
Abstract
Cholesterol is an essential structural component of membranes that contributes to membrane integrity and fluidity. Cholesterol homeostasis plays a critical role in the maintenance of cellular activities. Recently, increasing evidence has indicated that cholesterol is a major determinant by modulating cell signaling events governing the hallmarks of cancer. Numerous studies have shown the functional significance of cholesterol metabolism in tumorigenesis, cancer progression and metastasis through its regulatory effects on the immune response, ferroptosis, autophagy, cell stemness, and the DNA damage response. Here, we summarize recent literature describing cholesterol metabolism in cancer cells, including the cholesterol metabolism pathways and the mutual regulatory mechanisms involved in cancer progression and cholesterol metabolism. We also discuss various drugs targeting cholesterol metabolism to suggest new strategies for cancer treatment.
Similar content being viewed by others
Introduction
Cholesterol is an important lipid in mammals and an essential component of the membrane. In addition to its role as a membrane constituent, cholesterol is a precursor of bile acids and steroid hormones. Cholesterol and its derivatives are critical for cellular functions. Dyshomeostasis of cholesterol is one of the hallmarks of cancer1. In cancer cells, cholesterol uptake and synthesis rates are usually increased, leading to abnormal metabolism2,3. The efflux and esterification of cholesterol affect the formation of tumor cells4,5. Oxysterols, derivatives of cholesterol, are also involved in tumorigenesis6. Recent studies have shown that the regulation of cholesterol level with the respect to the tumor microenvironment (TME) plays an important role in tumor cell proliferation7. In addition, ferroptosis, autophagy, tumor cell, and immune cell stemness, and the cellular DNA damage response (DDR) are regulated through cholesterol metabolism8,9,10. Cholesterol metabolism plays an important role in tumorigenesis and progression, and therefore, targeting cholesterol metabolism has become a new direction in the treatment of cancer.
In this review, we summarize recent literature regarding cholesterol metabolism in cancer cells, including discussion on the cholesterol metabolism process and metabolites, the functional significance of cholesterol metabolism in cancer cells and mutual regulatory mechanisms. We also discuss various drugs targeting cholesterol metabolism to establish new strategies for cancer treatment.
Overview of cholesterol metabolism
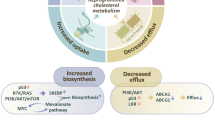
The process of cholesterol metabolism involves an acetyl coenzyme A (acetyl-CoA)-based synthesis pathway, cholesterol uptake, efflux, and esterification (Fig. 1).

Major pathways of cholesterol metabolism in cancer cells include cholesterol biosynthesis, uptake, efflux, and esterification. Cholesterol is synthesized through the mevalonate pathway in the ER in an extremely complex process: It starts with acetyl-CoA, then mevalonate and squalene are synthesized, and finally cholesterol is generated. Cholesterol uptake depends on the LDLR-mediated endocytosis pathway. Cholesterol efflux is mediated by ABCA1 and ABCG1. Cholesterol is a precursor of CEs, which are generated via the action ACAT1. The transcription of cholesterol metabolism-related genes (such as HMGCR, LDLR, ABCA1, and ABCG1) is mediated by mature SREBP-2. SREBP-2 is cleaved to form mature SREBP-2 in the Golgi apparatus.
De novo cholesterol biosynthesis
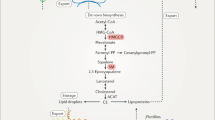
Cholesterol is synthesized through the mevalonate pathway via an extremely complex process involving the synthesis of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), mevalonic acid (MVA), and squalene and their subsequent conversion into other molecules. These biosynthetic processes, for which acetyl-CoA is the starting material, are energy consuming and depend on ATP and NADPH (Fig. 2). Generally, two molecules of acetyl-CoA are condensed into acetoacetyl-CoA in a process catalyzed by cytosolic thiolase, which binds with one molecule of acetyl-CoA in a reaction catalyzed by HMG-CoA synthase to generate HMG-CoA. HMG-CoA reductase (HMGCR), the primary rate-limiting enzyme, catalyzes the reduction of HMG-CoA to MVA, which requires the consumption of two NADPH molecules. MVA undergoes a three-step enzymatic reaction involving phosphorylation and decarboxylation to yield isopentenyl pyrophosphate (IPP). A series of enzymatic reactions in the cytoplasm converts IPP to farnesyl pyrophosphate (FPP), and two molecules of FPP are condensed into squalene in a reaction catalyzed by squalene synthase. Squalene is oxidized to 2,3-epoxysqualene by squalene epoxidase (SQLE) and then cyclized to lanosterol, which is eventually converted to cholesterol through a complex reaction in the endoplasmic reticulum (ER). Newly synthesized cholesterol in the ER is directly or indirectly transported to the cell membrane through the Golgi apparatus11.

Cholesterol is synthesized through a series of ~30 reactions. The substrate in the mevalonate pathway is acetyl-CoA, which is condensed into acetoacetyl-CoA. Acetoacetyl-CoA engages with another molecule of acetyl-CoA in a reaction catalyzed by HMG-CoA synthase, forming HMG-CoA. HMGCR catalyzes the reduction of HMG-CoA to yield MVA. MVA undergoes a three-step enzymatic reaction involving phosphorylation and decarboxylation to produce isopentenyl pyrophosphate (IPP). A series of enzymatic reactions converts IPP to farnesyl pyrophosphate (FPP), and two molecules of FPP condense into squalene in a reaction catalyzed by squalene synthase. Squalene is oxidized to be 2,3-epoxysqualene by SQLE and then cyclized to lanosterol, which is eventually converted to cholesterol.
Sterol regulatory element binding proteins (SREBPs) are transcription factors that play central roles in modulating cholesterol biosynthesis12. SREBPs constitute a class of membrane proteins in the bHLH-Zip superfamily; they are anchored to the ER and nucleus through their “basic helix-loop-helix-leucine zipper” (bHLH-Zip) motif13,14. Three members of the SREBP family have been identified: SREBP-1a, SREBP-1c, and SREBP-2. SREBP-1a and SREBP-1c mainly regulate the expression of enzymes related to fatty acid, triglyceride, and glucose metabolism, while SREBP-2 mainly regulates cholesterol metabolism (Fig. 3). SREBP-2, synthesized in the ER, is a nonfunctional precursor that forms a complex with SREBP cleavage-activating protein (SCAP) to coregulate intracellular cholesterol levels. SREBP2 must be translocated from the ER to the Golgi apparatus, where it is released from the membrane by site 1 protease (S1P) and site 2 protease (S2P)15. After sequential processing by S1P and S2P, SREBP-2 loses the N-terminal fragment containing the bHLH-Zip region16. The processed SREBP2 enters the nucleus as a homodimer and attaches to the sterol regulatory element (SRE) sequence in the promoters of target genes, such as HMGCR and SQLE (encoding squalene monooxygenase)17,18.

SCAP interacts with SREBP and mediates the translocation of SREBP from the ER to the Golgi apparatus, where it is cleaved by proteases. Active fragments of SREBP in the nucleus bind to the SRE sequence in HMGCR or another cholesterol synthesis-related enzyme promotor region, thereby initiating gene transcription.
Cholesterol uptake
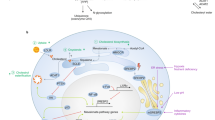
In addition to de novo cholesterol biosynthesis in the ER, cells mainly acquire cholesterol from the receptor-mediated uptake of exogenous cholesterol, including low-density lipoprotein (LDL) acquisition through the endocytosis pathway and high-density lipoprotein (HDL) acquisition through the selective uptake pathway (Fig. 4). These pathways jointly regulate cellular cholesterol levels19,20.

LDL particles combine with LDLR in the cell membrane and enter cells through endocytosis. Free cholesterol is then dissociated from the LDLR in the lysosome. Unbound LDLR is recycled to the cell membrane. LDLR is degraded in lysosomes in the presence of PCSK9. HDL particles bind to SR-B1 on the cell membrane. HDL-derived cholesterol and cholesteryl esters can be delivered into the cell via the hydrophobic channels mediated by SR-B1 and further to other organelles, such as the endoplasmic reticulum and mitochondria.
LDL receptor (LDLR)-mediated LDL endocytosis is the most important mechanism underlying plasma cholesterol clearance. LDL particles bind to an LDLR in the cell membrane and eventually reaches the lysosome after internalization where it releases free cholesterol through acid lipase hydrolysis. Receptor-mediated HDL cholesteryl ester uptake requires the involvement of HDL receptor scavenger receptor B-type I (SR-BI), which is a member of the class B family of scavenger receptors20. SR-BI is a lipophilic channel in the plasma membrane, and through its extracellular domain, SR-BI binds to HDL cholesteryl esters and selectively delivers them into the cell. The extracellular loop of SR-BI contains six highly conserved cysteine residues that are critical to SR-BI activity21,22. Notably, mice with SR-BI mutations presented with increased plasma cholesterol levels23, and inhibition of SR-BI glycosylation led to defective selective uptake of HDL cholesteryl esters24.
