
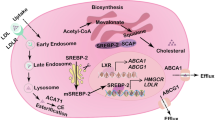
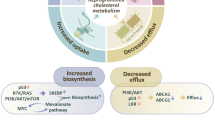
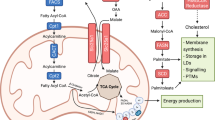
Abstract
Cholesterol metabolism produces essential membrane components as well as metabolites with a variety of biological functions. In the tumour microenvironment, cell-intrinsic and cell-extrinsic cues reprogram cholesterol metabolism and consequently promote tumourigenesis. Cholesterol-derived metabolites play complex roles in supporting cancer progression and suppressing immune responses. Preclinical and clinical studies have shown that manipulating cholesterol metabolism inhibits tumour growth, reshapes the immunological landscape and reinvigorates anti-tumour immunity. Here, we review cholesterol metabolism in cancer cells, its role in cancer progression and the mechanisms through which cholesterol metabolites affect immune cells in the tumour microenvironment. We also discuss therapeutic strategies aimed at interfering with cholesterol metabolism, and how the combination of such approaches with existing anti-cancer therapies can have synergistic effects, thus offering new therapeutic opportunities.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Degirolamo, C., Modica, S., Palasciano, G. & Moschetta, A. Bile acids and colon cancer: solving the puzzle with nuclear receptors. Trends Mol. Med. 17, 564–572 (2011).
Attard, G., Cooper, C. S. & de Bono, J. S. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell 16, 458–462 (2009).
Finlay-Schultz, J. & Sartorius, C. A. Steroid hormones, steroid receptors, and breast cancer stem cells. J. Mammary Gland Biol. Neoplasia 20, 39–50 (2015).
Porter, J. A., Young, K. E. & Beachy, P. A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259 (1996).
Xiao, X. et al. Cholesterol modification of smoothened is required for hedgehog signaling. Mol. Cell 66, 154–162.e10 (2017).
Sheng, R. et al. Cholesterol selectively activates canonical Wnt signalling over non-canonical Wnt signalling. Nat. Commun. 5, 4393 (2014).
Oneyama, C. et al. Transforming potential of Src family kinases is limited by the cholesterol-enriched membrane microdomain. Mol. Cell Biol. 29, 6462–6472 (2009).
Berndt, N., Hamilton, A. D. & Sebti, S. M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791 (2011).
Goldstein, J. L. & Brown, M. S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 29, 431–438 (2009).
Altmann, S. W. et al. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science 303, 1201–1204 (2004).
Li, P. S. et al. The clathrin adaptor Numb regulates intestinal cholesterol absorption through dynamic interaction with NPC1L1. Nat. Med. 20, 80–86 (2014).
Zhang, Y. Y. et al. A LIMA1 variant promotes low plasma LDL cholesterol and decreases intestinal cholesterol absorption. Science 360, 1087–1092 (2018).
Luo, J., Jiang, L. Y., Yang, H. & Song, B. L. Intracellular cholesterol transport by sterol transfer proteins at membrane contact sites. Trends Biochem. Sci. 44, 273–292 (2019).
Chang, T. Y., Chang, C. C., Ohgami, N. & Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157 (2006).
Wang, Y. J. et al. Cholesterol and fatty acids regulate cysteine ubiquitylation of ACAT2 through competitive oxidation. Nat. Cell Biol. 19, 808–819 (2017).
Brown, M. S., Radhakrishnan, A. & Goldstein, J. L. Retrospective on cholesterol homeostasis: the central role of Scap. Annu. Rev. Biochem. 87, 783–807 (2018).
Wang, B. & Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 14, 452–463 (2018).
Widenmaier, S. B. et al. NRF1 is an ER membrane sensor that is central to cholesterol homeostasis. Cell 171, 1094–1109.e15 (2017).
Voisin, M. et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl Acad. Sci. USA 114, E9346–E9355 (2017).
Chimento, A. et al. Cholesterol and its metabolites in tumor growth: therapeutic potential of statins in cancer treatment. Front. Endocrinol. (Lausanne) 9, 807 (2019).
