
Abstract
Phenotype-based compound screening has advantages over target-based drug discovery, but is unscalable and lacks understanding of mechanism of drug action. A chemical-induced gene expression profile provides a mechanistic signature of phenotypic response; however, the use of such data is limited by their sparseness, unreliability and relatively low throughput. Few methods can perform phenotype-based de novo chemical compound screening. Here we propose a mechanism-driven neural network-based method, DeepCE—which utilizes a graph neural network and multihead attention mechanism to model chemical substructure–gene and gene–gene associations—for predicting the differential gene expression profile perturbed by de novo chemicals. Moreover, we propose a novel data augmentation method that extracts useful information from unreliable experiments in the L1000 dataset. The experimental results show that DeepCE achieves superior performances to state-of-the-art methods. The effectiveness of gene expression profiles generated from DeepCE is further supported by comparing them with observed data for downstream classification tasks. To demonstrate the value of DeepCE, we apply it to drug repurposing of COVID-19 and generate novel lead compounds consistent with clinical evidence. DeepCE thus provides a potentially powerful framework for robust predictive modelling by utilizing noisy omics data and screening novel chemicals for the modulation of a systemic response to disease.
Similar content being viewed by others


Main
Target-based high-throughput screening dominates the conventional drug discovery process. It has been the focus of computer-aided drug discovery for decades, including recent applications of deep learning; however, the readout from the modulation of a single protein by a chemical is poorly correlated with organism-level therapeutic effects or side effects. As a result, the failure rate from a lead compound generated from the target-based screening to approved drug is high. Phenotype-based screening has created renewed interests for identifying cell-active compounds but suffered from low throughput and difficulty in target deconvolution. A high-throughput, mechanism-driven phenotype compound screening method will therefore facilitate drug discovery and development.
Gene expression profiling has been widely used to characterize cellular and organismal phenotypes. Systematic analysis of genome-wide gene expression of chemical perturbations on human cell lines has led to considerable improvements in drug discovery and systems pharmacology. In particular, gene expression profiling can be applied to drug repurposing1,2,3,4, discovering drug mechanisms5, lead compound identification6 and predicting side effects for preclinical compounds7. The use of genome-wide chemical-induced gene expression was initially made possible by the appearance of Connectivity Map (CMap)8, which consists of gene expression profiles of five human cancer cell lines perturbed by ~1,300 compounds after 6 h; however, the limited data availability across cell types restricts the performances of the above-mentioned analyses, which heavily depend on the coverage of chemicals and human cell lines. To overcome this limitation, a novel gene expression profiling method, L1000 (which is an extension of the CMap project), was developed by the National Institutes of Health (NIH) library of integrated network-based cellular signatures (LINCS) programme9. After Phase I of LINCS, the L1000 dataset consists of ~1,400,000 gene-expression profiles on the responses of ~50 human cell lines to one of ~20,000 compounds across a range of concentrations. The L1000 dataset and its normalization versions10 were recently widely used in drug repurposing and discovery11,12. Despite these successes, there are several major problems when utilizing L1000. First, although the number of gene expression profiles is much larger than that in CMap, many missing expression values remain in the vast combinatorial space of chemicals and cell lines. Second, there are hundreds of millions of drug-like, purchasable chemicals that are potential drug candidates13. It is infeasible to experimentally test all of these chemicals for their chemical-induced gene expression profiles across multiple cell lines. Finally, due to various experimental problems (for example, the batch effect), many experiment measurements are not reliable (as shown in Supplementary Fig. 1). These serious obstacles will limit the effectiveness and scope of utilizing L1000 dataset in drug discovery. Predicting gene expression values for unmeasured and unreliable experiments is therefore necessary.
Missing entries in the combinatorial space is not a problem exclusive to the L1000 dataset. Before the appearance of L1000, several methods of imputing missing values had been proposed for gene expression datasets. We categorize these methods into two main approaches that pivot on the dependence of other information beyond gene expression data. The first approach does not use any extra information. Works following this approach include k-nearest neighbour (kNN)14, singular value decomposition14, least mean square15,16,17, Bayesian principal component analysis18, Gaussian mixture clustering19 and support vector regression20. The second approach uses additional information to predict expression profiles. For example, chemical structures are used to predict chemical-induced gene expressions but that work does not consider cell-specific information21.
The approaches described above are designed for matrix-structured data (that is, gene × experiment), whereas the L1000 dataset is formulated as tensor-structured data (that is, gene × chemical × cell × dosage × time) and therefore cannot be applied to capture high-dimensional associations that help to impute missing values for L1000. Several methods were proposed to predict gene expression profiles in the L1000 dataset. In particular, to deal with high-dimensional structured data, an extension of a linear regression model named polyadic regression is developed to capture interactions emerging across features22. Matrix completion methods are also adapted to handle tensor-structured gene expression data23,24.
The above methods for the L1000 dataset just focus on imputing the missing values of some gene expression profiles or the whole gene expression profiles of some missing experiments. They are not very useful in the real setting of drug discovery where the chemical-induced gene expression profile of new chemicals needs to be identified. This motivates us to solve a more practical but more challenging problem: predicting gene expression profiles for de novo chemicals (chemicals that do not appear in training data). Solving this problem is necessary as it helps to infer gene expression profiles of new chemicals without conducting experiments that require time and human resources. More importantly, this problem can be expanded to predict gene expression profiles for new cell lines, which can be difficult for measuring in in vitro environments. However, current computational approaches for predicting gene expression values for L1000 cannot work well in a de novo setting. In particular, the tensor completion approach cannot predict gene expression profiles for new chemicals due to inaccessibility to chemical features. Polyadic regression, theoretically, can predict gene expression profiles for high-dimensional data in a de novo chemical setting due to using chemical features; however, in practice, it is not feasible due to the huge computational resources required for handling high-dimensional data (that is, this method fails when applied to data in dimensions greater than three). There is therefore a strong incentive to develop a new and effective method that exploits high-dimensional data for predicting gene expression profiles for a de novo chemical setting.
