
Abstract
Interlayer excitons in layered materials constitute a novel platform to study many-body phenomena arising from long-range interactions between quantum particles. Long-lived excitons are required to achieve high particle densities, to mediate thermalisation, and to allow for spatially and temporally correlated phases. Additionally, the ability to confine them in periodic arrays is key to building a solid-state analogue to atoms in optical lattices. Here, we demonstrate interlayer excitons with lifetime approaching 0.2 ms in a layered-material heterostructure made from WS2 and WSe2 monolayers. We show that interlayer excitons can be localised in an array using a nano-patterned substrate. These confined excitons exhibit microsecond-lifetime, enhanced emission rate, and optical selection rules inherited from the host material. The combination of a permanent dipole, deterministic spatial confinement and long lifetime places interlayer excitons in a regime that satisfies one of the requirements for simulating quantum Ising models in optically resolvable lattices.
Similar content being viewed by others


Introduction
Since the demonstration of Bose–Einstein condensation1,2, ultracold atoms have played a central role in the study and realisation of macroscopic-scale quantum phenomena, including Mott transition3, superfluidity4 and many-body localisation5. Dipolar particles have gained attention as they introduce long-range anisotropic interactions to the exploration of new states of quantum matter6,7,8. Atomic realisations of dipolar ensembles include Rydberg atoms9,10,11, high magnetic-moment atoms12,13,14,15,16 and ultracold polar molecules17,18,19. Exciton-polaritons in semiconductors are an analogous platform to atoms20,21,22,23,24,25,26. For semiconductors, long-range interactions can be achieved via spatially indirect excitons, where electrons and holes have finite separation27,28 yielding a permanent electric dipole moment29,30,31. Most progress in realising such states has been in AlGaAs/GaAs double quantum wells30,32,33,34,35, while layered materials heterostructures of transition metal dichalcogenide (TMD) monolayers36 are a promising alternative37,38,39,40. TMD monolayers are semiconductors offering optical access to orbital, spin and valley degrees of freedom41. The difference in band energies of two TMD monolayers can be exploited to create type-II interlayer excitons42,43 with static electric dipole. Moreover, their optical transition strength, energy, and selection rules can be engineered by controlling the layer separation44 and their relative stacking angle45,46,47,48,49,50.
The potential of TMD heterostructure devices to explore many-body effects is evidenced by reports of excitonic condensation51 and Mott–Hubbard physics with charge carriers in moiré superlattices52,53,54. The periodicity of these superlattices, typically up to a ~10-nm characteristic length scale, is determined by the interlayer lattice constant mismatch and stacking angle mismatch. Confining interlayer excitons in an arbitrary potential energy landscape independent of the TMD lattice opens up the prospect of exploiting the long-range nature of their dipole–dipole interactions, as well as their optical accessibility to explore many-body physics, such as quantum spin Ising models. Such a system could be viewed as a solid-state analogue to quantum simulators using Rydberg atoms55 and molecules56 in optical lattices or tweezers arrays. At the cost of reduced coherence time and site-to-site homogeneity in a solid-state device, they offer larger arrays and shorter refresh times.
For long-range interactions to emerge, excitons do not have to be indistinguishable, but their decay rate must be lower than the interaction rate between neighbouring lattice sites56. In an array with single-site optical addressability, the confining traps must be spaced by an optically resolvable distance ~0.5 μm. At this spacing, interlayer excitons, with an electric dipole of ~0.6 nm ⋅ qe (~29 Debye)40, have a dipole–dipole coupling rate of 1 MHz. This places a lifetime requirement of at least 1 μs on excitons. Here, we show the deterministic confinement of interlayer excitons with lifetimes exceeding 1 μs. This opens up the prospect of implementing confined interlayer excitons in quantum simulation applications.
Results and discussion
Optical characterisation of the device
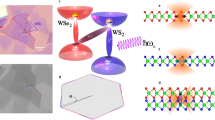
Figure 1a is an illustration of our devices, fabricated by exfoliating WSe2 and WS2. With the aim of creating long-lived excitons, we limit non-radiative decay57, which often dominates exciton loss51,58, by working with TMDs that have low point-defect density of 109–1010 cm−2 as estimated from X-Ray Diffraction (XRD) and Scanning Transmission Electron Microscopy (STEM) measurements (see Supplementary Note S1 and Supplementary Figs. S1 and S2). We transfer WSe2 and WS2 monolayers onto a SiO2/Si substrate patterned with an array of nanopillars 220–250 nm tall and 4 μm apart (see Methods and Supplementary Note S2). For the first device (device A), we choose an interlayer stacking angle of ~50°, away from high-symmetry angles of 0° or 60°, in order to introduce an interlayer momentum mismatch that reduces the direct optical exciton recombination rate59. Five distinct locations are labelled in Fig. 1a (L1–L5). These are marked by circles in the optical image of device A shown in Fig. 1b, where the WSe2 and WS2 monolayers are outlined in red and green, respectively, yielding a large region (>160 μm2) of WS2/WSe2 heterostructure.

