}}}}\) can then be defined as follows:
where
where Etype and Ecoords represent the embedding functions for the element type, and the corresponding coordinates, respectively. H is the size of the embedded vectors. The symbol ‘+’ represents the element-wise addition operator and the symbol '[ ]' represents the concatenation operator.
During the fine-tuning stage, the input is changed from perturbed molecules to pockets. The input feature Efine can be defined as follows:
where
where Emain/side, Eresidue, Ehbd/hba and ENCI/Anchor represent the embedding functions for the main or side chain, residue type, hydrogen bond donor or acceptor and NCI/anchor point, respectively.
Decoder
The molecule generation process is implemented by two decoders: one 2D topology decoder (D2D) generates FSMILES tokens and local coordinates, and the other decoder generates 3D global coordinates (D3D). First, the next token is predicted using D2D, then the latest 2D token is input to D3D. The 3D global coordinates decoder predicts the global coordinates of the new fragment in the molecule. Both decoders simultaneously predict the local coordinates r, θ and ϕ, particularly the radial distance (r), bond angle (θ) and dihedral angle (ϕ) of the molecule. The local coordinates prediction by D3D only serves as an auxiliary training task.
The input of the decoder network for a molecule can be defined as \({M}^{{{{\rm{dec}}}}}=({m}_{1}^{{{{\rm{dec}}}}},{m}_{2}^{{{{\rm{dec}}}}}\ldots {m}_{n}^{{{{\rm{dec}}}}})\), mi = (tokeni, global_coordsi) and Mbias = (D, J), where D is a distance matrix of size n × n and J is an edge vector matrix of size n × n, where n is the length of the sequence. Mdec is transformed into embeddings using the same process as equation (2). These embeddings are then fed into the 2D topology decoder and global coordinates decoder, respectively. For non-atom tokens, we assigned the same coordinates as those of the most recently generated atom.
The bias terms BD and BJ are derived from the distance and edge vector matrices D and J, respectively. Modified attention scores were calculated by incorporating the following bias terms:
For FSMILES, we used a multilayer perceptron projection head to predict the next token on the basis of D2D’s output. To predict the local coordinates, we established a local coordinates system with three atomic reference points: root1 is current position’s parent atom, root1’s parent atom is root2 and root2’s parent atom is root3. The parent atom is the atom that connects to the child atom.
To predict the radial distance r, we used features of root1 (hroot1) extracted from the D2D’s hidden representation, the current FSMILES token (Etype(cur)) and the molecule’s hidden representation (Htopo) from D2D, Htopo = (h1, h2…hi), where each h represents a FSMILES token’s hidden representation and i represents the number of the generated tokens. For the polar angle θ, we concatenated the hidden representations hroot1 and hroot2. To predict the dihedral angle ϕ, we concatenated the representations of all three roots.
We used multilayer perceptrons as projection heads to predict the local coordinates (r, θ, ϕ). The mathematical representations of these processes are as follows:
distance prediction r:
angle prediction θ:
dihedral angle prediction ϕ:
The predicted local coordinates (r, θ, ϕ) are obtained by taking the argmax operator of the corresponding softmax output.
When predicting global coordinates, as illustrated in Fig. 1a, D3D receives the hidden representation of D2D, and concatenate the predicted FSMILES token, and then predicts the global coordinate (x, y, z).
For pretraining and fine-tuning loss, in our model, the loss function is a combination of multiple components that evaluates different aspects of the predicted molecule. The overall loss function is defined as follows:
where:
-
LFSMILES measures the discrepancy between the predicted and ground-truth molecular topology.
-
Labs_coord evaluates the difference between the predicted and ground-truth atomic coordinates.
-
Lr and Lr_aux measure the error between the predicted and ground-truth radial distances.
-
Lθ and Lθ_aux assess the discrepancy between the predicted and ground-truth bond angles.
-
Lϕ and Lϕ_aux evaluate the difference between the predicted and ground-truth dihedral angles.
All the prediction tasks are treated as classification tasks. Therefore, the cross entropy loss is used for each individual loss component. Auxiliary prediction tasks r_aux, θ_aux and ϕ_aux are used only during training. They are not used during the actual inference process. For further details, please refer to the ‘Generation process’ section.