Cholesterol efflux and esterification
Dietary cholesterol is absorbed from the gastrointestinal tract, where cholesterol and triglycerides form chylomicrons. In the blood circulatory system, chylomicrons are modified into celiac remnants, which are then transported to the liver. In the liver, very low-density lipoprotein (VLDL) particles of lipids and cholesterol are secreted by hepatocytes, and these particles are further modified into LDL in the blood circulation and then transported to peripheral cells. Excess cholesterol in peripheral cells is released as HDL-C, which triggers the reverse transport of cholesterol back to the liver (Fig. 5).

ABCA1 mediates cellular HDL efflux, and HDL surface cholesterol is esterified by LCAT. With an increase in cholesterol esters, HDL is converted into HDL2 and HDL3. HDL is finally absorbed into hepatocytes in the presence of the HDL receptor SR-B1. In addition, the cholesterol esters in HDL can be transferred to VLDL by CETP and produce LDL.
Cholesterol efflux is the most important step in reverse cholesterol transport. There are four ways in which cholesterol is released from cells: (1) passive diffusion of cholesterol by mature HDL particles, (2) SR-B1-mediated facilitated diffusion, (3) ATP-binding cassette (ABC) transporter subfamily A member 1 (ABCA1)-mediated efflux with ApoA1, and (4) ABC subfamily G (ABCG)-mediated efflux with mature HDL25. Free cholesterol (FC) released from the plasma membrane undergoes passive aqueous diffusion mediated via HDL and is driven by the cholesterol gradient, which is maintained through esterification of HDL surface cholesterol through the action of lecithin–cholesterol acyltransferase (LCAT)25.
The plasma contains not only FC but also esterified cholesterol. Cholesterol esterification into cholesteryl esters is mediated by acyl-CoA: cholesterol acyltransferase (ACATs). Of the two ACATs ACAT1 and ACAT2, ACAT1 is expressed in most human tissue cells. Cholesterol esters synthesized by ACAT1 are usually stored in lipid droplets. In contrast, ACAT2 is expressed in a development- and species-specific manner. ACAT2 is highly expressed in the human intestine and in the infant liver, and the cholesteryl esters synthesized by ACAT2 are mainly transported by lipoproteins, which can be secreted out of cells26,27. Studies have shown that the expression and activity of ACAT-1 are increased in breast, pancreatic, and glioblastoma tumor cells and that they promote cholesteryl ester production28. Excessive cholesteryl ester production activates SREBP1, which promotes tumor metastasis. Inhibition of ACAT-1 production leads to inhibited glioblastoma growth and prostate cancer cell invasiveness5,29.
Cholesterol oxygenation and oxysterols
In addition to cholesteryl esters, cholesterol can be converted into oxysterols directly through autooxidation in the presence of reactive oxygen species (ROS) or via enzymatic processes. Oxysterols are found in low or very low concentrations in the human body and mainly exist in oxidized lipoproteins30, which modulate the fluidity of the cell membrane and exhibit other cellular functions. Cholesterol oxygenation usually occurs in the steroid backbone or aliphatic sidechain6. Oxidation in the side chain generates 27-hydroxycholesterol (27-HC), 25-hydroxycholesterol (25-HC), 24-hydroxycholesterol (24-HC), and 22-hydrocholesterol (22-HC). Backbone oxidation generates 7-ketocholesterol (7-KC), 5,6α/β-epoxycholesterol (5,6α-EC/5,6β-EC), and 7α/β-hydroxycholesterol (7α/β-HC). Oxysterol production on the side chain is mainly mediated through enzymatic processes, which are mainly catalyzed by the cytochrome p450 (CYP) family of enzymes31,32. 27-HC is generated by CYP27A1 and catabolized by CYP7B1. CYP46A1 catalyzes cholesterol metabolism to yield 24(S)-HC. 22(R)-HC is generated by CYP11A1. However, 25-HC is generated by cholesterol-25-hydroxylase, not the CYP family. In addition, lipid peroxidation indirectly generates oxysterols, such as B-ring oxysterols33.
The regulation of cholesterol metabolism in cancer
Internal and external factors regulation of cholesterol metabolism, driving altered cholesterol pathways in cancer cells is critical (Fig. 6). Internal factors such as some signaling pathway molecules or cholesterol itself affect cholesterol metabolism by regulating the activity of SREBP and LXR. External factors such as the acidification and inflammatory of the tumor microenvironment (TME) could also affect cholesterol metabolism.

The SCAP-SREBP2 complex is trafficked in COPII vesicles from the ER to the Golgi for proteolytic activation of SREBP2 when the intracellular cholesterol is low. PI3K/AKT, mTOR, and PTEN are critical for SREBP activation. MIEF2 enhances lipid biosynthesis by increasing mitochondrial reactive oxygen species (ROS) production and promoting the subsequent activation of the AKT/mTOR signaling pathway. The Wnt-β-catenin and p53 pathways are critical for the transcription of MVA pathway-related genes. c-Fos inhibits LXR signaling and increases the production of cholesterol. PCSK9 induces the degradation of LDLR. Cholesterol metabolites such as oxysterols function as endogenous ligands for LXRs, suppressing the transcriptional program to inhibit LDL uptake and promote cholesterol efflux, thereby maintaining cholesterol homeostasis. External factors such as low pH, LPS, and TNF can affect cholesterol metabolism.
Intercellular molecules mediate cholesterol metabolism
SREBP activation plays a central role in the abnormal cholesterol metabolism of tumor cells. SREBP is inhibited by ER cholesterol, Insig protein, and SCAP. Cholesterol concentrations regulate the evacuation of the SREBP2 precursor from the ER through SCAP, which binds SREBP2 via its C-terminal domain. SCAP detects and reacts to changes in ER cholesterol to control the switch between open and closed by modulating its binding to COPII-coated vesicles. The SCAP-SREBP2 complex is sorted into COPII vesicles and transported from the ER to the Golgi, where the proteolytic activation of SREBP2 is triggered when the intracellular cholesterol is low34. The dynamic balance in intracellular cholesterol levels regulated by SREBPs requires the joint contributions of SCAP, S1P, S2P, and SRE, and any abnormality, such as a mutation, in any one of these participants can lead to the disruption of cholesterol metabolism35. 25-Hydroxycholesterol and other oxysterols are much more effective than cholesterol in binding to Insig proteins and promoting Insig binding to SCAP, which triggers ER retention of the SCAP-SREBP2 complex36. Under sterol-deficient conditions, Insig1 degradation leads to the dissociation of the SCAP-Insig complex and activation of the SREBP2 pathway, permitting the transcription of downstream genes37.
In tumor cells, overactivation of the PI3K/AKT signaling and p53-mediated signaling pathways influence SREBP activity. The AKT/mTOR pathway is the most frequently studied de novo cholesterol synthesis pathway in cancer cells. Akt drives SREBP-2 activity and inhibits SREBP-2 degradation, thereby promoting the expression of cholesterol synthesis-related genes38. mTOR1 is an important upstream signaling molecule in Akt-induced SREBP activation. mTOR1 promotes nSREBP-2 proteins by phosphorylating and inhibiting the nuclear entrance of lipin 1 and regulating cholesterol trafficking from lysosomes to the ER39. Studies have shown that PTEN affects AKT and mTOR activation, thus regulating SREBP-2-mediated target transcription40. In addition to the AKT/mTOR pathway, p53 plays an important role in the MVA pathway (Fig. 7). Mutations in p53 promote the mevalonate pathway by interacting with SREBPs and increasing the activity of SREBPs41. These mutations induce mevalonate-5-phosphate (MVP) production, and in turn, MVP promotes mutant p53 stabilization, triggering positive feedback regulation of SREBPs42. Accumulation and stabilization of mature SREBP2 increase mevalonate pathway enzyme expression in the absence of p5343. In addition, p53 increases the expression of the cholesterol efflux transporter ABCA1, thereby repressing SREBP2 maturation and subsequently inhibiting the MVA pathway41. Additionally, SREBP2 transcriptional activity is promoted by PIK3CA mutations or growth factors through various mechanisms. In prostate cancer, androgen receptor (AR) signaling mediates SCAP upregulation and promotes SREBP activity44. Moreover, the Wnt-β-catenin pathway is activated upon Cilia repression, which promotes the expression of genes involved in the MVA pathway through SREBP-2 interactions45. MIEF2 (mitochondrial elongation factor 2) is one of the key regulators of mitochondrial fission. Studies have shown that MIEF2 enhances lipid biosynthesis by increasing mitochondrial reactive oxygen species (ROS) production and subsequent activation of the AKT/mTOR signaling pathway, upregulating the expression of SREBP1 and SREBP2 and their transcriptional targets, such as the lipogenic enzymes ACC1, FASN, SCD1, HMGCS1, and HMGCR. MIEF2 overexpression-mediated mitochondrial dysfunction plays a key role in the reprogramming of lipid metabolism in ovarian cancer cells46.

p53 increases the expression of the cholesterol efflux transporter ABCA1, repressing SREBP2 maturation and subsequently inhibiting the MVA pathway. Mutp53 promotes mevalonate pathway activation by interacting with SREBPs, increasing SREBP activity. The activated mevalonate pathway increases the levels of MVP, leading to mutp53 stabilization (positive-feedback loop).
