
Abstract
One of the most exciting new therapies for cancer involves the use of autologous T cells that are engineered to recognize and destroy cancerous cells. Patients with previously untreatable B cell leukaemias and lymphomas have been cured, and efforts are underway to extend this success to other tumours. Here, we discuss recent studies and emerging research aimed to extend this approach beyond oncology in areas such as cardiometabolic disorders, autoimmunity, fibrosis and senescence. We also summarize new technologies that may help to reduce the cost and increase access to related forms of immunotherapy.
Similar content being viewed by others
Main
The use of redirected T cells and chimeric antigen receptors (CARs) in therapeutic applications has a long history that has recently culminated in the extraordinary success of these interventions and several Food and Drug Administration (FDA)-approved therapies for cancer. While the use of CAR T cells did not start out targeting neoplastic cells, the therapeutic applications gained traction with the success of CAR T cell therapy for B cell malignancies. In 2017, the first of several CAR T cell therapies against B cell cancer was approved by the US FDA1, which sparked hundreds of clinical trials and countless preclinical programs, solidifying cell therapies as the ‘third pillar’ of medicine along with small-molecule drugs and biologics2.
More recently, a number of studies have built on the success of CAR T cell therapy in cancer to branch out to other disease areas such as cardiometabolic disorders, autoimmune disease, fibrosis and cellular senescence. Furthermore, infectious pathologies, which were some of the earliest targets of CAR therapy, are being revisited with an enhanced understanding of immune responses and improved tools. These include several generations of CAR constructs, improved manufacturing, the ability to target various subsets of immune cells including regulatory T cells (Treg cells) and macrophages3 and improvements in clinical practice.
The dramatic successes of CAR T cell therapy in oncology have spurred enormous recent interest and investment, but the first CAR T treatments date back decades and were focused on targeting infectious disease, specifically, human immunodeficiency virus (HIV). These first-generation CAR constructs consisted of a CD4 extracellular domain fused by a hinge to an intercellular CD3ζ signalling domain (CD4ζCAR). This CD4ζCAR was able to redirect T cells to HIV-infected CD4+ cells in vitro and in vivo, resulting in numerous clinical trials beginning in the late 1990s4. These initial studies demonstrated the safety and feasibility of this concept5 but were ultimately unable to durably control infection or lower viral burden. Since that time, there have been many generations of CAR design, such as the inclusion of a co-stimulatory domain like CD28 or 4-1BB, to improve function and persistence6. Recently, there have been notable advances in the treatment of HIV with CAR T cells, including various antigen-targeting strategies (HIV envelope protein moieties, broadly neutralising antibodies, bispecific targeting and others) and methods to overcome challenges such as virus escape and host antigen downregulation7,8,9, which have been recently reviewed10. Similar CAR T cell approaches are ongoing for other infectious diseases such as infections due to cytomegalovirus11,12, hepatitis B virus13,14,15,16, hepatitis C virus17, invasive Aspergillus18 and tuberculosis19. CAR T cell therapy is now poised to impact a host of disease areas beyond oncology and infectious disease, including autoimmunity, cardiometabolic disease, fibrosis and senescence.
CAR T cell therapy beyond cancer
Autoimmune disease
Recent advances suggest that CAR T therapy may hold enormous promise for the treatment of many types of autoimmune diseases, which occur when the immune system abnormally attacks host cells or tissues. In some cases, individuals produce autoantibodies against a nonpathogenic endogenous protein through B cells. There are dozens of described autoimmune conditions, only some of which are due to autoantibodies, and, in most of these cases, the specific antigenic targets of the autoantibodies are unknown. In other cases, autoimmune disease is thought to be due to abnormal T cell responses (which may also be dependent on B cells through antigen presentation) or other aspects of the immune system. A common strategy for treatment is immunosuppression; however, this is not curative and can have safety concerns and side effects.