a Illustration of our devices. The SiO2 substrate with nanopillars is in blue, the WSe2 monolayer in red and the WS2 monolayer on top in green. Representative locations on the device are indicated: location L1 is the WS2 monolayer, location L2 is the WSe2 monolayer, location L3 is the flat WS2/WSe2 heterostructure, location L4 is the WSe2 monolayer on a nanopillar and location L5 is the WS2/WSe2 heterostructure on a nanopillar. b Optical image of device A. WSe2 and WS2 monolayers are outlined in red and green, respectively. Scale bar: 4 μm. c Integrated photoluminescence (PL) intensity map of device A under 2.33-eV continuous-wave (CW) excitation at 4 K. Scale bar: 4 μm. d Representative PL spectra under 2.33 eV CW excitation taken at the five locations highlighted in b. The colour coding indicates the origin of PL emission, where green (red) comes from intralayer excitons in WS2 monolayer (WSe2 monolayer), and yellow from interlayer excitons in the WS2/WSe2 heterostructure. In subpanel (i), the PL between 1.3 and 1.5 eV originates from interlayer exciton emission leaking into the detection spot. The multiplicative factors of each PL spectrum are relative to subpanel (iv), WSe2 monolayer spectrum at L4. e PL intensity map under 1.50 eV CW excitation at 4 K, to which only WS2/WSe2 heterostructure regions contribute. Localised PL enhancement and spectrally sharp emission peaks are also seen in device B, for which we choose a smaller stacking angle <7° with respect to high-symmetry alignments (0° or 60°). Scale bar: 4 μm.
Figure 1c shows a photoluminescence (PL) map taken with continuous-wave (CW) 2.33-eV (532-nm) laser excitation at a temperature T = 4 K. This energy is greater than both WSe2 and WS2 monolayer optical bandgaps of 1.7360 and 2.08 eV61, respectively. Consequently, PL emission is observed from both WSe2 and WS2 monolayers and WS2/WSe2 heterostructure regions. The spectral emission from the flat WSe2 and WS2 monolayer regions (L1 and L2 in Fig. 1b) is shown in Figs. 1d (i) and (ii), respectively. The PL emission between 1.65 and 2.0 eV in panel (i) (filled in green) is the intralayer exciton recombination in monolayer WS261,62, and that between 1.6 and 1.75 eV in panel (ii) (filled in red) is the intralayer exciton recombination in monolayer WSe260,63. Figure 1d panel (iii) shows the spectrum from the flat part of the heterostructure, L3 in Fig. 1b. A lower-energy feature at 1.4 eV (filled in yellow) is found only in the heterostructure region and is consistent with interlayer exciton emission from stacked heterostructures reported previously50,64. Figure 1d (iv) is the PL spectrum from a monolayer WSe2 on a nanopillar, displaying a ~50-fold brighter emission with respect to flat monolayer WSe2 and a sub-meV full-width at half-maximum emission peak. Given that monolayer WSe2 on such nanopillars results in the strong quantum confinement of intralayer excitons62,65, we look for similar signatures in the interlayer emission spectra. Figure 1d (v) is the PL spectrum of the heterostructure on a nanopillar. We observe a 20-fold brighter interlayer emission in the low optical excitation regime (<1 μW), and sharp spectral features (~2 meV or less full-width at half-maximum) between 1.35 and 1.50 eV on eight nanopillar locations, in contrast to the weak and spectrally broad emission from the flat heterostructure. These remain after a full thermal cycle with only minor spectral variations (see Supplementary Note S10 and Supplementary Figs. S17–S19).
To isolate interlayer excitations from any effects coming from the monolayers, such as free charge carriers66 or intralayer excitons67, we use an optical excitation energy of 1.50 eV for all remaining measurements. This energy is at the onset of the interlayer exciton PL spectrum, Fig. 1d (iii), and below the optical bandgaps of WSe2 and WS2 monolayers. Figure 1e displays the PL intensity map under 1.50 eV excitation. The emission is only observed from the heterostructure region with both monolayer regions remaining dark, demonstrating that only interlayer excitons are generated.