Ding, X., Zhang, W., Li, S. & Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 9, 219–227 (2019).
Wang, Y., Liu, C. & Hu, L. Cholesterol regulates cell proliferation and apoptosis of colorectal cancer by modulating miR-33a-PIM3 pathway. Biochem. Biophys. Res. Commun. 511, 685–692 (2019).
Liu, Z., Liu, X., Liu, S. & Cao, Q. Cholesterol promotes the migration and invasion of renal carcinoma cells by regulating the KLF5/miR-27a/FBXW7 pathway. Biochem. Biophys. Res. Commun. 502, 69–75 (2018).
Costa, G. A. et al. Tumor cell cholesterol depletion and V-ATPase inhibition as an inhibitory mechanism to prevent cell migration and invasiveness in melanoma. Biochim. Biophys. Acta Gen. Subj. 1862, 684–691 (2018).
Lyu, J. et al. Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett. 409, 91–103 (2017).
Wen, Y. A. et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 9, 265 (2018).
Li, N. et al. Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 65, 1936–1947 (2017).
Lewis, C. A. et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34, 5128–5140 (2015).
Cai, D. et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 10, 4621 (2019).
Kim, W. Y. Therapeutic targeting of lipid synthesis metabolism for selective elimination of cancer stem cells. Arch. Pharm. Res. 42, 25–39 (2019).
Ehmsen, S. et al. Increased cholesterol biosynthesis is a key characteristic of breast cancer stem cells influencing patient outcome. Cell Rep. 27, 3927–3938.e6 (2019).
Mullen, P. J., Yu, R., Longo, J., Archer, M. C. & Penn, L. Z. The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731 (2016).
Konstantinopoulos, P. A., Karamouzis, M. V. & Papavassiliou, A. G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555 (2007).
Kaymak, I. et al. Mevalonate pathway provides ubiquinone to maintain pyrimidine synthesis and survival in p53-deficient cancer cells exposed to metabolic stress. Cancer Res. 80, 189–203 (2020).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Doll, S. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019).
Garcia-Bermudez, J. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019).
Stopsack, K. H. et al. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis 38, 806–811 (2017).
Schörghofer, D. et al. The HDL receptor SR-BI is associated with human prostate cancer progression and plays a possible role in establishing androgen independence. Reprod. Biol. Endocrinol. 13, 88 (2015).
Wang, B. et al. Phospholipid remodelling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220.e204 (2018).
Walther, T. C. & Farese, R. V. Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714 (2012).
Li, J. et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 35, 6378–6388 (2016).
Mulas, M. F. et al. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. 44, 360–371 (2011).
Yue, S. et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 (2014).
Antalis, C. J., Uchida, A., Buhman, K. K. & Siddiqui, R. A. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 28, 733–741 (2011).
Geng, F. et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 22, 5337–5348 (2016).
Jiang, Y. et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 (2019).
Wang, J. et al. Lysosomal acid lipase promotes cholesterol ester metabolism and drives clear cell renal cell carcinoma progression. Cell Prolif. 51, e12452 (2018).
Kloudova, A., Guengerich, F. P. & Soucek, P. The role of oxysterols in human cancer. Trends Endocrinol. Metab. 28, 485–496 (2017).
Griffiths, W. J. & Wang, Y. Oxysterol research: a brief review. Biochem. Soc. Trans. 47, 517–526 (2019).
Olkkonen, V. M., Béaslas, O. & Nissilä, E. Oxysterols and their cellular effectors. Biomolecules 2, 76–103 (2012).
Wu, Q. et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 5, 637–645 (2013).
Nelson, E. R. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098 (2013).
Raza, S. et al. The cholesterol metabolite 27-hydroxycholesterol regulates p53 activity and increases cell proliferation via MDM2 in breast cancer cells. Mol. Cell Biochem. 410, 187–195 (2015).
Zhu, D. et al. The ROS-mediated activation of STAT-3/VEGF signaling is involved in the 27-hydroxycholesterol-induced angiogenesis in human breast cancer cells. Toxicol. Lett. 264, 79–86 (2016).