Chimeric autoantibody receptor T cells
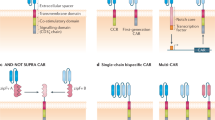
One example of an autoimmune disorder for which the target of autoantibodies is understood in many cases is pemphigus vulgaris. This is a skin-blistering condition that is life threatening, frequently caused by the production of autoantibodies against desmoglein 3 (DSG3). Clinical trials attempting to nonspecifically deplete the source of these antibodies, B cells, using the anti-CD20 monoclonal antibody rituximab resulted in short-term remission in almost all patients but very high levels of relapse and notable safety concerns20. In 2016, Payne and colleagues published a report that cleverly re-engineered the CAR to selectively deplete DSG3-autoreactive B cells to avoid the risks of nonspecific B cell aplasia21. This construct came to be known as a chimeric autoantibody receptor (CAAR). Instead of a single-chain variable fragment (scFv)-specific against an antigen as in a traditional CAR, CAARs instead have the target of the autoantibodies, in this case, recombinant DSG3, fused to CD137–CD3ζ signalling domains (CD137 is also known as 4-1BB). DSG3-specific CAAR T cells selectively ablate only B cells that expressed B cell receptors specific for DSG3 while sparing the rest (Fig. 1). This therapy ameliorated the skin phenotype in a mouse model of pemphigus vulgaris without off-target toxicity. A phase 1 clinical trial in humans is currently underway (NCT04422912).

Second-generation CARs consist of an scFv fused to a co-stimulatory domain (CS, for example, CD28 or 4-1BB) and a CD3ζ signalling domain, while CAARs have a recombinant autoantigen substituted in place of the scFv. Pictured are examples of therapy for fibrotic diseases (anti-fibroblast activation protein (FAP) CAR), autoimmune diseases (anti-CD19 CAR and autoantigen CAAR) and diseases arising from cell senescence (anti-uPAR CAR). TM, transmembrane.
A similar approach was reported in a study targeting B cells producing factor VIII (FVIII)-specific autoantibodies22. A substantial portion of haemophiliacs develop anti-FVIII antibodies during the course of FVIII infusions, making treatment difficult. In what is essentially the same concept as that of CAARs, which the authors term B cell antibody receptors (BARs), the FVIII domain is fused to CD28–CD3ζ and expressed on cytotoxic CD8+ T cells. Adoptive transfer of these cells into haemophilic mice leads to significantly reduced anti-FVIII antibody levels. Success of this CAAR–BAR strategy would suggest that this type of cell therapy can be applied to additional autoimmune disorders for which the autoantibody target is known.
Anti-B cell CAR T cells
In most cases of autoimmune disorders, the autoantibody target is not known, and other strategies need to be used. Nonspecific depletion of B cells is one such strategy, although two separate clinical trials using rituximab for the treatment of systemic lupus erythematosus failed in achieving efficacy23,24. In both cases, this was most likely due to incomplete depletion of B cells and failure to ablate autoantibody-producing clones. However, unlike monoclonal antibodies, anti-B cell CAR T cells have been shown to result in complete and sustained depletion of B cells, as is the case with CD19-specific CARs (Fig. 1). B cell ablation by CD19-specific CAR T cells in mouse models of systemic lupus erythematosus (SLE) was effective in both preventing symptoms of the disorder but also in the treatment of established disease and increasing lifespan25,26. CD19-specific CAR T cells have already been approved by the FDA for treatment of B cell malignancies, making this an attractive approach for SLE, and clinical trials are currently underway (NCT03030976). Promising results from a patient with SLE receiving this therapy have recently been reported27. A similar approach of whole-B cell depletion is being taken to treat myasthenia gravis, a neuromuscular disorder, by using anti-B cell maturation antigen CAR T cells, which is also in clinical trials (NCT04146051). More targeted approaches, such as those targeting muscle-associated receptor tyrosine kinase (MuSK), are currently in preclinical development28.
CAR Treg cells
Until this point, we have been discussing redirecting cytotoxic T cells using CARs or modified CAR constructs to target and ablate specific cells. An alternative approach with great therapeutic potential is the targeting of Treg cells to areas affected by autoreactive immunity to locally suppress the immune response. Three examples of preclinical work on autoimmune disorders using CAR Treg cells are those of colitis29,30, multiple sclerosis31 and type 1 diabetes (T1D)32. Additionally, human leucocyte antigen (HLA)-targeted CAR Treg cells have been proposed to help prevent immune rejection of a transplanted organ33,34,35,36,37, which is discussed further below.