NCI/anchor prediction model
In this work, we aimed to enhance the generation model by integrating NCI and anchor point information during the fine-tuning and inference stage. To achieve this, we used an NCI/anchor prediction model, which mirrors the generation model’s architecture. This prediction model was initialized using the generation model’s pretrained parameters. Equipped with a specialized output head, the encoder can predict whether a pocket atom will form an NCI with the ligand or act as an anchor point.
This approach allowed us to enhance the generation model in two ways. First, we enriched the model’s input by incorporating the predicted NCI and anchor point data as distinct features of the pocket atoms. Second, we started the molecule generation process by predicting the first atom of the molecule near a chosen NCI site. Specifically, we sampled an NCI site within a pocket and generated the first small-molecule atom within a 4.5 Å radius of this site.
There are three main implications of our approach: (1) We can enhance the perception of NCI and pocket shape, which is critical for generating 3D molecules that can effectively interact with the target protein. (2) By positioning the first atom near the atoms within the NCI pocket, we can increase the likelihood of obtaining a correct NCI pair with a high degree of certainty. Although our model is designed to generate molecules that are prone to forming NCI pairs, it cannot ensure the exact positioning of the generated molecule in relation to the NCI pocket atoms. This is because the model does not explicitly enforce the coverage of all predicted NCI pocket positions, thereby allowing for some degree of variability in the positioning of the generated molecule. (3) Obtaining a good starting position: the autoregressive generation can be seen as a sequential decision-making problem. If the initial step is not chosen appropriately, it can affect every subsequent step. Our empirical study suggests that the NCI position as a starting point is a better choice than a random sample from the predicted 3D coordinates distribution or by selecting the coordinates with highest possibility.
For NCI/anchor prediction loss, we defined two loss functions: The NCI loss measures the difference between the predicted NCI sites and the ground-truth NCI sites, and the anchor loss measures the difference between the predicted anchor points and the ground-truth anchor points. Both loss functions use binary cross entropy.
The total loss for the model is the sum of the NCI loss, anchor loss and all other auxiliary losses from the original 3D generation task.
where LNCI and LAnchor represent the NCI and anchor losses, respectively. Lgen corresponds to the losses from the original 3D generation task that served as only an auxiliary training objective.
Generation process
Here we present the process of generating the final 3D molecule, as shown in Extended Data Fig. 5.
Atom generation
First, the NCI/anchor prediction model predicts the NCI sites and anchor atoms. We then sampled one NCI site from these predictions, and generated the first ligand atom. This atom was positioned at the global coordinates (x, y, z) with the highest predicted joint probability, provided it lies within a 4.5 Å radius of the sampled NCI site. The subsequent atomic positions were generated iteratively. For each step i, D2D predicts the (i + 1)th FSMILES token and local coordinates (r, θ, ϕ). On the basis of the (i + 1)th FSMILES token, we identified the indices of the root1, root2 and root3 atoms. The 3D global coordinates decoder D3D was used to predict the probability distribution of the (i + 1)th 3D coordinates (x, y, z) by incorporating information from the (i + 1)th FSMILES token and the local coordinates.
Within a molecule, bond lengths and angles are largely fixed, and FSMILES fragments are consistently rigid and replicable. As a result, the prediction of local spatial positions will get easier by using local coordinates, which include bond length, bond angle and dihedral angle. By contrast, the global coordinates offer a robust global 3D perception, which is essential for assessing the overall structural context.
We proposed a fusion method that combines the local and global coordinates. Particularly, this method defines a flexible search space around the predicted local coordinates, then selects the global coordinates with the highest probability. The search space is defined as follows:
-
r ± 0.1 Å (angstrom) for distance.
-
θ ± 2° for the angle.
-
ϕ ± 2° for the dihedral angle.
Within this search space, we determined the position with the highest joint probability in the predicted global coordinate distributions. The generation process was repeated to extend the molecular structure progressively. The pseudocode is shown in Supplementary Algorithm 2.
Sampling strategy
We used State(t) = (pocket, ligand(t)) to characterize the generative status at step t, where ligand(t) represents the ligand state after step t is completed. The Action(t) consists of fragments generated by the encoder or decoder model on the basis of State(t). Thus, within the framework of this sampling strategy, the encoder or decoder model functions as an action generator under the context of State(t). When the system adopts a certain Action(t) under the condition State(t) = (pocket, ligand(t)), it moves to the next state State(t + 1) = (pocket, ligand([t + 1)) with a probability of 1, as P(State(t + 1)∣State(t), Action(t)) = 1. It is particularly noted that State(0) = (pocket, ).