Shen, Z. et al. 27-Hydroxycholesterol induces invasion and migration of breast cancer cells by increasing MMP9 and generating EMT through activation of STAT-3. Environ. Toxicol. Pharmacol. 51, 1–8 (2017).
Guo, F. et al. Upregulation of 24(R/S),25-epoxycholesterol and 27-hydroxycholesterol suppresses the proliferation and migration of gastric cancer cells. Biochem. Biophys. Res. Commun. 504, 892–898 (2018).
Warns, J., Marwarha, G., Freking, N. & Ghribi, O. 27-hydroxycholesterol decreases cell proliferation in colon cancer cell lines. Biochimie 153, 171–180 (2018).
Levy, D. et al. Oxysterols selectively promote short-term apoptosis in tumor cell lines. Biochem. Biophys. Res. Commun. 505, 1043–1049 (2018).
Porstmann, T. et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24, 6465–6481 (2005).
Wang, X. et al. MYC-regulated mevalonate metabolism maintains brain tumor-initiating cells. Cancer Res. 77, 4947–4960 (2017).
Bakiri, L. et al. Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation. J. Exp. Med. 214, 1387–1409 (2017).
Moon, S. H. et al. p53 Represses the mevalonate pathway to mediate tumor suppression. Cell 176, 564–580.e519 (2019).
Liu, D. et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 10, eaap9840 (2018).
Kato, Y. et al. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 13, 89 (2013).
Kondo, A. et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 18, 2228–2242 (2017).
Nakamura, K. & Smyth, M. J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 95, 325–332 (2017).
He, M. et al. Pro-inflammation NF-κB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J. Exp. Clin. Cancer Res. 36, 15 (2017).
Kusnadi, A. et al. The cytokine TNF promotes transcription factor SREBP activity and binding to inflammatory genes to activate macrophages and limit tissue repair. Immunity 51, 241–257.e249 (2019).
Xu, C., Bailly-Maitre, B. & Reed, J. C. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 115, 2656–2664 (2005).
Wang, M. & Kaufman, R. J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 (2014).
Jakobsen, C. H. et al. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J. Lipid Res. 49, 2089–2100 (2008).
Størvold, G. L. et al. Docosahexaenoic acid activates some SREBP-2 targets independent of cholesterol and ER stress in SW620 colon cancer cells. Lipids 44, 673–683 (2009).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Turley, S. J., Cremasco, V. & Astarita, J. L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 15, 669–682 (2015).
Dumitru, C. A., Moses, K., Trellakis, S., Lang, S. & Brandau, S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol. Immunother. 61, 1155–1167 (2012).
Moses, K. & Brandau, S. Human neutrophils: their role in cancer and relation to myeloid-derived suppressor cells. Semin. Immunol. 28, 187–196 (2016).
Raccosta, L. et al. The oxysterol-CXCR2 axis plays a key role in the recruitment of tumor-promoting neutrophils. J. Exp. Med. 210, 1711–1728 (2013).
Soncini, M. et al. 24-Hydroxycholesterol participates in pancreatic neuroendocrine tumor development. Proc. Natl Acad. Sci. USA 113, E6219–E6227 (2016).
Baek, A. E. et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 8, 864 (2017).
Condamine, T. et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, aaf8943 (2016).
Goossens, P. et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 29, 1376–1389.e1374 (2019).
Eibinger, G. et al. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines: implications for chemotactic monocyte recruitment. Exp. Cell Res. 319, 1828–1838 (2013).
Wang, S., Yao, Y., Rao, C., Zheng, G. & Chen, W. 25-HC decreases the sensitivity of human gastric cancer cells to 5-fluorouracil and promotes cells invasion via the TLR2/NF-κB signaling pathway. Int. J. Oncol. 54, 966–980 (2019).
Kidani, Y. et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 14, 489–499 (2013).
Bensinger, S. J. et al. LXR signalling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111 (2008).
Yang, W. et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016).
Kidani, Y. & Bensinger, S. J. Modulating cholesterol homeostasis to build a better T cell. Cell Metab. 23, 963–964 (2016).