Liver X receptors (LXRs), which form obligate heterodimers with retinoid X receptors (RXRs), are other transcription factors that regulate the expression of ABCA1 and ABCG147. As sensors, LXRs are activated by a high intracellular cholesterol content. The oncogene c-Fos in hepatocytes inhibits LXR signaling and increases the production of cholesterol and cholesterol-derived metabolites, such as oxysterols and bile acids. These effects are associated with increased hepatocellular carcinogenesis48. Oxysterols can function as endogenous ligands for LXRs, suppressing a transcriptional program to inhibit LDL uptake and promote cholesterol efflux, thereby maintaining cholesterol homeostasis49. Several oxysterols, such as 22(R)-HC, 25-HC, 27-HC, and 24(S)-HC, modulate the activity of LXRs49,50. In addition, some oxysterols exert effects by targeting other nuclear receptors, such as RAR-related orphan receptors (RORs), estrogen receptors (ERs), and the glucocorticoid receptor (GR). Different oxysterols show various functions, acting as both agonists or inverse agonists for RORs and thus participate in cholesterol metabolism. 27HC functions as an agonist of RORγ. However, 24(S)-HC and 25-HC have been reported as inverse agonists of RORα and RORγ51. Interestingly, a study reported that 25-HC and 22-HC agonistic activity restored RORγ transcriptional activity that had been suppressed by a synthetic inhibitor52. In addition, 22(R)-HC, 24(S)-HC, 25-HC, 27-HC, and 7 KC have been reported to regulate ER activities52. In addition, 6-oxo-cholestan-3β,5α-diol (OCDO) generated from 5,6-EC has been identified as a GR ligand53. OCDO has also reported as a dual GR and LXR ligand. 27-HC is the most abundant oxysterol in the cell membrane and blood. Therefore, it easily crosses the blood‒brain barrier and functions as a “cerebrosterol”. Increasing evidence suggests that 27-HC is important in cancer cells, especially in breast cancer cells54. Studies have elucidated that 27-HC activates LXRs to promote the metastasis of ER-positive breast cancer cells54,55. However, in endometrial cancer, 27-HC promotes cell proliferation by activating ERs but not LXRs56. 25-HC also promotes cell migration and invasion in lung, gastric, and brain cancers by activating a G protein-coupled receptor or the Toll-like receptor 2 (TLR2)/NF-kB pathways and the LXR/interleukin-1B (IL-1B) signaling pathway56,57,58.
In addition to that at the transcriptional level, cholesterol synthesis is also regulated at the translational level. In sterol-rich environments where HMGCR transcription is inhibited, HMGCR synthesis is controlled by mevalonate derivatives. FPP and GGPP are downstream of mevalonate in the cholesterol synthesis pathway, and depletion of mevalonate via HMG-CoA reductase inhibitors reduces the availability of FPP and GGPP35, thereby regulating cholesterol synthesis. Cholesterol derivatives, such as 25-hydroxycholesterol or oxygenated cholesterol derivatives such as 27-HC have been shown to reduce the activity of HMGCR55.
Cholesterol synthesis-related enzymes, such as HMGCR, are regulated via posttranslational modification, including acetylation, ubiquitination, and phosphorylation. Gp78, TRC8, HRD1, MARCHF6, and RNF145 are E3 ubiquitin ligases that lead to HMGCR degradation59,60,61,62. HMGCR degradation is induced by sterols, including C4-dimethylated sterols and lanosterol and its C24-saturated derivative 24,25-dihydrolanosterol; however, cholesterol itself does not affect HMGCR itself63,64,65,66. In addition to ubiquitination, HMGCR is regulated via phosphorylation. In humans, AMPK-mediated HMGCR Ser872 phosphorylation inhibits the activity of HMGCR and decreases cholesterol production67.
Modulation of cholesterol metabolism by the tumor microenvironment (TME)
In addition to its influence on intercellular molecules, the TME is involved in the regulation of cholesterol metabolism. Low pH conditions promote cholesterol biosynthesis68. For example, in pancreatic cancer cells at pH 6.8, low-pH-responsive genes mediate SREBP2 nuclear localization, upregulating target gene expression. Under hypoxic conditions and low nutrient concentrations, TME-induced ER stress affects cholesterol metabolism because cholesterol is synthesized in the ER69. Moreover, lipopolysaccharide (LPS) and the cytokine TNF enhance cholesterol accumulation by activating SREBP270. HGFs in the liver microenvironment activate cholesterol metabolism through c-Met/PI3K/AKT/mTOR signaling pathways71.
Noncoding RNAs (ncRNAs) and cholesterol homeostasis in cancer
Numerous studies have shown that ncRNAs affect cholesterol homeostasis by influencing cholesterol transport, uptake, and efflux, and these ncRNAs play critical roles in cancer progression. Table 1 shows the ncRNAs affecting gene expression in cholesterol metabolism. MicroRNAs (miRNAs), such as miR-128-1, miR-148a, miR-130b, and miR-301b, which are highly conserved in vertebrates and expressed in many tissues, have been found to play critical regulatory roles in cholesterol homeostasis and transport72,73,74. The target proteins of miRNAs include LDLR, ABCA1 and the insulin receptor (InsR). MiR-185, miR-96, and miR-223 have been reported to regulate the selective uptake of HDL-C by targeting hepatic SR-B175. Specifically, miR-24 targets the SR-B1 3′ untranslated region (UTR) and inhibits SR-B1 expression. miR-24 has been found to promote the expression of genes involved in cholesterol synthesis in an indirect manner76. miR-33, encoded by an intron within the SREBF2 gene, is located upstream of ABCA177,78. miR-183 functions as an oncogene targeting the 3′UTR of ABCA1, and reportedly regulates cholesterol efflux in many malignancies79,80. miR-26, miR-20a/b, miR-758, and miR-19b have also been reported to regulate cholesterol efflux by targeting ABCA181,82,83,84. For example, downregulation of miR-129-5p expression promoted ABCG1 expression and led to chemoresistance in gastric cancer cells85. The long noncoding RNA (lncRNA) lncARSR, a regulator of AKT signaling associated with hepatocellular carcinoma (HCC) and renal cell carcinoma (RCC), regulates SREBP-2 expression86. Furthermore, in plasma, a high level of the lncRNA HULC, which downregulates miR-9 expression and upregulates RXRA expression during cholesterol synthesis, has been identified as a biomarker of liver cancer87. NEAT1 and ANRIL, which are also associated with cholesterol synthesis, are considered biomarkers of non-small cell lung cancer88,89. In addition, MALAT1, known to be involved in cholesterol efflux, has been suggested to be a diagnostic indicator of lung cancer90.
Functional significance of cholesterol metabolism in cancer
The reprogramming of lipid metabolism is a hallmark of cancer. Cholesterol, an important component of lipids, is thought to be necessary for cancer cell proliferation and survival. Mechanistically, cholesterol affects tumor cells by modulating immune responses, ferroptosis and autophagy, tumor cell stemness, and the DNA damage response (Fig. 8).

Cholesterol is related to various immune cells, including DCs, CD8+ cells, neutrophils, NK cells, macrophages, and γδ-T cells, to regulate the cellular immune response. IPP regulates GPX4 activity and ferroptosis by promoting Sec-tRNA maturation. Cholesterol activates cellular signaling pathways downstream of sonic hedgehog, Notch and receptor tyrosine kinases and promotes stem cell development. In addition, ZMYND8/SREBP2-coordinated enhancer–promoter interactions activate the MVA pathway and promote intestinal stemness. FDPS is critical for cell stemness. Statins, inhibitors of HMGCR, affect the DDR by inhibiting CHK1 and p53 activity.
Cholesterol metabolism regulates immune responses in the cancer context
The interplay between tumor metabolic reprogramming and immune cells is a determining factor in the antitumor immune response. Increasing evidence has suggested that cholesterol metabolism not only plays a key role in tumorigenesis and patient survival but also influences the function of immunity-related molecules91,92. High cholesterol levels in tumor cells facilitates tumor cell escape from immune surveillance, and tumor cell-induced increases in the cholesterol level promote the expression of suppressive immune checkpoint genes, thereby inhibiting antitumor effects. Indeed, high cholesterol disrupts the homeostasis of the lipid metabolic network in immune cells, suppressing the immune response. In total, cholesterol metabolism regulates immune responses in three ways: inhibition of antigen presentation, enrichment of immunosuppressive cells (neutrophils and tumor-associated macrophages (TAMs), and regulation of immune-effector cells.
Inhibition of antigen presentation and enrichment of immunosuppressive cells
Conditional medium obtained from several cancer cell cultures activated LXR-α signaling on the dendritic cell surface, thereby decreasing CC chemokine receptor-7 (CCR7) expression in these DCs93. Cholesterol metabolites are also related to the enrichment of immunosuppressive cells. Cholesterol metabolites, such as 22-HC, 24S-HC, 24-HC, and 27-HC, recruit neutrophils. One study showed that 27-HC promoted breast cancer metastasis by affecting immune cells94. Elimination or inhibition of the CYP27A1 enzyme, which is critical to 27-HC biosynthesis, significantly reduced cancer metastasis. The strong effect of 27-HC on metastasis depended on the function of bone marrow immune cells. 27-HC increased the number of polymorphonuclear neutrophils and γδ-T cells at distant metastatic sites and decreased the number of CD8+ T cells94.