The Reward(t) is defined to evaluate State(t) by using two metrics: the model’s predictive confidence and the degree of anchor fulfilment. The computation of the model’s predictive confidence involves averaging the conditional probabilities of each token involved in Action(t). The degree of anchor fulfilment was measured by calculating the proportion of anchors that are within 4 Å of a ligand atom.
To sample at step t, the encoder or decoder model uses an independent and identically distributed approach based on State(t) to generate N instances of Action(t). Then, instances containing atoms with less than 2.5 Å distance from non-hydrogen pocket atoms were discarded to avoid potential clashes. From the remaining set of Action(t), we individually summed the normalized model’s confidence and the degree of anchor fulfilment to calculate their respective Rewards. We then retained the top 0.2 × N instances of Action(t) with the highest Rewards.
The entire sampling process is executed through a depth-first search methodology, ensuring a coherent and systematic progression throughout the entire sampling procedure. The pseudocode is shown in Supplementary Algorithm 3.
Data availability
The evaluation dataset CrossDocked2020 are from the previous study TargetDiff18 and is available at their GitHub https://github.com/guanjq/targetdiff, and the DUD-E31 dataset is a publicly available dataset and is available on our GitHub https://github.com/stonewiseAIDrugDesign/Lingo3DMol. The PDBbind29 dataset is publicly available at http://pdbbind.org.cn/. The NCI training dataset’s NCI label are labelled using an open-source tool, ODDT33. The protein–ligand complex structures used for model fine-tuning, the generated molecules used for evaluation, and a part of the molecules used for pretraining are accessible via figshare repository49. The full pretraining dataset is a private in-house dataset including molecules sourced from both commercial databases and publicly available databases. Due to contractual obligations with the commercial database vendors, we are unable to share the full pretraining dataset publicly. Nonetheless, we are pleased to offer partial data, specifically 1.4 million molecules, which were obtained from publicly available databases. To request access to additional data, we kindly ask interested researchers to contact the corresponding authors with a proposal outlining their non-commercial research intentions. On receipt of a research proposal, we will review it on a case-by-case basis and work towards finding a suitable solution that adheres to the contractual obligations while promoting scientific progress.
Code availability
The source code for inference and model architecture is publicly available on GitHub. The pretraining, fine-tuning and NCI model checkpoints are also available on our GitHub https://github.com/stonewiseAIDrugDesign/Lingo3DMol and figshare repository50. The model is also available as an online service at https://sw3dmg.stonewise.cn.
References
Anderson, A. C. The process of structure-based drug design. Chem. Biol. 10, 787–797 (2003).
Bjerrum, E. J. & Threlfall, R. Molecular generation with recurrent neural networks (RNNs). Preprint at https://arxiv.org/abs/1705.04612 (2017).
Kusner, M. J., Paige, B. & Hernández-Lobato, J. M. Grammar variational autoencoder. Preprint at https://arxiv.org/abs/1703.01925 (2017).
Segler, M. H., Kogej, T., Tyrchan, C. & Waller, M. P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131 (2018).
Xu, M., Ran, T. & Chen, H. De novo molecule design through the molecular generative model conditioned by 3D information of protein binding sites. J. Chem. Inform. Model. 61, 3240–3254 (2021).
Li, Y., Vinyals, O., Dyer, C., Pascanu, R. & Battaglia, P. Learning deep generative models of graphs. Preprint at https://arxiv.org/abs/1803.03324 (2018).
Liu, Q., Allamanis, M., Brockschmidt, M. & Gaunt, A. L. Constrained graph variational autoencoders for molecule design. Preprint at https://arxiv.org/abs/1805.09076 (2018).
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. Preprint at https://arxiv.org/abs/1802.04364 (2018).
Shi, C. et al. GraphAF: a flow-based autoregressive model for molecular graph generation. Preprint at https://arxiv.org/abs/2001.09382 (2020).
Francoeur, P. G. et al. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inform. Model. 60, 4200–4215 (2020).
Skalic, M., Sabbadin, D., Sattarov, B., Sciabola, S. & De Fabritiis, G. From target to drug: generative modeling for the multimodal structure-based ligand design. Mol. Pharm. 16, 4282–4291 (2019).