Ma, X. et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 30, 143–156.e145 (2019).
Ma, X. et al. Cholesterol negatively regulates IL-9-producing CD8+ T cell differentiation and antitumor activity. J. Exp. Med. 215, 1555–1569 (2018).
Villablanca, E. J. et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 16, 98–105 (2010).
Ramakrishnan, R. et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 192, 2920–2931 (2014).
Gruenbacher, G. & Thurnher, M. Mevalonate metabolism in immune-oncology. Front. Immunol. 8, 1714 (2017).
Thurnher, M., Gruenbacher, G. & Nussbaumer, O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim. Biophys. Acta 1831, 1009–1015 (2013).
Armitage, J. The safety of statins in clinical practice. Lancet 370, 1781–1790 (2007).
Athyros, V. G. et al. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet 376, 1916–1922 (2010).
Poynter, J. N. et al. Statins and the risk of colorectal cancer. N. Engl. J. Med. 352, 2184–2192 (2005).
Nielsen, S. F., Nordestgaard, B. G. & Bojesen, S. E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 367, 1792–1802 (2012).
Cardwell, C. R., Hicks, B. M., Hughes, C. & Murray, L. J. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. J. Clin. Oncol. 32, 3177–3183 (2014).
Larsen, S. B. et al. Postdiagnosis statin use and mortality in Danish patients with prostate cancer. J. Clin. Oncol. 35, 3290–3297 (2017).
Sanfilippo, K. M. et al. Statins are associated with reduced mortality in multiple myeloma. J. Clin. Oncol. 34, 4008–4014 (2016).
Malik, M., Britten, J., Borahay, M., Segars, J. & Catherino, W. H. Simvastatin, at clinically relevant concentrations, affects human uterine leiomyoma growth and extracellular matrix production. Fertil. Steril. 110, 1398–1407.e1 (2018).
Xia, Y. et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell 175, 1059–1073.e21 (2018).
Christie, C. F. et al. Statin-dependent modulation of mitochondrial metabolism in cancer cells is independent of cholesterol content. FASEB J. 33, 8186–8201 (2019).
Cirmena, G. et al. Squalene epoxidase as a promising metabolic target in cancer treatment. Cancer Lett. 425, 13–20 (2018).
Maione, F. et al. The cholesterol biosynthesis enzyme oxidosqualene cyclase is a new target to impair tumour angiogenesis and metastasis dissemination. Sci. Rep. 5, 9054 (2015).
Lanterna, C. et al. The administration of drugs inhibiting cholesterol/oxysterol synthesis is safe and increases the efficacy of immunotherapeutic regimens in tumor-bearing mice. Cancer Immunol. Immunother. 65, 1303–1315 (2016).
Torres-Adorno, A. M. et al. Eicosapentaenoic acid in combination with EPHA2 inhibition shows efficacy in preclinical models of triple-negative breast cancer by disrupting cellular cholesterol efflux. Oncogene 38, 2135–2150 (2019).
Yang, Z. et al. Cholesterol inhibits hepatocellular carcinoma invasion and metastasis by promoting CD44 localization in lipid rafts. Cancer Lett. 429, 66–77 (2018).
Bandyopadhyay, S. et al. Cholesterol esterification inhibition and imatinib treatment synergistically inhibit growth of BCR-ABL mutation-independent resistant chronic myelogenous leukemia. PLoS One 12, e0179558 (2017).
Lee, H. J. et al. Cholesterol esterification inhibition suppresses prostate cancer metastasis by impairing the Wnt/β-catenin pathway. Mol. Cancer Res. 16, 974–985 (2018).
Lee, S. S. et al. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. ACS Nano. 9, 2420–2432 (2015).
Shim, S. H. et al. Disrupting cholesterol esterification by bitter melon suppresses triple-negative breast cancer cell growth. Mol. Carcinog. 57, 1599–1607 (2018).
Zhao, L. et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J. Immunother. 41, 45–52 (2018).
Pan, J. et al. Potentiation of Kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine 49, 72–81 (2019).