In recent years, considerable clinical and preclinical evidence has shown that TAMs play important roles in tumor growth, invasion, and metastasis. Researchers have conducted in-depth studies on the reprogramming mechanism in TAMs in the TME. They proposed that tumor cell secretion promotes membrane cholesterol efflux from TAMs, which drives TAM reprogramming and promotes tumor growth95. Researchers first performed a subgroup analysis to detect the phenotypes of TAMs in a tumor model and found that peripheral blood mononuclear cell-derived (MN-derived) TAMs were gradually replaced by embryonic precursor cell-derived resident macrophages. Moreover, a transcriptome analysis showed that the expression of cholesterol metabolism- and efflux-related genes in TAMs was significantly upregulated during tumor progression. Further experiments revealed that ovarian cancer cells promoted membrane cholesterol efflux by secreting hyaluronic acid (HA), an extracellular matrix component, resulting in the absence of cholesterol-enriched lipid rafts in the cell membrane. These cholesterol-related alterations promoted the onset of IL-4-mediated reprogramming in macrophages. Thus, macrophages acquired an IL-4-induced phenotype, and IFN-γ-related signaling was disrupted, which resulted in the suppression of IFN-γ-induced expression of related genes. Further functional experiments demonstrated that knocking out ABCs prevented TAM reprogramming and inhibited tumor growth.
Regulation of immune effector cells
The regulation of cholesterol metabolism-related enzymes has a profound impact on immune effector cells. A typical example in tumors involves the immunomodulatory function of PCSK9, which is related to cholesterol endocytosis into tumor cells. Knocking down or blocking PCSK9 activity through treatment with a specific antibody enhanced the effect of tumor immunotherapy in mice. PCSK9, a serine protease produced predominantly in the liver, induces the degradation of LDLR96. Overexpression of PCSK9, which binds to LDLR, resulted in a much lower ability of a patient to eliminate LDL-cholesterol (LDL-C)97. In this case, the effects of PCSK9 inhibition in conjunction with an anti-PD-1 antibody treatment lead to a synergistic therapeutic outcome91. Knocking out PCSK9 significantly inhibited tumor growth in immunocompetent mice, but this outcome was not observed in immunodeficient mice. Recently, another team found that PCSK9 inhibited LDLR expression in CD8+ T cells, thereby repressing the antitumor activity of these CD8+ T cells. These data strongly suggest that PCSK9 inhibition may significantly enhance the immune response and suppress tumor growth98. Cholesterol metabolism and transporter enzymes exert important effects on the production and activity of immune cells. For example, ACAT1 has been shown to be a regulatory target in the T-cell metabolic pathway99. The activation and production of CD8+ T cells are required for maintaining the membrane cholesterol content, which is partially regulated by ACAT1. Notably, knocking out ACAT1 in CD8+ T cells increased the level of membrane cholesterol, increased T-cell receptor aggregation, promoted T-cell proliferation and activation, and ultimately enhanced their antitumor effects99. These studies suggested that cholesterol at high levels exerts immunosuppressive effects. However, the question remains: how does high cholesterol promote the expression of immune checkpoint genes? To answer this question, researchers treated T cells with different concentrations of cholesterol and then used gene chips to analyze the effect. They found that high cholesterol disrupted the lipid metabolic network, which induced ER stress100. ER stress led to upregulated expression of XBP1, an ER stress receptor101. XBP1 activation, in turn, promoted the expression of immune checkpoint genes, causing T-cell suppression100. Recent studies have revealed that XBP1 is a transcription factor involved in cholesterol biosynthesis. ER stress induces XBP1, driving cholesterol biosynthesis by directly binding to the promoters of key genes102.
In addition to metabolism-related enzymes, some studies have found that 25-HC inhibits trogocytosis and promotes the antitumor activities of cytotoxic T lymphocytes103. In addition, NK cell functions are regulated by cholesterol104,105. SREBP is a key factor in regulating energy production in NK cells, which affects NK cell activation105.
Abnormal cholesterol metabolism induces ferroptosis and autophagy in cancer cells
Depletion of glutathione, decreased glutathione peroxidase (GPX4) activity, or reduced cellular antioxidant capacity results in lipid peroxidation and metabolic dysfunction and increased levels of lipid reactive oxygen species (ROS), which trigger ferroptosis106. The susceptibility of cells to ferroptosis is closely related to many biological processes, including amino acid, ferritin, and polyunsaturated fatty acid metabolism.
GPX4 is a selenium-containing protein with a selenocysteine active center, and it is required for the function of a specific selenocysteine transporter, selenocysteine transfer RNA (Sec-tRNA)107. The addition of IPP to specific adenine sites in Sec-tRNA precursors is necessary for Sec-tRNA maturation; hence, IPP exerts an important effect on ferroptosis. FPP, dolichol, squalene, and coenzyme Q (CoQ), which are IPP derivatives, are also involved in the Sec-tRNA maturation process and exert multiple effects on ferroptosis108. The mechanism underlying p53 regulation of ferroptosis may involve its recently recognized role as a key mediator of the MVA pathway. Under metabolic stress, p53 mediates the expression of ABCA1, which is critical for the reverse translocation of cholesterol from the plasma membrane to the ER. This leads to the inactivation of SREBP2 and gene transcription and ultimately inhibits the production of several metabolites, such as squalene and CoQ.
Recently, researchers have found that high cholesterol results in resistance to ferroptosis and increased tumorigenicity and metastasis in breast cancer8. Migrating cancer cells can engulf cholesterol in response to stress. Generally, most cancer cells die when exposed to stress, but surviving cancer cells contain high concentrations of cholesterol to counteract the onset of ferroptosis. Cells chronically exposed to 27-HC were shown to cell exhibiting increased cellular uptake and/or biosynthesis of cholesterol8. These cells exhibit significantly increased tumorigenic and metastatic capacity, and GPX4 prevents ferroptosis and promotes their metastasis.
Statins, which block the rate-limiting enzymes in the ferroptosis pathway, interfere with the efficient translation of GPX4, increasing cell susceptibility to ferroptosis. Squalene is thought to exert an antiferroptotic effect on cancer cells109. Cholesterol is also involved in ferroptosis, and exogenous cholesterol hydroperoxide has been shown to induce cell death in a dose-dependent manner110. GPX4 inhibition accelerates ferroptosis induced by cholesterol hydroperoxide, while GPX4 overexpression greatly enhances cell resistance to cholesterol hydroperoxide-induced ferroptosis.
Recent research has found that decreases in cell membrane cholesterol facilitates autophagy initiation protein recruitment and promotes autophagosome synthesis. The cholesterol transporter protein GRAMD1C may inhibit autophagosome synthesis and decrease mitochondrial bioenergetics by regulating cholesterol transport between the cell membrane and endoplasmic reticulum or between the endoplasmic reticulum and mitochondria. Although the study explained the function of the cholesterol transporter protein in autophagy, the roles of cholesterol regulation in autophagosome development and cancer progression remain unknown111.
Cholesterol metabolism and cell stemness in cancer
Cholesterol biosynthesis plays an important role in maintaining tumor stem cells112. It can activate cellular signaling pathways downstream of sonic hedgehog, Notch and receptor tyrosine kinases and promote stem cell development. To capitalize on their own robust self-renewal properties, intestinal stem cells require an adequate supply of lipids and cholesterol. Excess dietary cholesterol or enhanced intracellular cholesterol synthesis accelerated tumorigenesis in mice. Zinc finger MYND type 8-containing (ZMYND8), an epigenetic reader, interacts with SREBP2 to activate the MVA pathway and promotes intestinal stemness and tumorigenesis113,114. A research group has provided insight into this relationship from the perspective of the regulation of lipid and cholesterol dynamic homeostasis9. Inhibition of the phospholipid remodeling enzyme LPCAT3 increased cholesterol saturation in the membrane and stimulated cholesterol biosynthesis, which in turn promoted small intestine stem cell proliferation. Pharmacological blockade of cholesterol synthesis inhibited the development of intestinal crypts in LPCAT3-deficient mice. In contrast, increasing intracellular cholesterol levels stimulated the development of intestinal crypts. Feeding a diet with excess cholesterol or increasing endogenous cholesterol synthesis through SREBP-2 expression promoted small intestine stem cell proliferation in vivo. In addition, the group found that disrupting the LPCAT3-mediated dynamic balance between phospholipid and cholesterol levels significantly increased the tumorigenesis rate in Apc-/- mice.
Recent research has revealed a mechanism through which hypercholesterolemia-induced production of oxidized low-density lipoprotein (ox-LDL) leads to bladder cancer progression through ox-LDL regulation of tumor cell stemness115. Using two hypercholesterolemic mouse models (induced by feeding the mice high-fat high-cholesterol chow or knocking out the Ldlr gene), researchers demonstrated that excessive serum cholesterol increased tumor cell stemness and promoted bladder cancer development. In contrast, mice with hormone-induced hypercholesterolemia treated with the selective cholesterol uptake inhibitor ezetimibe showed significantly reduced tumor cell stemness and slower tumorigenesis, suggesting that cholesterol was critical for the increased malignancy of bladder cancer in these mice. Further validation and signaling pathway analyses revealed that ox-LDL binds to the bladder cancer cell membrane receptor CD36 and affects the intracellular JAK2-STAT3 signaling pathway, thereby regulating tumor cell stemness-related genes and promoting bladder cancer cell proliferation.