To address the aforementioned problems, we design a mechanism-driven neural network-based model, DeepCE25, which captures high-dimensional associations among biological features, as well as non-linear relationships between biological features and outputs, to predict gene expression profiles when given a new chemical compound. Our proposed DeepCE considerably outperforms state-of-the-art models for predicting gene expression profiles in L1000, not only in a de novo chemical setting but also a traditional imputation setting. Several novelties in the architecture of the model contribute to the success of DeepCE. First, we leverage a graph convolutional network (GCN) to automatically extract chemical substructure features from data. Second, an attention mechanism is used to capture associations among chemical substructures and genes, and among genes in cell lines. Finally, gene expression values of all L1000 genes are predicted simultaneously from hidden features by a multi-output, multilayer feed-forward neural network. Aside from developing this neural network-based model, we propose a data augmentation method by which we can extract useful information from unreliable experiments in L1000 to improve the prediction performance of our model. We also verify the effectiveness of DeepCE by comparing the performances of several classification models trained on gene expression profiles generated from DeepCE and those trained on original gene expression profiles in L1000 for two downstream tasks: predicting the targets and indications of drugs. Finally, we assess the value of our proposed method for a challenging and urgent problem: finding treatment for COVID-19 by in silico screening all chemical compounds in DrugBank against COVID-19 patient clinical phenotypes. The prioritized lead compounds are consistent with existing clinical evidences. The source code of DeepCE and the generated gene expression profiles of all chemical compounds in DrugBank are publicly available for research purposes, which could make an important contribution to drug discovery and development in particular, and computational chemistry and biology research in general.
Chemical-induced gene expression prediction models and datasets
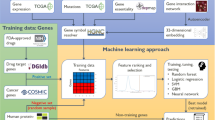
In this section we present datasets used in our study and our proposed model, DeepCE, as well as baseline models for predicting gene expression profiles, such as linear models, a vanilla neural network, kNN and tensor-train weight optimization (TT-WOPT) models. The general framework for training and testing these computational models for L1000 gene expression profile prediction is shown in Fig. 1. Basically, computational models take L1000 experimental information (that is, the chemical compound, cell line, time stamp and chemical dosage) from L1000 as inputs, transform them into numerical representations and then predict L1000 gene expression profiles on the basis of these representations. The details of the numerical feature transformation process for chemical and biological objects used in our study and model implementation of DeepCE and other baselines are shown in the Supplementary Notes 2 and 4. We also present the data augmentation method that extracts useful information from unreliable experiments in L1000 to improve the prediction performance of our models, and the evaluation method for our models.

θ is the set of model parameters, f is the function of θ that maps experiment information to gene expression profiles, and l is the function of θ that computes the differences between predicted and ground-truth gene expression profiles. The objective for the learning process is to minimize the loss between predicted profiles and ground–truth profiles in the L1000 dataset. After training, the models are used to generate profiles for new chemicals in an external molecular database (DrugBank). These profiles are then used for in silico screening (comparing with patient gene expression) to find potential drugs for COVID-19 treatment.
Datasets
In the following paragraphs we present the details and usages of several biological datasets in our study, including L1000, STRING, DrugBank and transcriptome data of COVID-19 patients. We also provide a summary of these datasets in Supplementary Table 1.
Bayesian-based peak deconvolution of L1000 dataset
After the original version of L1000 was released9, many efforts have been made to improve the quality of this dataset. For example, instead of using a k-means clustering algorithm as per the original version, some works propose to use a Gaussian mixture model to enhance the accuracy of the peak deconvolution step26,27. One work, alternatively, develops a multivariate method called characteristic direction to compute gene signatures instead of using the moderated z-score from the original version10. In our study we conduct experiments on a Bayesian-based peak deconvolution L1000 dataset, which has been shown to generate more robust z-score profiles from L1000 assay data and therefore gives better representation for perturbagens28. In particular, we train and evaluate our proposed methods on level 5 data of this dataset. The gene expression profiles that result from experiments featuring the seven most frequent cell lines and six most frequent chemical dosages in the L1000 dataset are used to construct our gene expression dataset. We then select high-quality experiments from our dataset and split them into a high-quality training set as well as a development and testing set. We also construct the original training set by keeping unreliable experiments in our gene expression dataset and the augmented training set generated by our data augmented algorithm. The details of constructing these sets are described in the Supplementary Note 1. The statistics of these training, development and testing sets are shown in Supplementary Table 2.
STRING database for human protein–protein interactions
STRING29 is a multisource database of protein–protein interactions. These interactions—which can be known or predicted, direct (physical) or indirect (functional)—are collected from five main sources, including genomics context prediction, high-throughput laboratory experiments, conserved co-expression, automated text-mining and past knowledge databases. In our setting we extract the human protein–protein interaction network (that is, ~19,000 nodes (proteins) and ~12,000,000 edges (interactions)) from this database to compute vector representations for L1000 genes. The drug-target vector representations for chemical compounds used in our study are also computed from this human protein–protein interaction network. The details of generating these representations from the STRING database are shown in the Supplementary Note 2.
DrugBank database for drug–target interactions and disease predictions
DrugBank is a well-known, comprehensive database used in many bioinformatics and cheminformatics tasks30. This database consists of information about drugs and their targets. In our experiments we extract Anatomical Therapeutic Chemical (ATC) labels derived from the first level of the ATC tree and targets of drugs that appear in the L1000 dataset from DrugBank. There are 698 drug targets and 14 ATC labels in the extracted dataset. We select the most frequent ATC labels and drug targets—on the basis of their frequency as drug labels in this dataset—to form drug-target and ATC prediction datasets, respectively. These datasets are used to evaluate the performance of gene expression profiles generated from our models. We also predict gene expression profiles for all drugs in DrugBank and use them to screen potential candidates for COVID-19 treatment.