Tissue transplantation
Tissue rejection and graft-versus-host disease are serious immunologic complications following organ transplantation. Treg cells engineered with a CAR to recognize the donor tissue may have the potential to locally suppress host immunologic response to a transplant. Administration of Treg cells expressing a CAR against HLA antigen A*02 (A2-CAR) administered before human skin xenograft improved mouse tolerance to the transplant and avoided graft-versus-host disease34,37. Similar benefits were observed following co-injection of A2-CAR Treg cells with HLA-mismatched peripheral blood mononuclear cells35. A2-CAR Treg cells can be further engineered to constitutively express interleukin 10, which helps maintain their immunosuppressive phenotype over time36. These preclinical data suggest exciting opportunities for future application of CAR T cell technology to augment transplantation, which may especially benefit patients with complex HLA types.
Metabolic disease
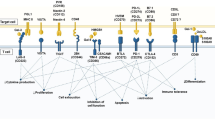
CAR T cell therapy is emerging as a potential approach for a number of metabolic disorders, including those with an underlying autoimmune pathogenesis and those due to hyperactivity of a specific cell type that could be targeted and depleted by cytotoxic T cells. T1D is an autoimmune condition resulting in the destruction of insulin-producing beta cells in the pancreas, necessitating lifelong insulin treatment. There are currently no approved therapies for the prevention or reversal of T1D. Here we explore two potential CAR T cell strategies for the treatment of T1D. First, it may be possible to target and destroy the autoimmune cells responsible for beta cell destruction. A 2019 study described a CAR T cell against an insulin peptide–major histocompatibility complex (MHC) class II complex on antigen-presenting cells (Fig. 1a) in non-obese diabetic (NOD) mice that are responsible for development of diabetes in a mouse model38. When adoptively transferred, this CAR T cell treatment significantly delayed the onset of T1D; however, the cells were not long lived and were undetectable by 15 weeks. In 2020, a group developed a new first-generation biomimetic five-module CAR (5MCAR) targeting autoimmune CD4+ T cells in a mouse model of diabetes39. The 5MCAR cytotoxic lymphocytes were capable of eliminating pathogenic autoimmune T cells (Fig. 1a) and were able to both prevent and treat T1D in mice. Persistence of these cells was observed in a subset of treated mice for up to a year.
A second strategy employed has been the use of Treg cells to suppress the immune system in the context of T1D. Because of the known Treg dysfunction in patients with T1D, cell therapy to increase the number or function of Treg cells have been explored. There have been at least two clinical trials transferring polyclonal Treg cells into patients with T1D with mixed results40,41,42. There are several possible reasons for the modest efficacy observed in these trials, but one important factor may be that antigen-specific Treg cells, which have been shown to be effective for autoimmunity, were not used. To overcome this, a group has attempted to localize Treg cells to the pancreas (Fig. 2b) by engineering them to express a CAR against insulin. Coexpression of forkhead box (FOX)P3 with the CAR in T effector cells was used to transform the cells into functional Treg cells. These Treg cells were successfully localized, suppressive and long lived in vivo, but this localisation was not able to delay progression of T1D in a mouse model32.

a, CAR T cells can be used to ablate insulin autoantigen-presenting cells (APCs) by targeting the insulin peptide–MHC class II complex and pathogenic CD4+ T cells responsible for beta cell destruction by using a biomimetic 5MCAR. b, CAR Treg cells can be targeted to the pancreas to locally suppress the immune system and prevent T1D. c, Anti-beta cell CAR T cells can potentially be used to deplete insulin-producing cells in congenital hyperinsulinism.
Additional cardiometabolic indications for T cell therapy that have been proposed include treatments for atherosclerosis using Treg cells43 or by targeting senescent cells within the vessel wall44. Investigators are also exploring whether targeting of oxidized low-density lipoprotein may be beneficial in atherosclerosis, as has been suggested by studies using anti-oxidized low-density lipoprotein antibodies45. Furthermore, congenital hyperinsulinism may be amenable to CAR T cell therapy. Hyperinsulinism is a rare genetic disorder (caused by mutation of at least 11 different genes) characterized by inappropriate secretion of insulin by beta cells in the pancreas of affected infants, resulting in debilitating hypoglycaemia. Current strategies to control blood sugar and insulin production include partial or total pancreatectomy. In theory, a cytotoxic CAR T cell directed against beta cells (Fig. 2c) could have a similar effect without sacrificing the entire pancreas (including its important exocrine functions). It may also be possible to titrate the dose of transient cytotoxic CAR T cells based on need, thus preserving some endocrine function as well.