Confined and delocalised interlayer excitons
Figure 2a shows the interlayer exciton PL spectra from the flat WS2/WSe2 heterostructure for 0.05-μW (black curve), 1-μW (red curve) and 100-μW (blue curve) laser excitation power, P, at 4 K. The spectrally integrated PL intensity of the interlayer exciton with respect to P is shown in Fig. 2b. While the interlayer exciton emission starts with a linear dependence on P, it converges to a P0.3 scaling for P > 0.3 μW. Sublinear behaviour was also previously reported for MoSe2/WSe2 heterostructures36,68. This can be caused by density-dependent mechanisms, such as exciton–exciton annihilation, dipolar repulsion and phase-space filling. The interlayer exciton energy also undergoes a blue-shift with increasing P (see Supplementary Fig. S5 for device A and Supplementary Fig. S15 for device B), which is typically attributed to dipolar interactions36,68,69.

a Photoluminescence (PL) spectra at 4 K from the flat WS2/WSe2 heterostructure under 1.50 eV excitation at 0.05 μW (black curve), 1 μW (red curve) and 100 μW (blue curve). b Spectrally integrated intensity of interlayer exciton PL as a function of excitation power, P. The dashed grey curve follows a linear dependence in P, while the solid grey curve follows P0.3. Sub-linear PL intensity is also seen in device B, see Supplementary Fig. S15. The error bars are smaller than the data points. c PL spectra from the WS2/WSe2 heterostructure on a nanopillar at 0.05 μW (black curve), 1 μW (red curve) and 100 μW (blue curve). d Spectrally integrated PL intensity of the three emission peaks shaded with green, grey and blue bands in c as a function of P. Data (filled squares) are colour-coded to the spectral bands of c. Solid curves are fits to data using the saturation function A × Pn/(Psat + Pn), from where we determine n = 0.94 ± 0.12, 1.06 ± 0.08, 1.04 ± 0.06 and Psat = 0.1, 0.6, 0.9 μW for the green, grey and blue data, respectively. The error bars are smaller than the data points. For the saturation curves of sharp emission peaks on device B see Supplementary Fig. S16.
Figure 2c presents PL spectra from the heterostructure on a nanopillar for P = 0.05 μW (black curve), 1 μW (red curve) and 100 μW (blue curve). For the three emission peaks highlighted in green, grey and blue we plot in Fig. 2d the integrated PL intensity as a function of P. For the analysis of emission peaks at other nanopillar locations in devices A and B, see Supplementary Figs. S7 and S16, respectively. Each emission peak shows a linear P-dependence, followed by saturation, consistent with previous reports of interlayer exciton localisation68,70. This behaviour is characteristic of a quantum-confined system71,72, and contrasts that of delocalised interlayer excitons in the flat heterostructure (Fig. 2a, b). The fact that these emission peaks saturate at different powers (see Supplementary Note S4) indicates multiple confining potentials at the nanopillar, possibly because the heterostructure does not conform smoothly to the nanopillar (see Supplementary Fig. S4a)62. The emission peaks are spectrally stable within 1 meV over a minute timescale (see Supplementary Note S5) and unchanged after repeated excitation power-dependent measurements (see Supplementary Fig. S9). In our time series, some emission peaks show intensity anticorrelation (see Supplementary Fig. S10), often referred to as spectral jumps, which is another signature of quantum-confined systems.
Optical selection rules of confined interlayer excitons
Figure 3a and b presents confined interlayer exciton PL spectra above a nanopillar (L5 of Fig. 1b) at 4 K, for out-of-plane magnetic field B from 0 to 9 T. The excitation is linearly polarised and the collection is right-hand σ+ (left-hand σ−) circularly polarised in panel a (panel b). The red-shifting Zeeman state from each emission peak is only seen for σ+ detection, while the blue-shifting Zeeman state is only seen for σ− detection. This demonstrates that the two magnetic configurations of confined interlayer excitons sustain optical selection rules with an in-plane transition dipole moment73.

a Photoluminescence (PL) spectrum at 4 K as a function of magnetic field B on a nanopillar under linearly polarised excitation and circularly polarised σ+ detection. b Same measurement as a, but with σ− detection. The intensity of the components in a increases with B, and the blue-shifting components in b decrease with B, consistent with exciton thermalisation of Zeeman states at 4 K63. c Measured Zeeman splittings of confined interlayer excitons as a function of B. The red (blue) circles correspond to confined interlayer excitons with the largest (smallest) g-factor of 15.4 ± 1.5 (11.8 ± 1.4) extracted from the linear fits (dashed curves). The mean g-factor value of all measured splittings is 13.2 with a standard deviation 1.1. The error bars are smaller than the data points. d PL spectra of confined interlayer excitons collected on the same nanopillar as a and b at 0 T for σ+ excitation and co-polarised (σ+) and cross-polarised (σ−) detection, shown as black and red curves, respectively.