Patient expression in response to SARS-CoV-2 infection
Patient expression datasets for this study are downloaded from National Genomics Data Center (NGDC, PRJCA002273)31 and the National Center for Biotechnology Information (NCBI, GSE147507)32. While the former includes eight SARS-CoV-2 patients and twelve healthy samples, the latter has only one SARS-CoV-2 patient and two healthy samples. For each dataset, we use expression profiles from both SARS-CoV-2 patients and healthy negative controls for differential expression analysis. The first dataset can be thus considered as population-based gene expression analysis whereas the second dataset a patient-specific gene expression analysis. The DESeq233 package is used to generate the differential gene expression profiles of the patients. Not all L1000 genes appear in the result of DESeq2 package and therefore we only consider genes that appear in both the L1000 dataset and DESeq2 package when comparing with chemical-induced gene expression profiles.
Overall architecture of DeepCE
Our neural network-based model for L1000 gene expression profile prediction, DeepCE, consists of several components. First, we use a GCN to learn the numerical representation of a chemical compound from its graph structure, and a feed-forward neural network to learn numerical representations of the cell line and chemical dosage. We also use numerical representations for L1000 genes, which are derived from the human protein–protein interaction network (described in the Supplementary Note 2). These vector representations are then put into the interaction component to capture high-level feature associations including chemical substructure–gene and gene–gene feature associations. Finally, the prediction component takes the outputs of the interaction component as inputs to predict the gene expression values for all L1000 genes simultaneously. The overall architecture of DeepCE and its hyperparameters used in our experiments are shown in Fig. 2 and Supplementary Table 4, respectively. The following paragraphs describe each component of DeepCE in detail.

This model consists of three main components are as follows: the feature transformation component that uses GCN to generate features for chemical compound, pre-trained information to represent L1000 genes, and feed-forward neural network to generate features for cell and dosage; the interaction network that learns high-level feature associations (the details of the second layer, which has similar architecture to the first layer in the interaction network, are omitted to save space); the prediction network that predicts gene expression profiles from high-level features.
GCN for neural fingerprints
Data-driven chemical fingerprints were recently shown to be more effective than predefined chemical fingerprints (for example, PubChem, Extended Connectivity Fingerprint (ECFP)) for many biological prediction problems. We therefore propose to use a GCN to capture the chemical substructure information. The original GCN model for chemical fingerprints34 takes a graph structure of a chemical compound as input and updates vector representations for each node (atom) in the graph (chemical compound) from its neighbourhoods by convolutional operation. The vector for each node after convolutional operation can thus be seen as the representation of chemical substructures. The final vector (which is the sum of vectors of every node) is used as the chemical fingerprint. The GCN model used in our experiments is primarily based on that model but with a minor modification. In particular, we output vector representations for every node instead of one vector representation for the chemical compound as we want to model the associations of chemical substructure features with gene features. In our settings we use the GCN model with two convolutional layers (radius, R = 2). It means that the output vector from GCN for each atom represents the chemical substructure, which is a span of two-hop from that atom. The initial representation of the atom, which captures the symbol, degree, number of Hydro neighbourhoods and aromaticity of atoms, and the initial representation of bond, which captures type of bond are multi-hot vectors that have lengths of 62 and 6, respectively. The details of GCN model used in our experiments are shown in Supplementary Algorithm 1.
Multihead attention for gene–gene and chemical substructure–gene feature associations
Attention mechanisms where an element of one set selectively focuses on a subset of another set (attention) or its set (self-attention) on the basis of attention weights are used widely in neural network-based models and are effectively applied to many artificial intelligence tasks, including computer vision and natural language processing. In our experiments we propose to apply the attention method named multihead attention for modelling associations among gene features, and gene and chemical substructure features. Multihead attention was first proposed in the transformer model, which achieves state-of-the-art results for many natural language processing tasks35. Basically, each element in sets can be represented by a set of three vectors: query, key and value. An individual attention module is a function of mapping queries and sets of key–value pairs to output matrix computed by:
where Q, K, V are matrices (sets) of queries, keys, values, respectively, T is a transpose operation, and dk is a scaling factor. Multihead attention focuses on different representation subspaces by concatenating several individual attention modules:
where \({\mathrm{hea{d}}}_{i}={\mathrm{Attention}}({Q}{{W}}_{i}^{Q},{K}{{W}}_{i}^{K},{V}{{W}}_{i}^{V})\). WO, WQ, WK, WV are learned parameters and h is the number of heads.
This multihead attention mechanism is the main ingredient used to construct the interaction component of DeepCE. In particular, the interaction component consists of two identical layers where outputs of the first layer are used as inputs for the second layer. For each layer, we use two separate multihead attention modules with four heads for each module to model associations among genes in gene set and among elements in gene set and chemical substructure set. The lengths of the query, key and value vectors are set to 512. Outputs from these two multihead attention modules are concatenated and put into normalization layer followed by feed-forward layer and another normalization layer. The abstract architecture of interaction component is shown in Fig. 2.
Multi-output prediction
The multi-output prediction component—a two-layer feed-forward neural network with a rectified linear unit (ReLU) activation function—takes input as the concatenation of the chemical neural fingerprint, the gene feature generated by the interaction component, the cell line and chemical dosage features, to predict gene expression values for all L1000 genes together as follows:
where W1, W2, b1, b2 are the weight matrices and bias vectors of this network. The output size of this feed-forward neural network is set at 978, which is the number of L1000 genes.
Objective function
The objective function used in DeepCE model is the mean squared error (MSE) between predicted and ground-truth gene expression values and is computed as follows:
where Θ is the set of parameters in the DeepCE model; N and M are the number of gene expression profiles in the dataset and number of L1000 genes, respectively; and zi,j and yi,j are ground-truth and predicted gene expression values, respectively, of the jth gene in the ith gene expression profile.
Baseline models
In this section we describe several baseline models used in our experiments including linear models, a vanilla neural network, kNN and TT-WOPT24.