Gebauer, N. W. A., Gastegger, M. & Schütt, K. T. Symmetry-adapted generation of 3D point sets for the targeted discovery of molecules. Preprint at https://arxiv.org/abs/1906.00957 (2019).
Ragoza, M., Masuda, T. & Koes, D. R. Generating 3D molecules conditional on receptor binding sites with deep generative models. Chem. Sci. 13, 2701–2713 (2022).
Luo, S., Guan, J., Ma, J. & Peng, J. A 3D generative model for structure-based drug design. Adv. Neural Inf. Process. Syst. 34, 6229–6239 (2021).
Liu, M., Luo, Y., Uchino, K., Maruhashi, K. & Ji, S. Generating 3D molecules for target protein binding. Preprint at https://arxiv.org/abs/2204.09410 (2022).
Peng, X. et al. Pocket2Mol: efficient molecular sampling based on 3D protein pockets. Preprint at https://arxiv.org/abs/2205.07249 (2022).
Li, Y., Pei, J. & Lai, L. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 12, 13664–13675 (2021).
Guan, J. et al. 3D equivariant diffusion for target-aware molecule generation and affinity prediction. Preprint at https://arxiv.org/abs/2303.03543 (2023).
Garcia, S. V., Hoogeboom, E. & Welling, M. E(n) equivariant graph neural networks. Preprint at https://arxiv.org/abs/2102.09844 (2021).
Hoogeboom, E., Garcia, S. V., Vignac, C. & Welling, M. Equivariant diffusion for molecule generation in 3D. Preprint at https://arxiv.org/abs/2203.17003 (2022).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8 (2009).
Polishchuk, P. G., Madzhidov, T. I. & Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 27, 675–679 (2013).
Weininger, D. Smiles, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28, 31–36 (1988).
Corso, G., Stärk, H., Jing, B., Barzilay, R. & Jaakkola, T. DiffDock: diffusion steps, twists, and turns for molecular docking. Preprint at https://arxiv.org/abs/2210.01776 (2022).
Ding, K. et al. Observing noncovalent interactions in experimental electron density for macromolecular systems: a novel perspective for protein–ligand interaction research. J. Chem. Inf. Model. 62, 1734–1743 (2022).
Lewis, M. et al. BART: denoising sequence-to-sequence pre-training for natural language generation, translation, and comprehension. Preprint at https://arxiv.org/abs/1910.13461 (2019).
Irwin, R., Dimitriadis, S., He, J. & Bjerrum, E. J. Chemformer: a pre-trained transformer for computational chemistry. Mach. Learn. Sci. Technol. 3, 015022 (2022).
Wang, R., Fang, X., Lu, Y. & Wang, S. The PDBbind database:collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 47, 2977–2980 (2004).
Watts, K. S. et al. Confgen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model 50, 534–546 (2010).
Mysinger, M. M., Carchia, M., Irwin, J. J. & Shoichet, B. K. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594 (2012).
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J. & Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Wojcikowski, M., Zielenkiewicz, P. & Siedlecki, P. Open drug discovery toolkit (ODDT): a new open-source player in the drug discovery field. J. Cheminform. 7, 26 (2015).
Friesner, R. A. et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 (2004).
Su, M. et al. Comparative assessment of scoring functions: the CASF-2016 update. J. Chem. Inf. Model. 59, 895–913 (2018).
Shen, C. et al. Beware of the generic machine learning-based scoring functions in structure-based virtual screening. Brief. Bioinform. 22, bbaa070 (2021).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46, D1074–D1082 (2017).
Jain, A. N., Brueckner, A. C., Cleves, A. E., Reibarkh, M. & Sherer, E. C. A distributional model of bound ligand conformational strain: from small molecules up to large peptidic macrocycles. J. Med. Chem. 66, 1955–1971 (2023).
Gu, S., Smith, M. S., Yang, Y., Irwin, J. J. & Shoichet, B. K. Ligand strain energy in large library docking. J. Chem. Inf. Model. 61, 4331–4341 (2021).
Ryde, U. & Soderhjelm, P. Ligand-binding affinity estimates supported by quantum-mechanical methods. Chem. Rev. 116, 5520–5566 (2016).
Wang, L. et al. A pocket-based 3D molecule generative model fueled by experimental electron density. Sci. Rep. 12, 15100 (2022).
Ma, W. et al. Using macromolecular electron densities to improve the enrichment of active compounds in virtual screening. Commun. Chem. 6, 173 (2023).