A comparison of the mRNA expression patterns in patient-derived glioblastoma spherical cells, which maintain stemness, and their differentiated counterparts, which lose stemness, revealed that the majority of the changed genes were networked in cholesterol metabolism pathway116. Among these differentially expressed genes, the isoprenoid biosynthesis enzyme farnesyl diphosphate synthase (FDPS) is critical for the survival of glioblastoma stem cells. Although the mechanism is unclear, cholesterol metabolism plays an important role in maintaining cell stemness.
Cholesterol metabolism and the DNA damage response in cancer cells
Dyshomeostasis of cholesterol metabolism and the DDR are hallmarks of cancer. Few studies have addressed the relationship between cholesterol metabolism and the DNA repair process. However, one study reported that lovastatin, which blocks cholesterol biosynthesis, may inhibit gallbladder cancer (GBC) cell proliferation by attenuating the DNA repair process117. This investigation showed that lovastatin sensitized GBC to cisplatin-induced apoptosis by impairing the DDR, which involved inhibiting CHK1, CHK2 and γ-H2AX activation. Treatments using statins with differing chemical properties to eliminate HMGCR or adding MVA reversed cisplatin resistance. RNA sequencing of GBC cells stimulated by statins revealed enrichment with DDR and G2/M checkpoint signaling factors. Treatment with statins or MVA, in fact, inhibited DDR and CHK1 signaling; specifically, DDR activation was suppressed by the inhibition of CHK1 and γ-H2AX expression. Another study revealed that pravastatin, an inhibitor of cholesterol biosynthesis, induced Mdm2 Ser166 phosphorylation, which increased Mdm2 ubiquitin ligase activity, attenuated p53 degradation and inhibited the DDR in hepatocytes118. Although a correlation between cholesterol metabolism and the DDR was identified, the mechanism underlying this relationship remains unknown.
Targeting cholesterol metabolism in cancer therapy
Targeting cholesterol synthesis for cancer treatment
Many basic studies and several clinical trials have examined the potential therapeutic efficacy of targeting cholesterol metabolism in cancer therapy (Table 2). The potential therapeutic involve targeting cholesterol synthesis, from the inhibition of specific enzymes to the blockade of the feedback mechanism regulated by SREBPs, have been clinically investigated119. In some of these studies, statins, which are HMGCR inhibitors, were developed and then used as the standard therapy for patients with high cholesterol levels. Statins induced multiple antitumor effects in tumor cell lines and preclinical animal models through a variety of distinct mechanisms, including the promotion of apoptosis, induction of cell cycle arrest, and inhibition of cell proliferation and invasion120. Statins also decreased the cholesterol level and inhibited proteasome action as well as the production of nitric oxide and ROS121,122,123. In addition, statins affected signaling pathways associated with tumorigenesis, including the p53, Akt, mTOR, Myc, p38, and VEGF pathways42,123,124,125. Although early trials reported an increased incidence of cancer in statin drug users, recent studies have shown a 20–28% reduction in the risk of using statins in cancer treatment, and the use of statins has been shown to reduce the risk of recurrence in prostate cancer patients who underwent radical prostatectomy126,127. The specific chemical properties of different statins determine their mechanisms of action. Some studies have shown that lipophilic statins are more likely to enter extrahepatic cells, while hydrophilic statins show greater affinity for the liver128. Pravastatin is a hydrophilic statin that is characterized as a sodium-independent organic anion translocator expressed only in the liver. In contrast, the lipophilic HMGCR inhibitor simvastatin is a hydrophobic statin that enters cells through various mechanisms. Pravastatin significantly reduces the risk of breast cancer recurrence, whereas hydrophilic statins do not129,130. Simvastatin reduces the level of geranylgeranyl pyrophosphate (GGPP) produced in the MVA pathway and the isoprenylation rate of the small GTPase Rab5 in antigen-presenting cells, thereby blocking endosome maturation, prolonging antigen retention, enhancing antigen presentation and T-cell activation, and ultimately enhancing antitumor immunity. In addition, simvastatin potently enhances tumor vaccine efficacy and synergizes with anti-programmed death receptor-1 (PD-1) antibodies in cancer treatment131.
Statin therapy prolongs the survival of patients with MM, colorectal cancer or metastatic pancreatic cancer who have been treated with a combination of first-line chemotherapeutic agents132,133,134. Perioperative trials with patients in the early stage of invasive breast cancer have shown that high doses of neoadjuvant atorvastatin and fluvastatin reduced tumor proliferation135,136. Atorvastatin or fluvastatin also induced MM cell apoptosis by targeting HMGCR137. Long-term statin therapy prolonged the survival of patients with glioblastoma multiforme138. Notably, the results of Phase II clinical trials for patients with non-small cell lung cancer indicated that a combination of simvastatin and the EGFR inhibitor gefitinib exerted better antitumor effects than treatment with the EGFR inhibitor alone135,139. Of course, some conflicting results do not corroborate the findings suggesting statin-conferred protection against cancer. The individual clinical effects of statins may depend on the specific chemical properties of the statin analyzed, the heterogeneity of the tumor treatment, duration of statin use, and other factors.
Other enzymes in the cholesterol biosynthesis pathway have been identified as drug targets. Bisphosphonate, a farnesyl diphosphate synthase (FDPS) inhibitor, has been approved for treating bone metastases because it may improve bone density and thus reduce skeletal-related complications140,141,142. RO48-8071, an inhibitor of oxidative oxytocin cyclase (OSC), significantly inhibits the growth and metastasis of colorectal and pancreatic tumors143. Application of this inhibitor increases the cell apoptosis rate and reduces cell proliferation. More importantly, RO48-8071 in combination with 5-fluorouracil showed enhanced antitumor effects. SQLE plays an important role in cholesterol synthesis and has been used as an antitumor target143. Several drugs targeting SQLE, such as terbinafine, have been clinically approved as antifungal agents; however, whether they can be used as antitumor agents remains to be investigated144.
Targeting cholesterol uptake and cholesterol efflux in cancer treatment
Numerous small molecules, including the cationic sterol U18666A and a class of triazole antifungal drugs, including itraconazole, can block NPC1-mediated cholesterol export and inhibit DHCR24145,146. Ezetimibe is an NPC-1L1 inhibitor that significantly reduces intestinal absorption of cholesterol, decreases plasma cholesterol levels, and promotes cholesterol clearance from plasma147. CETP is a plasma glycoprotein synthesized in the liver that binds to HDL and mediates the transfer of cholesterol from HDL to VLDL and LDL, and CETP inhibitors block this process and exert antiatherogenic and cardiovascular risk-reducing effects. Although CETP inhibitors, including torcetrapib, dalcetrapib, and evacetrapib, increase plasma HDL-C levels, these inhibitors are ineffective and cause adverse effects in patients148,149,150. As a result, CEPT inhibitor development has been halted. Niacin, which can increase HDL-C by 20–25% while reducing triacylglycerol and LDL-C levels151, has not shown significant tumor-suppressing effects. In diffuse large B-cell lymphoma (DLBCL) cells, mimicking natural HDL in the presence of synthetic HDL nanoparticles, altered cholesterol homeostasis and induced apoptosis152. Hence, the effects of HDL-based lipid-lowering drugs on cancer prevention and treatment remain unclear, and further preclinical and clinical trials are needed.
The inverse agonist SR9243 recruits LXR coinhibitors and inhibits LXR activity, thereby inducing cancer cell apoptosis153. In addition, the effects of the LXR agonist LXR623 and inverse agonist SR9243 on CCR have been investigated154. Both drugs were shown to be effective in inhibiting cancer cell proliferation and inducing apoptosis. SR9243 inhibits lipogenesis mainly by inhibiting some key enzymes, such as acetyl-CoA carboxylase, fatty acid synthase and stearoyl coenzyme A desaturase 1. LXR623 significantly reduces intracellular cholesterol levels by promoting cholesterol efflux and limiting cholesterol uptake. In addition, the LXR agonist T0901317 significantly increased ABCA1 expression in human lung epithelial cell lines155. In general, LXR agonists have shown clear results in the treatment of various cancers mainly by inhibiting cancer cell proliferation and inducing apoptosis; however, they can also modulate immune cell activity. RGX-104, an LXR agonist, effectively inhibited the growth of various mouse and human tumors and inhibited myeloid-derived suppressor cell (MDSC) activity by upregulating APOE, which is an LRX target gene, subsequently increasing T-cell activation156. Importantly, this LRX activity was further confirmed in a Phase I clinical trial in which an LXR agonist increased T-cell activation.