Linear models
We experiment with a multi-output linear regression model and its regularization versions, including Lasso regression (L1 regularization) and ridge regression (L2 regularization) models. Similar to DeepCE, input for these models is the concatenation of numerical representations for chemical, gene, cell line and chemical dosage features, but we use predefined chemical fingerprints and drug-target features instead of data-driven representations derived from GCN for chemicals. The details of these representations are described in the Supplementary Information. Multi-output linear models can be seen as one-layer feed-forward neural network without activation function.
Vanilla neural network
The vanilla neural network used in our experiments can be seen as a simpler version of the DeepCE model that does not include the interaction network component for modelling gene–gene and gene–chemical substructure feature associations and GCN for generating neural fingerprints. Input for this vanilla neural network is similar to its for linear models. The following layers in this network are similar to the prediction network component in DeepCE model, which is a two-layer feed-forward neural network with an ReLU activation function.
kNN
We also propose a kNN-based approach for gene expression prediction within a de novo chemical setting. In particular, a gene expression profile for a new chemical compound in one particular setting (that is, cell line, chemical dosage) is generated by averaging gene expression profiles of its nearest neighbourhoods in the training set in the same setting. In our research we experiment with different numbers of neighbourhoods from one to fifteen and different similarity measures including cosine, correlation, Jaccard and Tanimoto, as well as euclidean distance.
Tensor-train weight optimization
Tensor-train weight optimization (TT-WOPT) is a tensor completion approach proposed to retrieve missing values in tensor data from existing values. It has been shown to be effective for predicting values missing from the L1000 dataset, which can be formulated as a tensor-structure object without using additional information24. In our research, we conduct experiments to compare TT-WOPT with our proposed model, especially in a de novo chemical setting. As this model does not require additional information, inputs are therefore L1000 gene expression values formulated as a tensor.
Data augmentation
We can see from Supplementary Fig. 1 that only a small number of experiments in L1000 are reliable (average pearson correlation (APC) score ≥ 0.7) and thus it would be wasteful if we cannot exploit useful information from a large number of unreliable experiments. We show in Table 1 that simply adding unreliable experiments to the high-quality training set (original training set) makes the performances of our models worse. We thus propose the data augmentation method by which we can effectively exploit unreliable experiments to improve the performances of our models. We argue that although an experiment (level 5 data) is unreliable, not all of its bioreplicate experiments (level 4 data) are also unreliable and we will extract these reliable bioreplicate experiments by our proposed data augmentation method. The basic idea is that we first train our model on the high-quality training set and then generate predicted gene expression profiles for unreliable experiments. These predicted gene expression profiles are compared with their bioreplicate gene expression profiles and we incorporate bioreplicate gene expression profiles that have the similarity scores with their predicted gene expression profiles larger than the threshold. Supplementary Algorithm 2 presents this data augmentation method in detail. In our settings, the similarity score is Pearson correlation.
Performance evaluation
The Pearson correlation coefficient is used as the main metric to evaluate performances of models in our experiments. Correlation scores that measure the relationship between ground-truth and predicted gene expression profiles have been shown to be more effective than error measures for microarray data analysis36,37. Moreover, using Pearson correlation allows us to conduct unbiased evaluation for our models that are optimized for MSE. We calculate the average Pearson correlation for a dataset as follows:
where \({z}_{i,j},{y}_{i,j},{\bar{z}}_{i},{\bar{y}}_{i}\) are ground-truth and predicted gene expression values of the jth gene in the ith gene expression profile, and ground-truth and predicted mean values of the ith gene expression profile, respectively.
Aside from the Pearson correlation, we also report the performances of models by other metrics including root mean squared error (r.m.s.e.), gene set enrichment analysis (GSEA)38,39 and Precision@k. Although Pearson correlation and r.m.s.e. capture the variations among all L1000 genes, GSEA and P@k (including both positive and negative P@k) only focus on the most important up- and down-regulated genes. Using multiple metrics thus allows us to measure the performances of models in different aspects. The details of these additional metrics are shown in the Supplementary Note 3.
Furthermore, we use the area under the receiver operating characteristic curve (AUC) to verify the effectiveness of these predicted profiles for downstream binary classification tasks including drug-target and ATC code predictions.
Results and discussions
The results and discussions below are mainly based on Pearson correlation; we also observe the same patterns via other metrics.
DeepCE considerably outperforms baseline models in the novel chemical setting
In this experiment we compare DeepCE and its simpler variants constructed by removing either the whole interaction component or just one part of it (that is, chemical substructure–gene or gene–gene feature association modules) with several baseline models including a vanilla neural network, kNN, linear models and TT-WOPT. Although TT-WOPT predicts output on the basis of gene expression values only, other models learn the relationship between experimental information and gene expression profiles to make predictions. For DeepCE, we use neural fingerprints whereas for other models, we use predefined fingerprints including PubChem and circular (ECFP6) fingerprints, and drug-target information including latent target interaction profile (LTIP)40 and our proposed drug-target feature to represent chemicals. All models are trained on the high-quality training set and are evaluated on the test set.
As listed in Table 1, the DeepCE model and its variants achieve order-of-magnitude improvements over baseline models. In particular, the DeepCE model considerably outperforms other models including a vanilla neural network, kNN, linear models and TT-WOPT by achieving a Pearson correlation of 0.4907 on the testing set (paired t-test, P-value < 4.63 × 10−15). In comparison with its simpler variants whose interaction components are removed, DeepCE also achieves better performance, indicating that the effectiveness of modelling chemical substructure–gene and gene–gene feature associations. Specifically, the performance of DeepCE decreases to 0.4620, 0.4477 and 0.4418 when removing the chemical substructure–gene feature association part (Deep CE−drug−gene attn), gene–gene feature association part (Deep CE−gene−gene attn) and the whole interaction component (Deep CE−attn) (paired t-test, P-value < 2.25 × 10−5), respectively. We also delve deeper into the performance of DeepCE by looking it over cell lines, chemical dosages and L1000 genes. The results of this analysis is shown in Supplementary Figs. 2 and 3. For baseline models, vanilla neural network and kNN achieve pretty good performances. Linear models including linear regression, Lasso and ridge regression do not work well for our problem. It indicates that the linear relationship is not sufficient to model the dependencies among variables in this dataset. TT-WOPT, which, as expected, does not leverage additional features beyond gene expression values to make predictions, does not work well for de novo chemical setting. In particular, it achieves a Pearson correlation of 0.0144, which is similar to randomness. We also provide error estimate for these performances by conducting cross-validation on the high-quality dataset. The results are shown in Supplementary Table 5.