Fibrosis
Another area of enormous medical need that may be possible to address with engineered T cell therapy is fibrosis, which can affect every organ and tissue and underlies a wide range of human diseases. Fibrosis is characterized by the activation and proliferation of fibroblasts and the accumulation of excess extracellular matrix in response to injury, infection or other known or unknown disease processes. Despite the enormous medical burden of fibrotic diseases, antifibrotic therapies have been limited.
A pair of papers in 2016 found that inducible genetic ablation of activated cardiac fibroblasts in mice after injury resulted in significant resolution of cardiac fibrosis and, importantly, improvement of cardiac function46,47. Several studies had successfully targeted and ablated activated fibroblast in the tumour stroma of desmoplastic tumours with CAR T cells48,49. Thus, we adapted this approach to target activated fibroblasts in the heart after cardiac injury50. By comparing gene expression profiles in human patients with transplantation and either hypertrophic or dilated cardiomyopathies to those in healthy donor hearts, we were able to identify a target, encoding fibroblast activation protein (FAP), that was significantly upregulated in diseased hearts and was expressed at low levels, if at all, in healthy human hearts. FAP is a good target for CAR T cell targeting because it is a cell surface molecule that is only expressed during pathology and is not expressed at high levels elsewhere in the body under normal conditions. By using a mouse model of hypertensive heart disease, we were able to induce cardiac fibrosis with resultant impairment of cardiac function to test whether CAR T cells directed against FAP could target and ablate activated cardiac fibroblasts. We found that FAP-specific CAR T cell immunotherapy significantly reduced fibrosis in these hearts compared with that in injured controls and rescued both systolic and diastolic function. This proof of concept of using CAR T cells to target cardiac fibrosis is important as it not only opens the door to targeting other forms of cardiac fibrosis but potentially also many fibrotic conditions outside of the heart.
Cellular senescence
Cellular senescence, first described by Leonard Hayflick in 1961, is a state of permanent cell cycle arrest, distinct from that of quiescent cells, which are capable of re-entering the cell cycle, and that of terminally differentiated cells. In addition, these cells undergo marked changes in chromatin structure, metabolic activity and transcription, resulting in a pro-inflammatory phenotype known as the senescence-associated secretory phenotype51. Senescent cells are normally cleared from the body by the immune system; however, accumulation of senescent cells in organs and tissues has been shown to contribute to many age-related pathologies including inflammatory diseases, tissue degeneration and cancer. Previous reports demonstrate that removal of senescent cells in mice via diphtheria toxin-mediated ablation results in the amelioration of several age-related pathologies52. Building on these findings, Lowe and colleagues identified a cell surface molecule, urokinase-type plasminogen activator receptor (uPAR), that is widely expressed in senescent cells and designed a CAR against this antigen44. Adoptive transfer of uPAR-specific CAR T cells was able to efficiently ablate senescent cells in vitro and in vivo, resulting in prolonged survival in a mouse model of lung adenocarcinoma. Additionally, in both carbon tetrachloride-induced and nonalcoholic steatohepatitis mouse models of liver fibrosis, uPAR-specific CAR T cell adoptive transfer resulted in therapeutic efficacy. This report suggests that CAR T cells are a promising senolytic that can be used to treat many age-related conditions such as tissue fibrosis, osteoarthritis, diabetes and atherosclerosis, although much work will need to be done to assess whether targeting uPAR will be safe and effective in humans.
Future applications
The growing body of preclinical evidence for the effectiveness of CAR T cell therapy in non-oncologic conditions indicates a rapidly expanding field that holds much promise. Many of the proof-of-concept studies of cardiometabolic disorders, autoimmunity, fibrosis and senescence can potentially be applied to a wide array of disease processes that would benefit from the removal of pathogenic cells. As mentioned before, fibrosis can negatively affect nearly every organ system in the body and is an important factor in disease progression. Target organs for antifibrotic CAR T cell therapy include the heart (hypertrophic cardiomyopathy, coronavirus disease 2019 (COVID-19) myocarditis, ischaemic cardiomyopathies47), the liver (cirrhosis, nonalcoholic steatohepatitis, hepatitis C-induced liver fibrosis, primary biliary cholangitis), lungs (idiopathic pulmonary fibrosis, infectious pulmonary fibrosis including COVID-19), kidneys (diabetic and hypertensive nephropathy, polycystic kidney disease), skin (keloids, wound healing)53, joints54,55, skeletal muscle (skeletal muscle fibrosis; Duchenne and other muscular dystrophies) and others (strictures, myelofibrosis, radiation-induced fibrosis, sarcoidosis).