The distribution of Zeeman splitting with respect to B across different nanopillars is given in Fig. 3c as a blue-shaded region, where the data corresponding to the smallest and largest measured g-factor (11.9 and 15.4) are plotted in blue and red circles, respectively. The similarity in g-factors, mean value 13.2 and standard deviation 1.1, across the nanopillar confining traps and for different emission peaks (see Supplementary Fig. S8) suggests that the confined excitons have the same microscopic origin. The variation in g-factors, similar to that observed in confined intralayer excitons in WSe2 monolayers72,74,75,76, is one order of magnitude larger than observed for moiré-confined interlayer excitons49. Our average g-factor agrees with the expected g-factor of 1477,78,79 for electrons and holes residing in opposite valleys, corresponding to the \({K}^{\prime}-K\) transition. This is consistent with our heterostructure alignment close to 60°.
Figure 3d presents the PL spectra under σ+ excitation at B = 0 T for σ+ (black) and σ− (red) circularly polarised collection. The overlap of the two spectra demonstrates that excitation polarisation is not maintained for confined interlayer excitons, and the same is observed in delocalised excitons (see Supplementary Note S3 and Supplementary Fig. S6a). One dominant mechanism for the loss of exciton valley polarisation is the electron–hole exchange interaction80. The polarisation retention diminishes when the exchange interaction rate γEI exceeds the exciton decay rate γd. Relative to intralayer excitons, the increased electron–hole separation of interlayer excitons reduces γEI and γd by the same factor and, consequently, angle-aligned heterostructures exhibit valley polarisation49,64 similar to monolayers63, typically >10%. In our device, the momentum mismatch introduced by our large stacking angle further suppresses γd, while leaving γEI unaffected, resulting in a suppression of valley polarisation.
We also confirm that the g-factor of the delocalised exciton has the same sign as the confined excitons. To do this, we monitor the circular polarisation of the delocalised interlayer excitons under magnetic field (see Supplementary Note S3 and Supplementary Fig. S6b). In a positive (negative) magnetic field the σ+ (σ−) emission becomes dominant irrespective of excitation polarisation, owing to exciton thermalisation to the lower-energy Zeeman state63,81.
Lifetimes of confined and delocalised interlayer excitons
Figure 4 presents lifetime measurements on interlayer excitons under 1.5-eV laser excitation with ~3 ps pulses at 4 K (see Methods). Figure 4a shows an example emission intensity histogram of delocalised interlayer excitons (grey bars) of device A as a function of time after excitation, and an exponential fit (solid red curve) reveals a decay time of 180.6 ± 3.5 μs. Across the flat heterostructure region, the average lifetime is 175 ± 5 μs. The spatial and momentum separation of electrons and holes of interlayer excitons, and the use of low defect density WSe2 and WS2 monolayers, are likely responsible for these values. In device B, which has a smaller twist angle (<7°), the lifetime drops to 10 ± 2 μs for delocalised excitons, highlighting the role of momentum mismatch in exciton recombination (see Supplementary Fig. S15d). The lifetime of delocalised excitons shows a strong excitation dependence: increasing the excitation power reduces the exciton decay time (see Supplementary Note S6 and Supplementary Fig. S11), as expected from density-dependent interactions and loss channels, even in the regime where PL intensity depends linearly on the excitation power (P < 0.3 μW in Fig. 2b).

a Lifetime measurement of interlayer excitons in the flat WS2/WSe2 heterostructure region at 4 K. The data are fitted by a single exponential (solid red curve) with a lifetime τ = 180.6 ± 3.5 μs. b Lifetime measurement of a confined interlayer exciton (1.39 eV emission peak from Fig. 3d). The solid red curve is a biexponential fit with two lifetimes τs = 59.4 ± 0.9 ns and τl = 389.1 ± 1.2 ns. The inset presents a three-level model with coupling between a bright state \(\left|b\right\rangle\) and a shelving state \(\left|s\right\rangle\), as discussed in Supplementary Note S8. c τs (dark grey bars) and τl (light grey bars) values extracted from biexponential fits to the lifetime measurements at the eight nanopillar locations in device A (indexed a–h). Error bars are fitting errors. The average value of τs (τl) across all nanopillars is 80 ns (2 μs). The inset maps the physical location of the nanopillars on the WS2/WSe2 heterostructure region. The reduction of lifetime for confined interlayer excitons as compared to delocalised is also seen in device B, see Supplementary Figs. S15d and S16d.