DeepCE outperforms state-of-the-art methods in the imputation setting
We further investigate the performance of DeepCE for the traditional imputation setting that does not require the chemicals in the testing set to be different from those in the training set, and compare it with TT-WOPT, which has been shown to be effective for this setting. To do that, we randomly split the high-quality dataset to the new training, development and testing sets, and conduct the experiment on these sets. Note that, at this time, we split the dataset by gene expression profile instead of chemical compound. The details of the training, development and testing set for imputation setting are shown in Supplementary Table 3.
For the traditional imputation setting, we observe that DeepCE outperforms TT-WOPT by a large margin. In particular, DeepCE achieves a Pearson correlation of 0.7010 versus 0.5113 for TT-WOPT. This result indicates that DeepCE consistently achieves the best performances for both de novo chemical and traditional imputation settings by effectively leveraging features of chemical and biological objects including chemical compounds and genes.
Chemical similarity has an impact on prediction performance
To thoroughly investigate the prediction performance of our models, we explore the impact of chemical similarity between the testing and training sets. In particular, we compute the distance between one experiment in the testing set and its nearest-neighbour experiments in the training set, which are induced by the most similar chemicals (determined by comparing their fingerprints with the fingerprint of the chemical compound induced the experiment in the testing set) on the same cell line. The distance between the two experiments is the Tanimoto coefficient of PubChem fingerprints of their two chemicals, and the distance between the experiment in the testing set with its nearest-neighbour experiments in the training set is the average of distances between that experiment and each of its nearest neighbours. After computing the distances to the training set for all experiments in the testing set, we sort them by ascending order and compare the Pearson correlation scores of these experiments. We calculate the average Pearson correlation scores of all experiments in the testing set that have their distances to the training set smaller than the first quartile (Q1), from Q1 to the second quartile (Q2), from Q2 to the third quartile (Q3) and larger than Q3 of the sorted list. Figure 3 shows the average Pearson correlation scores with these distances of three models including DeepCE, a vanilla neural network and kNN; we can see the same pattern for all models that the prediction performances are higher when the experiments in the testing set are more similar to their nearest neighbour experiments on the training set. We also recognize that DeepCE achieves better performances than the vanilla neural network and kNN for all distance categories, especially for experiments that have their distances to the training set smaller than Q1.

Performances of DeepCE, vanilla neural network, and kNN with different distances among chemicals in the training and testing sets. d is the distance measured by the Tanimoto coefficient between chemical compounds in training and testing sets, and Q1, Q2, and Q3 are the first, second, and third quartiles of the sorted list of distances.
Data quality has a significant impact on prediction performance
Aside from the sparseness problem, the L1000 dataset also includes many unreliable gene expression profiles. To investigate the impact of noisy profiles on the prediction performances of our models, we train two baseline models (including a neural network and kNN) on different training sets generated by filtering unreliable gene expression profiles with different APC thresholds varying from –1 (original training set) to 0.7 (high-quality training set). The PubChem fingerprint is the chemical feature used in this experiment.
As shown in Fig. 4, all models have the same pattern. Starting at the threshold of 0.1, they achieve better performances on the testing set when the threshold is higher and the best setting is training our models on the high-quality training set (that is, a Pearson correlation of 0.3923 for the vanilla neural network and 0.3903 for kNN). For training on the original training set and other training sets generated by filtering unreliable experiments with thresholds <0.1, the ground-truth and predicted gene expression profiles are uncorrelated showing the randomness of the model predictions. These results indicate that unreliable data have a severely negative impact on prediction performances and removing this part from the dataset is necessary for achieving good performances.

Pearson correlation scores of vanilla neural network and kNN at different APC threshold settings. These models are trained on training sets generated by filtering unreliable experiments with different APC thresholds and then are evaluated on the high-quality testing set.
A novel data augmentation method improves the model performance
We propose the data augmentation method (described in detail in Supplementary Algorithm 2) to effectively exploit useful information from unreliable gene expression profiles. In this experiment we evaluate the impact of this method on our models. In particular, DeepCE trained on high-quality training set are used to generate gene expression profiles and the threshold for selecting bioreplicate profiles is 0.5, which is similar to the performance of DeepCE. The statistics of this augmented training set are shown in Supplementary Table 1.
The experimental results for training vanilla neural network, kNN and DeepCE on the augmented training set are shown in Table 1. We can see that the performances of all models trained on this augmented training set are improved in most cases. For example, the Pearson correlation of DeepCE increased from 0.4907 to 0.5014 (paired t-test, P-value <0.05). These results indicate that information extracted from unreliable gene expression profiles by our data augmentation method is effective for gene expression prediction.
The selection of chemical feature affects model performance
In this experiment we investigate the effectiveness of several chemical feature representations for our models. Models used in this experiment are a vanilla neural network for PubChem, ECFP fingerprints, our proposed drug-target features, and LTIP, and DeepCE model without interaction component for neural fingerprint. These models are trained on the high-quality training set. We also create random chemical features by generating random binary vectors whose size is similar to PubChem fingerprint from discrete uniform distribution.
Table 1 shows the performances measured by Pearson correlation of these models with different chemical feature representations. First, chemical features achieve much better performances than the random feature, indicating that chemical features capture important information about chemicals which is useful for predicting gene expression profiles. Second, DeepCE which uses neural fingerprint achieves the Pearson correlation of 0.4418 which is the best performance compared to other settings (paired t-test, P-value < 4.89 × 10−5). For other chemical features, biological-based features including drug-target feature and LTIP achieves slightly better performances than chemical-based features including PubChem and ECFP fingerprints. All of these observations are verified by the paired t-tests with P-values < 0.01. In fact, most of the P-values are much less than 0.01.