Muscular dystrophies such as Duchenne muscular dystrophy are an especially attractive target for this type of antifibrotic CAR T cell therapy. This monogenic disorder affects a relatively homogeneous population of patients (X linked, males affected) with a well-defined natural history of disease. Duchenne muscular dystrophy affects both skeletal muscle, with loss of ambulation usually within the first decade, and the heart, with cardiac fibrosis and impaired function usually by the teen years. Mortality is frequently the result of cardiopulmonary failure. Antifibrotic CAR T cell therapy in these patients has the potential to ‘kill two birds with one stone’ by addressing both skeletal and cardiac muscle fibrosis. Additionally, studies have suggested that antifibrotic therapies in patients with Duchenne muscular dystrophy may not only improve muscle function but also promote muscle regeneration56. While this approach would not address the underlying genetic disorder, one can conceive of using a CAR T approach to address existing fibrosis combined with a gene-therapy approach to restore dystrophin expression in muscle.
Additionally, there are several autoimmune diseases that result in fibrosis that could benefit from both ablation of autoantibody-producing B cells as well as activated fibroblasts. There are many factors to be considered with this type of dual targeting approach including stage of disease and level of fibrosis. An example of this type of therapy could be for treating the autoimmune disease rheumatoid arthritis (RA)57. The anti-B cell monoclonal antibody rituximab has shown clinical efficacy for the treatment of RA58, suggesting that targeting B cells with either nonspecific B cell ablation (for example, CD19-specific CAR) or specific depletion of autoreactive B cells (for example, CAARs or BARs) may be an effective approach to address the underlying cause of disease. A recent in vitro study has demonstrated the feasibility of using CAR T cells against RA autoreactive B cells59. However, depending on the stage of disease, there may be substantial joint fibrosis and potentially interstitial lung fibrosis associated with RA. Recently, Buckley, Croft and colleagues identified a subpopulation of destructive fibroblasts in RA that are characterized by FAP expression and lack of THY1 (ref. 60). By genetically ablating FAP+ fibroblasts in a mouse model of arthritis, they were able to inhibit bone erosion and inflammation in joints. Given the proof-of-concept evidence for using FAP-specific CAR T cells to target and eliminate pathogenic cardiac fibroblasts50, it is logical to conclude that this approach may also be effective to eliminate FAP+ bone-destructive fibroblast populations in RA. Similar approaches might be applied to autoimmune-induced fibrotic conditions in which FAP overexpression has been noted, such as systemic sclerosis61, idiopathic pulmonary fibrosis62,63 and Crohn’s disease64. This dual CAR targeting strategy offers an innovative way to both tackle the underlying cause of autoimmune disease and also address the cumulative insults that have already resulted from prior disease.
Arising technologies
The potential for CAR T cell therapies as treatments for a wide range of common disorder is exciting but also presents new challenges. Present CAR T cell approaches, such as those approved for B cell leukaemia and lymphoma, are appropriate for relatively few patients. Potential indications such as cardiac or liver fibrosis affect millions in the US alone. One of the major hurdles for this kind of therapy is manufacturing, scaling and costs of autologous cell products. Substantial efforts are being undertaken to produce ‘off-the-shelf’ cell therapy products to address some of these issues. Examples include allogeneic CAR T cells (gene editing, induced pluripotent stem cell derived, γδ T cells), CAR natural killer cells and CAR invariant natural killer T cells65,66. Efforts to automate, streamline and shorten the time required for manufacture of autologous cells are also underway, which could lower the cost and improve availability.