Targeting cholesterol esterification in cancer treatment
Avasimibe, an ACAT-1 inhibitor, suppresses cholesterol esterification, increases cellular FC levels and represses tumor metastasis, and the safety of avasimibe has been clinically verified in Phase III clinical trials157. In tumor cells, excessive FC can cause ER stress, which can disrupt metabolic homeostasis and lead to apoptosis. Studies have shown that avasimibe-induced inhibition of tumor cell cholesterol esterification effectively inhibited the proliferation, metastasis and invasion of pancreatic and prostate cancer cells158. In addition, avasimibe treatment effectively promoted CD8+ T-cell function and increased antitumor effects159. Cells treated with avasimibe efficiently killed CD19-expressing leukemic cells and promote the secretion of more INF-γ to regulate T-cell cholesterol homeostasis and enhance antitumor effects160. Therefore, avasimibe is expected to become an antitumor drug candidate. Studies on the treatment of chronic myelogenous leukemia (CML) have revealed that resistance to imatinib develops after long-term use and that inhibition of BCR-ABL with imatinib or MAPK/cholesterol esterification via avasimibe treatment alone led to insufficient effects. However, combination therapy significantly slowed tumor growth. Specifically, the combination of avasimibe and imatinib synergistically inhibited BCR-ABL mutation- and nonmutation-dependent imatinib CML cell proliferation by targeting cancer-specific CE accumulation, MAPK and natural BCR-ABL signaling161. This drug combination, combining relatively nontoxic metabolic inhibitors with existing therapies to overcome cancer cell resistance, is clinically relevant.
Conclusions
The diagnostic and therapeutic potential of targeting cholesterol metabolism for cancer has recently gained considerable attention. Numerous studies have shown that cholesterol metabolism plays a clear role in tumorigenesis, development, and metastasis; however, many questions remain to be answered. For example, which comes first: cholesterol metabolism disorder or tumorigenesis? Solid evidence for an association between cholesterol and tumorigenesis is lacking. Cholesterol homeostasis is regulated by complex feedback loops, and these mechanisms in tumors remain to be explored. Because of the complexity of the cholesterol metabolic network, inhibition of a single pathway in cholesterol metabolism may exert little therapeutic effect on cancer. Therefore, inhibiting cholesterol metabolism pathways simultaneously or in combination with other signaling pathways is likely to substantially improve cancer treatment. An increasing number of studies have indicated that the crosstalk between factors involved in cholesterol metabolism and TME molecules plays a critical role in tumor growth. Moreover, strategies to develop antitumor inhibitors that regulate the TME and cholesterol level are appealing study topics. Overall, cholesterol metabolism and cancer is an important topic for future anticancer research.
References
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Garcia-Bermudez, J. et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122 (2019).
Cai, D. et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 10, 4621 (2019).
Gu, Q. et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 363, 1085–1088 (2019).
Yue, S. et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 (2014).
Riscal, R., Skuli, N. & Simon, M. C. Even cancer cells watch their cholesterol. Mol. Cell 76, 220–231 (2019).
Priem, B. et al. Trained immunity-promoting nanobiologic therapy suppresses tumor growth and potentiates checkpoint inhibition. Cell 183, 786–801.e719 (2020).
Liu, W. et al. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat. Commun. 12, 5103 (2021).
Wang, B. et al. Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220.e204 (2018).
Zhang, Y. et al. Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle 18, 3337–3350 (2019).
Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138 (2008).
DeBose-Boyd, R. A. & Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 43, 358–368 (2018).
Yokoyama, C. et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 (1993).
Hua, X. et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc. Natl Acad. Sci. USA 90, 11603–11607 (1993).
Lee, S. H., Lee, J. H. & Im, S. S. The cellular function of SCAP in metabolic signaling. Exp. Mol. Med. 52, 724–729 (2020).
Luo, J., Yang, H. & Song, B. L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 21, 225–245 (2020).
Brown, M. S. & Goldstein, J. L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 (2002).
Meng, Y., Heybrock, S., Neculai, D. & Saftig, P. Cholesterol handling in lysosomes and beyond. Trends Cell Biol. 30, 452–466 (2020).
Meyer, J. M., Graf, G. A. & van der Westhuyzen, D. R. New developments in selective cholesteryl ester uptake. Curr. Opin. Lipido. 24, 386–392 (2013).
Papale, G. A., Hanson, P. J. & Sahoo, D. Extracellular disulfide bonds support scavenger receptor class B type I-mediated cholesterol transport. Biochemistry 50, 6245–6254 (2011).
Guo, L., Chen, M., Song, Z., Daugherty, A. & Li, X. A. C323 of SR-BI is required for SR-BI-mediated HDL binding and cholesteryl ester uptake. J. Lipid Res. 52, 2272–2278 (2011).
Rigotti, A. et al. A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc. Natl Acad. Sci. USA 94, 12610–12615 (1997).
Viñals, M., Xu, S., Vasile, E. & Krieger, M. Identification of the N-linked glycosylation sites on the high density lipoprotein (HDL) receptor SR-BI and assessment of their effects on HDL binding and selective lipid uptake. J. Biol. Chem. 278, 5325–5332 (2003).
Sharma, B. & Agnihotri, N. Role of cholesterol homeostasis and its efflux pathways in cancer progression. J. Steroid Biochem. Mol. Biol. 191, 105377 (2019).
Chang, T. Y., Chang, C. C. & Cheng, D. Acyl-coenzyme A:cholesterol acyltransferase. Annu. Rev. Biochem 66, 613–638 (1997).
Chang, T. Y., Chang, C. C., Ohgami, N. & Yamauchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol. 22, 129–157 (2006).
Xu, D. et al. The gluconeogenic enzyme PCK1 phosphorylates INSIG1/2 for lipogenesis. Nature 580, 530–535 (2020).
Vallejo, F. A. et al. The contribution of ketone bodies to glycolytic inhibition for the treatment of adult and pediatric glioblastoma. J. Neurooncol. 147, 317–326 (2020).
van Reyk, D. M., Brown, A. J., Hult’en, L. M., Dean, R. T. & Jessup, W. Oxysterols in biological systems: sources, metabolism and pathophysiological relevance. Redox Rep. 11, 255–262 (2006).
Vejux, A. & Lizard, G. Cytotoxic effects of oxysterols associated with human diseases: Induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Mol. Asp. Med. 30, 153–170 (2009).
Norlin, M. & Wikvall, K. Enzymes in the conversion of cholesterol into bile acids. Curr. Mol. Med. 7, 199–218 (2007).
Yoshida, Y., Umeno, A. & Shichiri, M. Lipid peroxidation biomarkers for evaluating oxidative stress and assessing antioxidant capacity in vivo. J. Clin. Biochem. Nutr. 52, 9–16 (2013).
Brown, M. S., Radhakrishnan, A. & Goldstein, J. L. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev. Biochem. 87, 783–807 (2018).
Kaneko, R. et al. Survivin down-regulation plays a crucial role in 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor-induced apoptosis in cancer. J. Biol. Chem. 282, 19273–19281 (2007).
Radhakrishnan, A., Ikeda, Y., Kwon, H. J., Brown, M. S. & Goldstein, J. L. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl Acad. Sci. USA 104, 6511–6518 (2007).
Horton, J. D. et al. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl Acad. Sci. USA 100, 12027–12032 (2003).
Luu, W., Sharpe, L. J., Stevenson, J. & Brown, A. J. Akt acutely activates the cholesterogenic transcription factor SREBP-2. Biochim. Biophys. Acta 1823, 458–464 (2012).
Eid, W. et al. mTORC1 activates SREBP-2 by suppressing cholesterol trafficking to lysosomes in mammalian cells. Proc. Natl Acad. Sci. USA 114, 7999–8004 (2017).
Jiang, Z. et al. Hepatic deficiency of Poldip2 in type 2 diabetes dampens lipid and glucose homeostasis. Metabolism 99, 90–101 (2019).
Moon, S. H. et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell 176, 564–580.e519 (2019).
Parrales, A. et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 18, 1233–1243 (2016).
Kaymak, I. et al. Mevalonate pathway provides ubiquinone to maintain pyrimidine synthesis and survival in p53-deficient cancer cells exposed to metabolic stress. Cancer Res. 80, 189–203 (2020).
Hashimoto, M. et al. Cyp3a deficiency enhances androgen receptor activity and cholesterol synthesis in the mouse prostate. J. Steroid Biochem. Mol. Biol. 163, 121–128 (2016).
Deng, Y. Z. et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J. Exp. Med. 215, 177–195 (2018).
Zhao, S. et al. MIEF2 reprograms lipid metabolism to drive progression of ovarian cancer through ROS/AKT/mTOR signaling pathway. Cell Death Dis. 12, 18 (2021).
Wang, B. & Tontonoz, P. Liver X receptors in lipid signalling and membrane homeostasis. Nat. Rev. Endocrinol. 14, 452–463 (2018).
Bakiri, L. et al. Liver carcinogenesis by FOS-dependent inflammation and cholesterol dysregulation. J. Exp. Med 214, 1387–1409 (2017).
Ma, L. & Nelson, E. R. Oxysterols and nuclear receptors. Mol. Cell Endocrinol. 484, 42–51 (2019).
Janowski, B. A. et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc. Natl Acad. Sci. USA 96, 266–271 (1999).
Mutemberezi, V., Guillemot-Legris, O. & Muccioli, G. G. Oxysterols: from cholesterol metabolites to key mediators. Prog. Lipid Res 64, 152–169 (2016).