We also conduct an ablation study to investigate the impact of other features (that is cell line, dosage) to the predictive performance by removing them from the feature vectors. The results in Supplementary Table 6 show that removing these features decreases the performance of DeepCE and the worst scenario is when removing both cell line and dosage information.
DeepCE is effective in predictive downstream tasks
In this section we design an experiment to answer a question about whether these predicted gene expression profiles can provide added values for downstream prediction tasks, especially in the case that original gene expression profiles in L1000 dataset are unreliable. We first extract gene expression profiles of chemicals that do not have reliable experiments in L1000 (original feature set) as well as use a DeepCE model trained on a high-quality training set to generate gene expression profiles for these drugs (predicted feature set). We then use these sets as the features for drugs to train classification models for two tasks: ATC code and drug-target predictions. The details of constructing these datasets are presented in the Supplementary Note 1 and Supplementary Table 7. Finally, we train four popular classification models, including logistic regression, support vector machine, kNN and a decision tree, using fourteen different versions of chemical features (seven cell-specific features for each original and predicted feature sets) for fourteen binary classification tasks (that is, ten ATC codes and four drug-targets). For each experiment setting, we use cross-validation and report the average results.
The differences in AUC between training classification models with predicted and original feature sets for drug-target and ATC prediction tasks are shown in Extended Data Fig. 1. The improvements in AUC when using predicted features instead of original features are recognized in all cell-specific profiles (Extended Data Fig. 1a), all classification models (Extended Data Fig. 1b), eight-tenths of ATC codes (Extended Data Fig. 1c) and three-quarters of drug-targets (Extended Data Fig. 1d), and these improvements are significant (paired t-test, P-value < 4.87 × 10−5). The details of AUC scores for predicted and original features for each setting (that is, per model, cell line, ATC code and drug-target) are shown in Supplementary Table 8. These results indicate that we can substitute unreliable gene expression profiles in L1000 dataset with gene expression profiles generated from DeepCE to achieve better performances on downstream prediction tasks.
Drug repurposing for COVID-19
To further demonstrate the value of DeepCE, we use a chemical-induced gene expression profile to discover potential drugs for COVID-19 treatment. As the disease state and symptom of COVID-19 patients vary dramatically depending on many factors such as age, gender, underlying conditions and so on, we therefore evaluate the drug repurposing for COVID-19 task under two settings including population- (group of patients) and individual-based (individual patient) analysis. In particular, we first use the trained DeepCE on the high-quality part of L1000 dataset to generate predicted gene expression profiles for all of 11,179 drugs in the Drugbank database at the largest chemical dosage. For patient gene expression profiles, we use SARS-COV-2 gene expression datasets from NGDC and NCBI to calculate the differential gene expression profiles of the patients under population- and individual-based settings, respectively. Specifically, the DESeq2 package is used to generate the patient profiles from eight SARS-CoV-2 patients and twelve healthy samples (population-based), and from one a SARS-CoV-2 patient and two healthy samples (individual-based). We then screen drugs in Drugbank by computing Spearman’s rank-order correlation scores between their gene expression profiles with the patient gene expression profiles and select drugs that give the most negative scores as the potential drugs. Here we incorporate the gene expression profiles of A549—the cancerous lung tissue—next to the main seven cell lines in the high-quality dataset. Aside from the predicted profiles, we also include the gene expression profiles extracted from the high-quality part of L1000 dataset. For each cell line, we extract the top 100 drugs that have the most negative correlation scores with the patient profile as the potential drugs. Finally, we output drugs that have potential for COVID-19 treatment at all cell lines as the result of our screening process.
The results for the population- and individual-based drug repurposing are shown in Table 2 and Extended Data Fig. 2, respectively. COVID-19-induced acute respiratory failure is thought to be related to both direct viral pathogenicity and dysregulated inflammatory host response. As shown in Table 2, among the ten drugs we identified for population-based analysis, three drugs are antiviral drugs used in hepatitis C treatment and two drugs are immunosuppressive agents. In particular, voclosporin and cyclosporine are immunosuppressant and calcineurin inhibitors that share similar structures. Cyclosporine has been used to prevent organ rejection and to treat T-cell-associated autoimmune diseases, and recently shows potential in preventing the uncontrolled inflammatory response, SARS-CoV-2 replication and acute lung injury caused by COVID-1941,42,43,44. Calcineurin inhibitors have also been shown to be promising treatment for severe COVID-19 cases45,46. Alisporivir, which is a non-immunosuppressive analogue of cyclosporine with potent cyclophilin inhibition properties, is shown effective in reducing SARS-CoV-2 RNA production in Vero E6 cells47. Moreover, valspodar inhibits P-glycoprotein, which affects the transportation of immunosuppressive agents, and ceftobiprole medocaril is used in hospital- and community-acquired pneumonia48.
For individual-based analysis, among the fifteen drugs we identified (Extended Data Fig. 2), nine drugs are antiviral drugs and seven of them are used for treating hepatitis C as a NS5A inhibitor. They are similar to the top-ranked drugs that are identified from the population-based analysis. Two from hepatitis C treatment (elbasvir and velpatasvir) in particular have been shown as potential candidates for COVID-19 treatment by using other approaches49,50,51. Moreover, two drugs show anti-inflammatory or immune-regulating function and have the potential to regulate the immune response under COVID-19 infection. Laniquidar can suppress the function of P-glycoprotein 1 and affect transportation of immunosuppressive agents. The individual-based analysis also recognizes the drugs with the similar mode of actions. AMG-487 targets chemokine receptor CXCR3, which can regulate leukocyte trafficking. It is noted that all potential drugs here are not available in L1000 dataset, showing the effectiveness of DeepCE for phenotype compound screening under both population-based and individual-based settings.