In situ reprogramming
A potentially disruptive technology that could be transformative to reduce cost and improve access is the emerging field of in situ cell reprogramming67. This entails targeting and reprogramming T cells in the body to express a CAR, obviating the need to perform any ex vivo manufacturing, thereby dramatically reducing costs and simplifying logistics while increasing scalability and access. The success of modified mRNA lipid nanoparticle (LNP) COVID-19 vaccines has validated the potential of mRNA therapies. Whereas the mRNA LNPs in the COVID-19 vaccines from Moderna and BioNTech–Pfizer are untargeted, mainly delivering mRNA cargo to the liver, Weissman and colleagues have recently published a report describing conjugation of a CD4-specific antibody to LNPs loaded with mRNA that specifically and efficiently targets T cells upon intravenous injection into mice68. Similarly, CD3-targeted nanocarriers loaded with anti-CD19 CAR mRNA resulted in the expression of the CAR on T cells after injection with resultant amelioration of B cell leukaemia in a mouse model69. In collaboration with the Weissman group, we have recently shown that T cell-targeted LNPs can deliver mRNA encoding an anti-activated fibroblast CAR in mice, producing functional CAR T cells in vivo70. In this study, we demonstrate that a single injection of an off-the-shelf, dosable product effectively reprogrammes the immune system and significantly improves heart function and reduces cardiac fibrosis. These technologies hold the promise of a drug-like delivery of CAR T cell therapy and a step toward scalability, lower cost and increased access (Fig. 3).

Targeted LNP (tLNPs) loaded with modified mRNA species encoding a CAR are decorated with antibodies targeted to T cells and, when bound, will be endocytosed and release their cargo. mRNA is subsequently directly translated in the cytoplasm, and CAR proteins are localized to the cell membrane, thus producing a CAR T cell in the body.
Conclusions
CAR T therapy has had enormous impact in some areas of oncology, and notable efforts are underway to extend the success to additional forms of cancer. However, the use of engineered T cells may be even more attractive for disorders other than cancer for several reasons. First, for a cancer therapy to be effective, every neoplastic cell must be eliminated to avoid recurrence. By contrast, lowering the fibrotic burden in diseases characterized by fibrosis would likely have substantial clinical benefits. Additionally, B cell malignancies and other cancers can consist of multiple pounds of tumour cells, the lysis of which is a major contributor to cytokine release syndrome. In many fibrotic indications, the number of pathogenic cells can be orders of magnitude fewer, thereby decreasing the risk of adverse effects such as cytokine release syndrome after cytotoxicity. Because of the need to avoid recurrence and the enormous number of neoplastic cells, CAR T cells in cancer therapy are required to expand in vivo and to be long lasting, necessitating the use of viral transduction. This poses risks such as insertional mutagenesis, overactivation and the need for lymphodepletion. Most non-cancer indications that could benefit from CAR T cell therapy do not have these requirements and are able to use safer approaches, such as transient mRNA CAR T cells, that negate the safety risks associated with viral transduction. Such ‘transient’ CAR T cells could also be delivered in multiple doses over time to titrate the necessary effect while reducing side effects.
The appreciation of the power and potential for engineering the immune system to combat disease is among the most exciting advances in modern medicine. The challenge lies ahead of us to extend these advances to a wider range of human afflictions and to use developing technologies to make these approaches safe, effective, widely available and affordable. The pace of advance is breathtaking, and early studies already predict the possibility of using CAR T cells or related products to treat autoimmune disorders, infections and the enormous unmet need of tissue fibrosis. Strategies for developing effective immune therapies for non-oncologic disease may diverge from those used to treat cancer and may benefit from mRNA and nanodelivery advances. These are still the early days of the ‘immuno-revolution’ in medicine.
References
Mullard, A. FDA approves first CAR T therapy. Nat. Rev. Drug Discov. 16, 669 (2017).
Fischbach, M. A., Bluestone, J. A. & Lim, W. A. Cell-based therapeutics: the next pillar of medicine. Sci. Transl. Med. 5, 179ps7 (2013).
Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 38, 947–953 (2020).
Mitsuyasu, R. T. et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood 96, 785–793 (2000).
Scholler, J. et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 4, p132ra53 (2012).
Tokarew, N., Ogonek, J., Endres, S., von Bergwelt-Baildon, M. & Kobold, S. Teaching an old dog new tricks: next-generation CAR T cells. Br. J. Cancer 120, 26–37 (2019).
Anthony-Gonda, K. et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl. Med. 11, eaav5685 (2019).
Hale, M. et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol. Ther. 25, 570–579 (2017).
Maldini, C., Ellis, G. & Riley, J. L. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol. 18, 605–616 (2018).
Kuhlmann, A.-S., Peterson, C. W. & Kiem, H.-P. CAR T cell approaches to HIV cure. Curr. Opin. HIV AIDS 13, 446–453 (2018).