Santori, F. R. et al. Identification of natural RORγ ligands that regulate the development of lymphoid cells. Cell Metab. 21, 286–298 (2015).
Voisin, M. et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc. Natl Acad. Sci. USA 114, E9346–e9355 (2017).
Huang, B., Song, B. L. & Xu, C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat. Metab. 2, 132–141 (2020).
Nelson, E. R. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098 (2013).
Gibson, D. A., Collins, F., Cousins, F. L., Esnal Zufiaurre, A. & Saunders, P. T. K. The impact of 27-hydroxycholesterol on endometrial cancer proliferation. Endocr. Relat. Cancer 25, 381–391 (2018).
Wang, S., Yao, Y., Rao, C., Zheng, G. & Chen, W. 25-HC decreases the sensitivity of human gastric cancer cells to 5-fluorouracil and promotes cells invasion via the TLR2/NF-κB signaling pathway. Int. J. Oncol. 54, 966–980 (2019).
Eibinger, G. et al. On the role of 25-hydroxycholesterol synthesis by glioblastoma cell lines. Implications for chemotactic monocyte recruitment. Exp. Cell Res. 319, 1828–1838 (2013).
Jo, Y., Lee, P. C., Sguigna, P. V. & DeBose-Boyd, R. A. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl Acad. Sci. USA 108, 20503–20508 (2011).
Menzies, S. A. et al. The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. Elife 7, https://doi.org/10.7554/eLife.40009 (2018).
Tsai, Y. C. et al. Differential regulation of HMG-CoA reductase and Insig-1 by enzymes of the ubiquitin-proteasome system. Mol. Biol. Cell 23, 4484–4494 (2012).
Zelcer, N. et al. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell Biol. 34, 1262–1270 (2014).
Chen, L. et al. Endogenous sterol intermediates of the mevalonate pathway regulate HMGCR degradation and SREBP-2 processing. J. Lipid Res. 60, 1765–1775 (2019).
Song, B. L., Javitt, N. B. & DeBose-Boyd, R. A. Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 1, 179–189 (2005).
Song, B. L. & DeBose-Boyd, R. A. Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J. Biol. Chem. 279, 28798–28806 (2004).
Sharpe, L. J., Coates, H. W. & Brown, A. J. Post-translational control of the long and winding road to cholesterol. J. Biol. Chem. 295, 17549–17559 (2020).
Soto-Acosta, R., Bautista-Carbajal, P., Cervantes-Salazar, M., Angel-Ambrocio, A. H. & Del Angel, R. M. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: a potential antiviral target. PLoS Pathog. 13, e1006257 (2017).
Kondo, A. et al. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 18, 2228–2242 (2017).
Wang, M. & Kaufman, R. J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 14, 581–597 (2014).
Dang, E. V., McDonald, J. G., Russell, D. W. & Cyster, J. G. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071.e1011 (2017).
Zhang, K. L. et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics 11, 6560–6572 (2021).
Wagschal, A. et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 21, 1290–1297 (2015).
Cheng, L. et al. MicroRNA-148a deficiency promotes hepatic lipid metabolism and hepatocarcinogenesis in mice. Cell Death Dis. 8, e2916 (2017).
Singaravelu, R. et al. MicroRNAs regulate the immunometabolic response to viral infection in the liver. Nat. Chem. Biol. 11, 988–993 (2015).
Wang, L. et al. MicroRNAs 185, 96, and 223 repress selective high-density lipoprotein cholesterol uptake through posttranscriptional inhibition. Mol. Cell Biol. 33, 1956–1964 (2013).
Wang, M. et al. Obesity-induced overexpression of miRNA-24 regulates cholesterol uptake and lipid metabolism by targeting SR-B1. Gene 668, 196–203 (2018).
Fernández-Hernando, C. & Moore, K. J. MicroRNA modulation of cholesterol homeostasis. Arterioscler Thromb. Vasc. Biol. 31, 2378–2382 (2011).
Zhao, L. et al. miR-33-5p knockdown attenuates abdominal aortic aneurysm progression via promoting target adenosine triphosphate-binding cassette transporter A1 expression and activating the PI3K/Akt signaling pathway. Perfusion 35, 57–65 (2020).
Tang, X. E. et al. IL-8 negatively regulates ABCA1 expression and cholesterol efflux via upregulating miR-183 in THP-1 macrophage-derived foam cells. Cytokine 122, 154385 (2019).
Bi, D. P., Yin, C. H., Zhang, X. Y., Yang, N. N. & Xu, J. Y. MiR-183 functions as an oncogene by targeting ABCA1 in colon cancer. Oncol. Rep. 35, 2873–2879 (2016).
Liang, B. et al. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 929–938 (2017).
Lv, Y. C. et al. MicroRNA-19b promotes macrophage cholesterol accumulation and aortic atherosclerosis by targeting ATP-binding cassette transporter A1. Atherosclerosis 236, 215–226 (2014).
Ramirez, C. M. et al. MicroRNA-758 regulates cholesterol efflux through posttranscriptional repression of ATP-binding cassette transporter A1. Arterioscler Thromb. Vasc. Biol. 31, 2707–2714 (2011).
Sun, D. et al. MiR-26 controls LXR-dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. 586, 1472–1479 (2012).
Wu, Q. et al. Methylation of miR-129-5p CpG island modulates multi-drug resistance in gastric cancer by targeting ABC transporters. Oncotarget 5, 11552–11563 (2014).
Huang, J., Chen, S., Cai, D., Bian, D. & Wang, F. Long noncoding RNA lncARSR promotes hepatic cholesterol biosynthesis via modulating Akt/SREBP-2/HMGCR pathway. Life Sci. 203, 48–53 (2018).
Cui, M. et al. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res. 75, 846–857 (2015).
Gast, M. et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc. Res. 115, 1886–1906 (2019).
Reichert, S., Seitter, L., Schaller, H. G., Schlitt, A. & Schulz, S. ANRIL polymorphisms (rs1333049 and rs3217992) in relation to plasma CRP levels among in-patients with CHD. Cytokine 127, 154932 (2020).
Liu, L., Tan, L., Yao, J. & Yang, L. Long non‑coding RNA MALAT1 regulates cholesterol accumulation in ox‑LDL‑induced macrophages via the microRNA‑17‑5p/ABCA1 axis. Mol. Med. Rep. 21, 1761–1770 (2020).
Liu, X. et al. Inhibition of PCSK9 potentiates immune checkpoint therapy for cancer. Nature 588, 693–698 (2020).
Kopecka, J., Godel, M. & Riganti, C. Cholesterol metabolism: at the cross road between cancer cells and immune environment. Int. J. Biochem. Cell Biol. 129, 105876 (2020).
Villablanca, E. J. et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 16, 98–105 (2010).
Baek, A. E. et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 8, 864 (2017).
Goossens, P. et al. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 29, 1376–1389.e1374 (2019).
Bhattacharya, A., Chowdhury, A., Chaudhury, K. & Shukla, P. C. Proprotein convertase subtilisin/kexin type 9 (PCSK9): a potential multifaceted player in cancer. Biochim. Biophys. Acta Rev. Cancer 1876, 188581 (2021).
Stoekenbroek, R. M., Lambert, G., Cariou, B. & Hovingh, G. K. Inhibiting PCSK9 - biology beyond LDL control. Nat. Rev. Endocrinol. 15, 52–62 (2018).
Yuan, J. et al. Potentiating CD8(+) T cell antitumor activity by inhibiting PCSK9 to promote LDLR-mediated TCR recycling and signaling. Protein Cell 12, 240–260 (2021).
Yang, W. et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016).
Ma, X. et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. 30, 143–156.e145 (2019).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Yang, Z. et al. Cancer cell-intrinsic XBP1 drives immunosuppressive reprogramming of intratumoral myeloid cells by promoting cholesterol production. Cell Metab. https://doi.org/10.1016/j.cmet.2022.10.010 (2022).
Lu, Z. et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. 34, 1342–1358.e1347 (2022).
Qin, W. H. et al. High serum levels of cholesterol increase antitumor functions of nature killer cells and reduce growth of liver tumors in mice. Gastroenterology 158, 1713–1727 (2020).
O’Brien, K. L. et al. De novo polyamine synthesis supports metabolic and functional responses in activated murine NK cells. Eur. J. Immunol. 51, 91–102 (2021).
Forcina, G. C. & Dixon, S. J. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19, e1800311 (2019).
Warner, G. J. et al. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J. Biol. Chem. 275, 28110–28119 (2000).
Bersuker, K. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
Shimada, K. et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016).
Hurst, R. et al. Hyperresistance to cholesterol hydroperoxide-induced peroxidative injury and apoptotic death in a tumor cell line that overexpresses glutathione peroxidase isotype-4. Free Radic. Biol. Med. 31, 1051–1065 (2001).
Ng, M. Y. W. et al. The cholesterol transport protein GRAMD1C regulates autophagy initiation and mitochondrial bioenergetics. Nat. Commun. 13, 6283 (2022).
Snaebjornsson, M. T., Janaki-Raman, S. & Schulze, A. Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 31, 62–76 (2020).