Conclusion
Deep learning has attracted a great attention in drug discovery. Past and existing efforts mainly focus on accelerating compound screening against a single target52. However, such a one-drug one-gene paradigm proved to be less successful in tracking complex diseases. A systematic compound screening approach—which both takes information on a biological system into consideration and uses a chemical-induced systematic response as readouts—will provide new opportunities on discovering safe and effective therapeutics that module the biological system. In this study we have proposed DeepCE, a novel and robust neural network-based model for predicting chemical-induced gene expression profiles from chemical and biological objects, especially in a de novo chemical setting. Our model achieves state-of-the-art results in predicting gene expression profiles compared with other models not only in de novo chemical setting but also in the traditional setting. Furthermore, we have addressed the unreliable measurement problem of L1000 by introducing the data augmentation method to effectively exploit useful information from unreliable gene expression profiles to improve the prediction performances of our models. Furthermore, the downstream prediction task evaluation shows that training classification models with gene expression profiles generated from DeepCE achieve better performances than training them with unreliable gene expression profiles in L1000, indicating the added values of DeepCE for downstream prediction. Finally, DeepCE is shown to be effective in the challenge and urgent problem, finding treatment for COVID-19, by in silico screening all chemical compounds in DrugBank against COVID-19 patient clinical phenotypes (that is, comparing chemical-induced gene expression profiles generated from DeepCE with the patient profiles). In summary, DeepCE could be a powerful tool for phenotype-based compound screening.
Data availability
The Bayesian-based peak deconvolution LINCS L1000 dataset is available at https://github.com/njpipeorgan/L1000-bayesian. The training, development, and testing gene expression sets used in our study, gene expression profiles generated from DeepCE for all drugs in DrugBank are available at https://github.com/pth1993/DeepCE.
Code availability
DeepCE source code and its usage instructions are available in Github (https://github.com/pth1993/DeepCE) and Zenodo (https://doi.org/10.5281/zenodo.3978774).
References
Lamb, J. et al. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006).
Hu, G. & Agarwal, P. Human disease-drug network based on genomic expression profiles. PLoS ONE https://doi.org/10.1371/journal.pone.0006536 (2009).
Dudley, J. T., Deshpande, T. & Butte, A. J. Exploiting drug–disease relationships for computational drug repositioning. Brief. Bioinform. 12, 303–311 (2011).
Kosaka, T. et al. Identification of drug candidate against prostate cancer from the aspect of somatic cell reprogramming. Cancer Sci. 104, 1017–1026 (2013).
Wei, G. et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 10, 331–342 (2006).
Hassane, D. C. et al. Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood 111, 5654–5662 (2008).
Stegmaier, K. et al. Gene expression-based high-throughput screening (GE-HTS) and application to leukemia differentiation. Nat. Genet. 36, 257–263 (2004).
Lamb, J. The connectivity map: a new tool for biomedical research. Nat. Rev. Cancer 7, 54–60 (2007).
Subramanian, A. et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171, 1437–1452 (2017).
Duan, Q. et al. L1000cds 2: LINCS L1000 characteristic direction signatures search engine. NPJ Syst. Biol. Appl. 2, 1–12 (2016).
Iwata, M., Sawada, R., Iwata, H., Kotera, M. & Yamanishi, Y. Elucidating the modes of action for bioactive compounds in a cell-specific manner by large-scale chemically-induced transcriptomics. Sci. Rep. 7, 40164 (2017).
Méndez-Lucio, O., Baillif, B., Clevert, D.-A., Rouquié, D. & Wichard, J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 11, 1–10 (2020).
Sterling, T. & Irwin, J. J. Zinc 15–ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Troyanskaya, O. et al. Missing value estimation methods for dna microarrays. Bioinformatics 17, 520–525 (2001).
Bø, T. H., Dysvik, B. & Jonassen, I. Lsimpute: accurate estimation of missing values in microarray data with least squares methods. Nucl. Acids Res. 32, e34–e34 (2004).
Kim, H., Golub, G. H. & Park, H. Missing value estimation for dna microarray gene expression data: local least squares imputation. Bioinformatics 21, 187–198 (2005).
Cai, Z., Heydari, M. & Lin, G. Iterated local least squares microarray missing value imputation. J. Bioinform. Comput. Biol. 4, 935–957 (2006).
Oba, S. et al. A bayesian missing value estimation method for gene expression profile data. Bioinformatics 19, 2088–2096 (2003).
Ouyang, M., Welsh, W. J. & Georgopoulos, P. Gaussian mixture clustering and imputation of microarray data. Bioinformatics 20, 917–923 (2004).
Wang, X., Li, A., Jiang, Z. & Feng, H. Missing value estimation for DNA microarray gene expression data by support vector regression imputation and orthogonal coding scheme. BMC Bioinform. 7, 32 (2006).
Lagunin, A., Ivanov, S., Rudik, A., Filimonov, D. & Poroikov, V. Digep-pred: web service for in silico prediction of drug-induced gene expression profiles based on structural formula. Bioinformatics 29, 2062–2063 (2013).
Perros, I. et al. Polyadic regression and its application to chemogenomics. In Proc. 2017 SIAM International Conference on Data Mining 72–80 (SIAM, 2017).
Hodos, R. et al. Cell-specific prediction and application of drug-induced gene expression profiles. In Pac. Symp. Biocomput Vol. 23, 32–43 (World Scientific, 2018).
Iwata, M. et al. Predicting drug-induced transcriptome responses of a wide range of human cell lines by a novel tensor-train decomposition algorithm. Bioinformatics 35, i191–i199 (2019).
Pham, T.-H. pth1993/DeepCE: First Release of DeepCE (Zenodo, 2020); https://doi.org/10.5281/zenodo.3978774
Liu, C. et al. Compound signature detection on LINCS L1000 big data. Mol. Biosyst. 11, 714–722 (2015).
Li, Z., Li, J. & Yu, P. l1kdeconv: an R package for peak calling analysis with LINCS L1000 data. BMC Bioinformatics 18, 356 (2017).