De Boussac, H. et al. Oxysterol receptors and their therapeutic applications in cancer conditions. Expert Opin. Ther. Targets 17, 1029–1038 (2013).
Lin, C. Y. & Gustafsson, J. A. Targeting liver X receptors in cancer therapeutics. Nat. Rev. Cancer 15, 216–224 (2015).
Russo, V. Metabolism, LXR/LXR ligands, and tumor immune escape. J. Leukoc. Biol. 90, 673–679 (2011).
Tavazoie, M. F. et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172, 825–840.e818 (2018).
Flaveny, C. A. et al. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell 28, 42–56 (2015).
Wu, G. Z. et al. Targeting the transcription factor receptor LXR to treat clear cell renal cell carcinoma: agonist or inverse agonist? Cell Death Dis. 10, 416 (2019).
Zhang, J. et al. Cholesterol content in cell membrane maintains surface levels of ErbB2 and confers a therapeutic vulnerability in ErbB2-positive breast cancer. Cell Commun. Signal. 17, 15 (2019).
Kong, Y. et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 293, 14328–14341 (2018).
Bhardwaj, A. et al. The isomiR-140-3p-regulated mevalonic acid pathway as a potential target for prevention of triple negative breast cancer. Breast Cancer Res. 20, 150 (2018).
McGregor, G. H. et al. Targeting the metabolic response to statin-mediated oxidative stress produces a synergistic antitumor response. Cancer Res. 80, 175–188 (2020).
Li, J., Qu, X., Tian, J., Zhang, J. T. & Cheng, J. X. Cholesterol esterification inhibition and gemcitabine synergistically suppress pancreatic ductal adenocarcinoma proliferation. PLoS One 13, e0193318 (2018).
Li, M. et al. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8+ T cells. Nanomedicine 14, 2541–2550 (2018).
Lei, J., Wang, H., Zhu, D., Wan, Y. & Yin, L. Combined effects of avasimibe immunotherapy, doxorubicin chemotherapy, and metal-organic frameworks nanoparticles on breast cancer. J. Cell Physiol. (2019).
Chen, X., Song, Q., Xia, L. & Xu, X. Synergy of dendritic cell vaccines and avasimibe in treatment of head and neck cancer in mice. Med. Sci. Monit. 23, 4471–4476 (2017).
Acknowledgements
We thank T. Horng at ShanghaiTech University for language editing, and X. He and L. Mu for figure design. C.X. is funded by CAS grants (Strategic Priority Research Program XDB29000000, Facility based Open Research Program QYZDB-SSW-SMC048, Fountain-Valley Life Sciences Fund of University of Chinese Academy of Sciences Education Foundation), NSFC grants (31861133009, 31621003), a MOST grant (2018YFA0800700) and the Ten Thousand Talent Program “Leading talent” of China.
Author information
Authors and Affiliations
Contributions
C. X. designed the framework. B.-L.S. wrote the first section, and B.H. wrote the rest of the manuscript. C.X. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.X. is a scientific co-founder of Hangzhou MetMed Therapeutics. The other authors declare no competing interests.
Additional information
Peer review information Primary Handling Editor: Christoph Schmitt.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Huang, B., Song, Bl. & Xu, C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab 2, 132–141 (2020). https://doi.org/10.1038/s42255-020-0174-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s42255-020-0174-0
This article is cited by
-
Cancer immunometabolism: advent, challenges, and perspective
Molecular Cancer (2024)
-
The novel molecular mechanism of pulmonary fibrosis: insight into lipid metabolism from reanalysis of single-cell RNA-seq databases
Lipids in Health and Disease (2024)
-
Oncolytic adenovirus encoding apolipoprotein A1 suppresses metastasis of triple-negative breast cancer in mice
Journal of Experimental & Clinical Cancer Research (2024)
-
Nanoparticles targeting mutant p53 overcome chemoresistance and tumor recurrence in non-small cell lung cancer
Nature Communications (2024)
-
Shaping immune landscape of colorectal cancer by cholesterol metabolites
EMBO Molecular Medicine (2024)