Pan, Q. et al. The ZMYND8-regulated mevalonate pathway endows YAP-high intestinal cancer with metabolic vulnerability. Mol. Cell 81, 2736–2751.e2738 (2021).
Li, N. et al. ZMYND8 reads the dual histone mark H3K4me1-H3K14ac to antagonize the expression of metastasis-linked genes. Mol. Cell 63, 470–484 (2016).
Yang, L. et al. Oxidized low-density lipoprotein links hypercholesterolemia and bladder cancer aggressiveness by promoting cancer stemness. Cancer Res. 81, 5720–5732 (2021).
Kim, H. Y. et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 50, 1–12 (2018).
Krüger, K. et al. Lovastatin prevents cisplatin-induced activation of pro-apoptotic DNA damage response (DDR) of renal tubular epithelial cells. Toxicol. Appl. Pharm. 292, 103–114 (2016).
Pääjärvi, G., Roudier, E., Crisby, M., Högberg, J. & Stenius, U. HMG-CoA reductase inhibitors, statins, induce phosphorylation of Mdm2 and attenuate the p53 response to DNA damage. FASEB J. 19, 476–478 (2005).
Xu, H., Zhou, S., Tang, Q., Xia, H. & Bi, F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Cancer 1874, 188394 (2020).
Longo, J., van Leeuwen, J. E., Elbaz, M., Branchard, E. & Penn, L. Z. Statins as anticancer agents in the era of precision medicine. Clin. Cancer Res. 26, 5791–5800 (2020).
Rao, S. et al. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc. Natl Acad. Sci. USA 96, 7797–7802 (1999).
Kotamraju, S., Williams, C. L. & Kalyanaraman, B. Statin-induced breast cancer cell death: role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 67, 7386–7394 (2007).
Qi, X. F. et al. HMG-CoA reductase inhibitors induce apoptosis of lymphoma cells by promoting ROS generation and regulating Akt, Erk and p38 signals via suppression of mevalonate pathway. Cell Death Dis. 4, e518 (2013).
Sahebkar, A., Ponziani, M. C., Goitre, I. & Bo, S. Does statin therapy reduce plasma VEGF levels in humans? A systematic review and meta-analysis of randomized controlled trials. Metabolism 64, 1466–1476 (2015).
Woodard, J., Sassano, A., Hay, N. & Platanias, L. C. Statin-dependent suppression of the Akt/mammalian target of rapamycin signaling cascade and programmed cell death 4 up-regulation in renal cell carcinoma. Clin. Cancer Res. 14, 4640–4649 (2008).
Swanson, K. M. & Hohl, R. J. Anti-cancer therapy: targeting the mevalonate pathway. Curr. Cancer Drug Targets 6, 15–37 (2006).
Prabhu, N. et al. Statin use and risk of prostate cancer biochemical recurrence after radical prostatectomy. Urol. Oncol. 39, 130.e139–130.e115 (2021).
Duncan, R. E., El-Sohemy, A. & Archer, M. C. Statins and the risk of cancer. Jama 295, 2720 (2006). author reply 2721-2722.
Ahern, T. P. et al. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J. Natl Cancer Inst. 103, 1461–1468 (2011).
Manthravadi, S., Shrestha, A. & Madhusudhana, S. Impact of statin use on cancer recurrence and mortality in breast cancer: a systematic review and meta-analysis. Int. J. Cancer 139, 1281–1288 (2016).
Xia, Y. et al. The mevalonate pathway is a druggable target for vaccine adjuvant discovery. Cell 175, 1059–1073.e1021 (2018).
Abdel-Rahman, O. Statin treatment and outcomes of metastatic pancreatic cancer: a pooled analysis of two phase III studies. Clin. Transl. Oncol. 21, 810–816 (2019).
Brånvall, E. et al. Statin use is associated with improved survival in multiple myeloma: a Swedish population-based study of 4315 patients. Am. J. Hematol. 95, 652–661 (2020).
Cardwell, C. R., Hicks, B. M., Hughes, C. & Murray, L. J. Statin use after colorectal cancer diagnosis and survival: a population-based cohort study. J. Clin. Oncol. 32, 3177–3183 (2014).
Lee, Y. et al. Randomized phase II study of afatinib plus simvastatin versus afatinib alone in previously treated patients with advanced nonadenocarcinomatous non-small cell lung cancer. Cancer Res Treat. 49, 1001–1011 (2017).
Garwood, E. R. et al. Fluvastatin reduces proliferation and increases apoptosis in women with high grade breast cancer. Breast Cancer Res. Treat. 119, 137–144 (2010).
Pandyra, A. et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 74, 4772–4782 (2014).
Gaist, D., Hallas, J., Friis, S., Hansen, S. & Sørensen, H. T. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol. 38, 722–727 (2014).
Han, J. Y. et al. A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 17, 1553–1560 (2011).
Göbel, A., Rauner, M., Hofbauer, L. C. & Rachner, T. D. Cholesterol and beyond—the role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta Rev. Cancer 1873, 188351 (2020).
Van Acker, H. H., Anguille, S., Willemen, Y., Smits, E. L. & Van Tendeloo, V. F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharm. Ther. 158, 24–40 (2016).
Gralow, J. R. et al. Phase III randomized trial of bisphosphonates as adjuvant therapy in breast cancer: S0307. J. Natl Cancer Inst. 112, 698–707 (2020).
Maione, F. et al. The cholesterol biosynthesis enzyme oxidosqualene cyclase is a new target to impair tumour angiogenesis and metastasis dissemination. Sci. Rep. 5, 9054 (2015).
Li, C. et al. Squalene epoxidase drives cancer cell proliferation and promotes gut dysbiosis to accelerate colorectal carcinogenesis. Gut 71, 2253–2265 (2022).
Trinh, M. N. et al. Triazoles inhibit cholesterol export from lysosomes by binding to NPC1. Proc. Natl Acad. Sci. USA 114, 89–94 (2017).
Lu, F. et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife 4, https://doi.org/10.7554/eLife.12177 (2015).
Rossebø, A. B. et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N. Engl. J. Med. 359, 1343–1356 (2008).
Lincoff, A. M. et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 376, 1933–1942 (2017).
Schwartz, G. G. et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 367, 2089–2099 (2012).
Barter, P. J. et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 357, 2109–2122 (2007).
Kamanna, V. S. & Kashyap, M. L. Mechanism of action of niacin. Am. J. Cardiol. 101, 20b–26b (2008).
Yang, S. et al. Biomimetic, synthetic HDL nanostructures for lymphoma. Proc. Natl Acad. Sci. USA 110, 2511–2516 (2013).
Flaveny, C. A. et al. Broad anti-tumor activity of a small molecule that selectively targets the warburg effect and lipogenesis. Cancer Cell 28, 42–56 (2015).
Wu, G. et al. Targeting the transcription factor receptor LXR to treat clear cell renal cell carcinoma: agonist or inverse agonist. Cell Death Dis. 10, 416 (2019).
He, P., Smith, A., Gelissen, I. C. & Ammit, A. J. The effect of statins and the synthetic LXR agonist T0901317 on expression of ABCA1 transporter protein in human lung epithelial cell lines in vitro. Pharm. Rep. 71, 1219–1226 (2019).
Liang, H. & Shen, X. LXR activation radiosensitizes non-small cell lung cancer by restricting myeloid-derived suppressor cells. Biochem. Biophys. Res. Commun. 528, 330–335 (2020).
Lei, J., Wang, H., Zhu, D., Wan, Y. & Yin, L. Combined effects of avasimibe immunotherapy, doxorubicin chemotherapy, and metal-organic frameworks nanoparticles on breast cancer. J. Cell Physiol. 235, 4814–4823 (2020).
Li, J. et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 35, 6378–6388 (2016).
Pan, J. et al. Potentiation of Kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine 49, 72–81 (2019).
Zhao, L. et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J. Immunother. 41, 45–52 (2018).
Bandyopadhyay, S. et al. Cholesterol esterification inhibition and imatinib treatment synergistically inhibit growth of BCR-ABL mutation-independent resistant chronic myelogenous leukemia. PLoS ONE 12, e0179558 (2017).
Acknowledgements
We thank American Journal Experts (AJE) for their assistance with language editing.
Funding
This study was jointly supported by the National Natural Science Foundation of China (81802352, 81902428, 82002541, and U21A20374), Shanghai Rising-Star Program (No. 20QA1402100), Shanghai Municipal Science and Technology Major Project (21JC1401500), Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1006A), Xuhui District Artificial Intelligence Medical Hospital Cooperation Project (2021-011), and the Shanghai Sailing Program (No. 19YF1409400 and 20YF1409000).
Author information
Authors and Affiliations
Contributions
M.M.X., J.X., and W.W. collected the related studies and drafted the manuscript. B.Z., J.L., J.L.L., H.X., and Y.J.Z. participated in the design of the review. S.S. and X.J.Y. initiated the study and revised the manuscript. All authors have read and agreed on the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, M., Xu, J., Wang, W. et al. Functional significance of cholesterol metabolism in cancer: from threat to treatment. Exp Mol Med 55, 1982–1995 (2023). https://doi.org/10.1038/s12276-023-01079-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01079-w