Xu, M. et al. GeoDiff: a geometric diffusion model for molecular conformation generation. Preprint at https://arxiv.org/abs/2203.02923 (2022).
Jing, B., Eismann, S., Soni, P. N. & Dror, R. O. Equivariant graph neural networks for 3D macromolecular structure. Preprint at https://arxiv.org/abs/2106.03843 (2021).
Deng, C. et al. Vector neurons: a general framework for SO(3)-equivariant networks. Preprint at https://arxiv.org/abs/2104.12229 (2021).
Simm, G. N. C., Pinsler, R., Csányi, G. & Hernández-Lobato, J. M. Symmetry-aware actor-critic for 3D molecular design. Preprint at https://arxiv.org/abs/2011.12747 (2020).
Landrum, G. et al. RDKit: open-source cheminformatics software. GitHub https://github.com/rdkit/rdkit (2016).
Ying, C. et al. Do transformers really perform badly for graph representation? Adv. Neural Inf. Process. Syst. 34, 28877–28888 (2021).
Feng, W. et al. Data for Lingo3DMol. figshare https://figshare.com/articles/dataset/Data_for_Lingo3DMol/24550351 (2023).
Feng, W. et al. Code for Lingo3DMol. figshare https://figshare.com/articles/software/Code_for_Lingo3DMo/24633084 (2023).
Bajusz, D., Racz, A. & Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 7, 20 (2015).
Acknowledgements
This study was funded by the National Key R&D Program of China (grant no. 2022YFF1203004 received by B.H.). This work was also supported by the Beijing Municipal Science and Technology Commission (grant no. Z211100003521001 received by J.Z. and W.Z.).
Author information
Authors and Affiliations
Contributions
W.Z. and B.H. conceived the study. W.Z. and J.Z. provided instructions for artificial intelligence modelling. B.H. and H.W. provided instructions on evaluation framework. W.F., L.W., Z.L., Y.Z., R.B., H.W. and J.D. developed the model. L.W. and R.B. prepared evaluation data. W.P. supported molecular docking tests.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Benoit Baillif and Sabina Podlewska for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Comparison of atom-atom distance distributions in reference and generated molecules.
(a) All atom pairs are considered in the analysis. (b) Only carbon-carbon atom pairs are considered.
Extended Data Fig. 2 Cases of generated molecules with good min-in-place GlideSP scores but not suitable as drug molecules.
Three generated compounds, namely Cpd. A, Cpd. B and Cpd. C, are showcased. These compounds exhibit good min-in-place GlideSP scores but have relatively poor QED or SAS. The generated binding poses used for min-in-place GlideSP scoring are also provided.
Extended Data Fig. 3 Comparison between Lingo3DMol and Pocket2Mol on the DUD-E dataset under varying degrees of information leakage.
Lingo3DMol has limited information leakage by excluding proteins that have more than 30% sequence identity with DUD-E targets from their training set. The assessment of information leakage is done from the perspective of Pocket2Mol. Specifically, DUD-E targets were categorized into three groups based on their sequency identity with Pocket2Mol training targets: severe (>90%, N = 74), moderate (30-90%, N = 19), and limited (<30%, N = 8) information leakage. The comparisons on average min-in-place GlideSP scores, GlideSP redocking scores, and RMSD vs. low energy conformers are listed in panel (a), (b), and (c), respectively.
Extended Data Fig. 4 FSMILES based molecule growing process.
Nine steps are listed to illustrate the fragment-by-fragment process of generating a molecule within a pocket.
Extended Data Fig. 5 Intuitive visualization of the 3D molecule generation process.
In Step 1, based on the precomputed pocket NCI information, we select an NCI as the starting position. Within a radius r, we select the position with the highest global coordinate probability. In each subsequent step, we predict the local coordinates r, θ, and φ, and combine them with the global coordinates to determine the final position.
Extended Data Fig. 6 Distribution of molecular weight for molecules generated by different methods.
This figure is provided as supplementary information for Table 1.
Supplementary information
Supplementary Information
Supplementary Tables 1–4, Algorithms 1–3 and Fig. 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Feng, W., Wang, L., Lin, Z. et al. Generation of 3D molecules in pockets via a language model. Nat Mach Intell 6, 62–73 (2024). https://doi.org/10.1038/s42256-023-00775-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-023-00775-6