Qiu, Y., Lu, T., Lim, H. & Xie, L. A Bayesian approach to accurate and robust signature detection on LINCS L1000 data. Bioinformatics 36, 2787–2795 (2020).
Szklarczyk, D. et al. String v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 47, D607–D613 (2019).
Wishart, D. S. et al. Drugbank: a comprehensive resource for in silico drug discovery and exploration. Nucl. Acids Res. 34, D668–D672 (2006).
Zhou, Z. et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890 (2020).
Blanco-Melo, D. et al. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. Cell 181, 1036–1045 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Duvenaud, D. K. et al. Convolutional networks on graphs for learning molecular fingerprints. In Proc. 28th International Conferene on Advances in Neural Information Processing Systems 2224–2232 (NIPS, 2015).
Vaswani, A. et al. Attention is all you need. In Proc. 30th Interrnational Conference on Neural Information Processing Systems 5998–6008 (NIPS, 2017).
Kotlyar, M., Fuhrman, S., Ableson, A. & Somogyi, R. Spearman correlation identifies statistically significant gene expression clusters in spinal cord development and injury. Neurochem. Res. 27, 1133–1140 (2002).
Allison, D. B., Page, G. P., Beasley, T. M. & Edwards, J. W. DNA Microarrays and Related Genomics Techniques: Design, Analysis, and Interpretation of Experiments (CRC, 2005).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Mootha, V. K. et al. Pgc-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003).
Ayed, M., Lim, H. & Xie, L. Biological representation of chemicals using latent target interaction profile. BMC Bioinform. 20, 674 (2019).
Cour, M., Ovize, M. & Argaud, L. Cyclosporine A: a valid candidate to treat COVID-19 patients with acute respiratory failure? Crit. Care 24, 276 (2020).
Rudnicka, L. et al. Cyclosporine therapy during the COVID-19 pandemic is not a reason for concern. J. Amer. Acad. Dermatol. 83, e151–e152 (2020).
Cure, E., Kucuk, A. & Cure, M. C. Cyclosporine therapy in cytokine storm due to coronavirus disease 2019 (COVID-19). Rheumatol. Int. 40, 1177–1179 (2020).
Kemmner, S., Guba, M. O., Schönermarck, U., Stangl, M. & Fischereder, M. Cyclosporine as a preferred calcineurin inhibitor in renal allograft recipients with COVID-19 infection. Kidney Int. 98, 507–508 (2020).
Hage, R., Steinack, C. & Schuurmans, M. M. Calcineurin inhibitors revisited: a new paradigm for COVID-19? Brazil. J. Infect. Dis. 24, 365–365 (2020).
Cavagna, L. et al. Calcineurin inhibitor-based immunosuppression and COVID-19: results from a multidisciplinary cohort of patients in northern Italy. Microorganisms 8, 977 (2020).
Softic, L. et al. Inhibition of SARS-CoV-2 infection by the cyclophilin inhibitor alisporivir (Debio 025). Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.00876-20 (2020).
Syed, Y. Y. Ceftobiprole medocaril: a review of its use in patients with hospital-or community-acquired pneumonia. Drugs 74, 1523–1542 (2014).
Mevada, V. et al. Drug repurposing of approved drugs elbasvir, ledipasvir, paritaprevir, velpatasvir, antrafenine and ergotamine for combating COVID19. Preprint at https://doi.org/10.26434/chemrxiv.12115251.v2 (2020).
Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 6, 3277–3286 (2020).
Shah, B., Modi, P. & Sagar, S. R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 252, 117652 (2020).
Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (NIGMS) of NIH (grant no. R01GM122845 to L.X.), the National Institute on Aging (NIA) of NIH (grant no. R01AD057555 to L.X.) and the National Science Foundation (NSF) (grant no. CBET-2037398 to P.Z.).
Author information
Authors and Affiliations
Contributions
L.X. and P.Z. conceived the project. T.H.P., Y.Q. and J.Z. extracted and preprocessed the data. T.H.P. and P.Z. developed the method. T.H.P. conducted the experiments. T.H.P., Y.Q. and J.Z. analysed the results. T.H.P., Y.Q., L.X. and P.Z. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Machine Intelligence thanks Jose Jimenez-Luna, Hao Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Improvement of predicted profiles over original profiles in AUC.
a, Per cell-specific profile, across experiments for different classification tasks and models. b, Per model, across experiments for different cell-specific profiles and classification tasks. c, Per ATC code, across experiments for different cell-specific profiles and models. d, Per drug-target, across experiments for different cell-specific profiles and models.
Extended Data Fig. 2
The chemical structures, status, and known uses of potential drugs for COVID-19 treatment. These potential drugs are selected based on their appearances in top 100 drugs for all eight cell lines determined by comparing their cell-specific predicted gene expression profiles with the individual-based patient profile by Spearman’s correlation.
Supplementary information
Supplementary Information
Supplementary Notes, Tables 1–8, Algorithms 1 and 2, and Supplementary Figs. 1–3.
Rights and permissions
About this article

Cite this article
Pham, TH., Qiu, Y., Zeng, J. et al. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat Mach Intell 3, 247–257 (2021). https://doi.org/10.1038/s42256-020-00285-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42256-020-00285-9
This article is cited by
-
Microfluidic high-throughput 3D cell culture
Nature Reviews Bioengineering (2024)
-
Drug discovery through Covid-19 genome sequencing with siamese graph convolutional neural network
Multimedia Tools and Applications (2024)
-
A Deep Neural Network for Predicting Synergistic Drug Combinations on Cancer
Interdisciplinary Sciences: Computational Life Sciences (2024)
-
Multitask joint strategies of self-supervised representation learning on biomedical networks for drug discovery
Nature Machine Intelligence (2023)
-
Machine Learning-Assisted Low-Dimensional Electrocatalysts Design for Hydrogen Evolution Reaction
Nano-Micro Letters (2023)