Full, F. et al. T cells engineered with a cytomegalovirus-specific chimeric immunoreceptor. J. Virol. 84, 4083–4088 (2010).
Proff, J., Brey, C. U., Ensser, A., Holter, W. & Lehner, M. Turning the tables on cytomegalovirus: targeting viral Fc receptors by CARs containing mutated CH2–CH3 IgG spacer domains. J. Transl. Med. 16, 26 (2018).
Bohne, F. et al. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 134, 239–247 (2008).
Festag, M. M. et al. Evaluation of a fully human, hepatitis B virus-specific chimeric antigen receptor in an immunocompetent mouse model. Mol. Ther. 27, 947–959 (2019).
Krebs, K. et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 145, 456–465 (2013).
Kruse, R. L. et al. HBsAg-redirected T cells exhibit antiviral activity in HBV-infected human liver chimeric mice. Cytotherapy 20, 697–705 (2018).
Sautto, G. A. et al. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 65, 512–523 (2016).
Kumaresan, P. R. et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl Acad. Sci. USA 111, 10660–10665 (2014).
Parida, S. K. et al. T-cell therapy: options for infectious diseases. Clin. Infect. Dis. 61, S217–S224 (2015).
Colliou, N. et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci. Transl. Med. 5, 175ra30 (2013).
Ellebrecht, C. T. et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016).
Parvathaneni, K. & Scott, D. W. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2, 2332–2340 (2018).
Gomez Mendez, L. M. et al. Peripheral blood B cell depletion after rituximab and complete response in lupus nephritis. Clin. J. Am. Soc. Nephrol. 13, 1502–1509 (2018).
Merrill, J. T. et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 62, 222–233 (2010).
Jin, X. et al. Therapeutic efficacy of anti-CD19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell. Mol. Immunol. 18, 1896–1903 (2021).
Kansal, R. et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 11, eaav1648 (2019).
Mougiakakos, D. et al. CD19-targeted CAR T cells in refractory systemic lupus erythematosus. N. Engl. J. Med. 385, 567–569 (2021).
Oh, S., O’Connor, K. & Payne, A. MuSK chimeric autoantibody receptor (CAAR) T cells for antigen-specific cellular immunotherapy of myasthenia gravis (2769). Neurology 94, 2769 (2020).
Blat, D., Zigmond, E., Alteber, Z., Waks, T. & Eshhar, Z. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol. Ther. 22, 1018–1028 (2014).
Elinav, E., Adam, N., Waks, T. & Eshhar, Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 136, 1721–1731 (2009).
Fransson, M. et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 9, 112 (2012).
Tenspolde, M. et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 103, 102289 (2019).
Bézie, S. et al. Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv. 3, 3522–3538 (2019).
Boardman, D. A. et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am. J. Transplant. 17, 931–943 (2017).
MacDonald, K. G. et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest. 126, 1413–1424 (2016).
Mohseni, Y. R. et al. Chimeric antigen receptor-modified human regulatory T cells that constitutively express IL-10 maintain their phenotype and are potently suppressive. Eur. J. Immunol. 51, 2522–2530 (2021).
Noyan, F. et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am. J. Transplant. 17, 917–930 (2017).
Zhang, L. et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J. Autoimmun. 96, 50–58 (2019).
Kobayashi, S. et al. A biomimetic five-module chimeric antigen receptor (5MCAR) designed to target and eliminate antigen-specific T cells. Proc. Natl Acad. Sci. 117, 28950–28959 (2020).
Bluestone, J. A. et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 7, 315ra189 (2015).
Marek-Trzonkowska, N. et al. Administration of CD4+CD25highCD127− regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 35, 1817–1820 (2012).
Marek-Trzonkowska, N. et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — results of one year follow-up. Clin. Immunol. 153, 23–30 (2014).
Ketelhuth, D. F. J., Gisterå, A., Johansson, D. K. & Hansson, G. K. T cell-based therapies for atherosclerosis. Curr. Pharm. Des. 19, 5850–5858 (2013).
Amor, C. et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020).
Li, S. et al. Targeting oxidized LDL improves insulin sensitivity and immune cell function in obese rhesus macaques. Mol. Metab. 2, 256–269 (2013).
Kanisicak, O. et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260 (2016).
Kaur, H. et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 118, 1906–1917 (2016).
Lo, A. et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 75, 2800–2810 (2015).
Wang, L.-C. S. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2, 154–166 (2014).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Rinkevich, Y. et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015).
Klimak, M. et al. Immunoengineering the next generation of arthritis therapies. Acta Biomater. 133, 74–86 (2021).
Marsh, L.-J., Kemble, S., Reis Nisa, P., Singh, R. & Croft, A. P. Fibroblast pathology in inflammatory joint disease. Immunol. Rev. 302, 163–183 (2021).
Zhou, L. & Lu, H. Targeting fibrosis in Duchenne muscular dystrophy. J. Neuropathol. Exp. Neurol. 69, 771–776 (2010).
Orvain, C., Boulch, M., Bousso, P., Allanore, Y. & Avouac, J. Is there a place for CAR-T cells in the treatment of chronic autoimmune rheumatic diseases? Arthritis Rheumatol. 73, 1954–1965 (2021).
Tak, P. P. et al. Inhibition of joint damage and improved clinical outcomes with rituximab plus methotrexate in early active rheumatoid arthritis: the IMAGE trial. Ann. Rheum. Dis. 70, 39–46 (2011).
Zhang, B. et al. In vitro elimination of autoreactive B cells from rheumatoid arthritis patients by universal chimeric antigen receptor T cells. Ann. Rheum. Dis. 80, 176–184 (2021).
Croft, A. P. et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251 (2019).
Canady, J., Arndt, S., Karrer, S. & Bosserhoff, A. K. Increased KGF expression promotes fibroblast activation in a double paracrine manner resulting in cutaneous fibrosis. J. Invest. Dermatol. 133, 647–657 (2013).
Acharya, P. S., Zukas, A., Chandan, V., Katzenstein, A.-L. A. & Puré, E. Fibroblast activation protein: a serine protease expressed at the remodeling interface in idiopathic pulmonary fibrosis. Hum. Pathol. 37, 352–360 (2006).
Kimura, T. et al. Loss of cells expressing fibroblast activation protein has variable effects in models of TGF-β and chronic bleomycin-induced fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 317, L271–L282 (2019).
Rovedatti, L. et al. Fibroblast activation protein expression in Crohn’s disease strictures. Inflamm. Bowel Dis. 17, 1251–1253 (2011).
Depil, S., Duchateau, P., Grupp, S. A., Mufti, G. & Poirot, L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 19, 185–199 (2020).
Simonetta, F. et al. Allogeneic CAR invariant natural killer T cells exert potent antitumor effects through host CD8 T-cell cross-priming. Clin. Cancer Res. 27, 6054–6064 (2021).
Parayath, N. N. & Stephan, M. T. In situ programming of CAR T cells. Annu. Rev. Biomed. Eng. 23, 385–405 (2021).
Tombácz, I. et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA–LNP. Mol. Ther. 29, 3293–3304 (2021).
Parayath, N. N., Stephan, S. B., Koehne, A. L., Nelson, P. S. & Stephan, M. T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 11, 6080 (2020).
Rurik, J. G. et al. CAR T cells produced in vivo to treat cardiac injury. Science 375, 91–96 (2022).
Acknowledgements
This work was supported by a grant from the Leducq Foundation, NIH R35 HL140018, the Cotswold Foundation and the WW Smith endowed chair to J.A.E.
Author information
Authors and Affiliations
Contributions
H.A. and J.A.E. wrote the manuscript. J.G.R. provided conceptualisation, editing and critical reading of the manuscript.
Corresponding author
Ethics declarations
Competing interests
H.A. and J.A.E. are cofounders and hold equity in Capstan Therapeutics, which develops therapeutics to reprogramme immune cells in vivo. At present, H.A. serves in a management role with Capstan Therapeutics. J.G.R. declares no competing interests.
Peer review
Peer review information
Nature Metabolism thanks Matthias Hardtke-Wolenski and Marko Radic for their contribution to the peer review of this work. Primary handling editor: Christoph Schmitt.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aghajanian, H., Rurik, J.G. & Epstein, J.A. CAR-based therapies: opportunities for immuno-medicine beyond cancer. Nat Metab 4, 163–169 (2022). https://doi.org/10.1038/s42255-022-00537-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-022-00537-5