Figure 4b is a lifetime measurement of the confined interlayer exciton emission peak at 1.39 eV from Fig. 3d (L5 in Fig. 1b). We fit a biexponential function of the form Ase\({}^{-t/{\tau }_{{\rm{s}}}}\) + Ale\({}^{-t/{\tau }_{{\rm{l}}}}\), where τl (τs) is the long (short) lifetime and Al (As) is the amplitude of the exponential with long (short) lifetime. This yields τs = 59.4 ± 0.9 ns and τl = 389.1 ± 1.2 ns. This biexponential behaviour, albeit with varying lifetimes, is observed throughout the entire spectral range for confined interlayer excitons (see Supplementary Note S7 and Supplementary Fig. S12). Figure 4c is a summary of the spectrally integrated lifetimes from the eight nanopillar confining traps. The extracted τs and τl are shown in Fig. 4c in black and grey bars, and lie in the ~10–175 ns and ~0.4–4 μs ranges, respectively. The reduction in interlayer exciton lifetime after confinement accompanies an enhancement in PL brightness (also for device B, see Supplementary Note S9), suggesting a modified oscillator strength under localisation. This likely arises from the relaxation of stacking-angle-induced momentum mismatch of the exciton transition under confinement that otherwise inhibits the recombination of delocalised interlayer excitons82,83.
One source of the observed biexponential decay is the presence of two excited states as shown in the energy level diagram of Fig. 4b: an optically active state, \(\left|b\right\rangle\), and an energetically similar shelving state that is dark, \(\left|s\right\rangle\). Lifetime measurements from 4 to 70 K (see Supplementary Fig. S13a–c) fitted with a three-level model, and diminishing PL intensity with increasing temperature (see Supplementary Fig. S13d), indicate that \(\left|s\right\rangle\) is higher in energy than \(\left|b\right\rangle\) (see Supplementary Note S8). We assume the radiative rate for the bright state, Γr, to be temperature independent and the coupling rates between bright and shelving states, Γbs (bright to shelving), Γsb (shelving to bright), and the non-radiative decay rate Γnr to be phonon mediated, hence temperature dependent. At 4 K we find that 1/τs ≈ Γr and 1/τl ≈ Γnr + Γbs, with average values across device A of 13 MHz and 500 kHz, respectively. The non-radiative loss rate is lower than all other rates for the confined interlayer excitons up to 70 K (see Supplementary Note S8). The rate Γbs increases with temperature and matches Γsb at 40 K, indicating that \(\left|s\right\rangle\) is ~5 meV higher in energy than \(\left|b\right\rangle\), comparable to the calculated ~10–29 meV conduction band splitting at the K-point of WS284 monolayer. A higher shelving state energy is also supported by a diminishing PL intensity with increasing temperature (see Supplementary Fig. S13d). These measurements indicate that the shelved state is the spin-forbidden \({K}^{\prime}-K\) transition.
Conclusions
In summary, we demonstrated deterministic confinement of long-lived interlayer excitons across multiple sites in devices with different stacking angles. The confined interlayer exciton lifetime up to ~4 μs is not limited by non-radiative decay and is sufficient to observe dipole-mediated interactions in an optically resolvable lattice with a spacing of ~0.5 μm. The next step is employing resonant excitation toward transform-limited linewidths85,86,87,88 and coherent optical control of excitons, leading ultimately to the detection of site-to-site interaction-induced spectral shifts. Further work will also focus on developing a full account of the photophysics of these confined interlayer excitons, including the influence of stacking angle, interlayer spacing, and electrostatic gating of the devices. Other confining geometries, such as ridges and rings, may also offer the opportunity to probe many-body phenomena in one dimension.
Methods
Fabrication
TMD bulk crystals were synthesised through flux zone growth89. Precursor powders were sourced from Alfa Aesar. Additional electrolytic purification is implemented to achieve purity ≥99.9999%. After purification, the powders were analysed using secondary ion mass spectroscopy to confirm the absence of metal impurities. Compared to crystals grown by chemical vapour transport, flux growth produces samples with much lower point and topological defects.
Flakes were then prepared by micromechanical cleavage90 on Nitto Denko tape, then exfoliated again for transfer to a polydimethylsiloxane (PDMS) stamp placed on a transparent glass slide, allowing bidirectional inspection of the flakes under an optical microscope. Optical contrast was utilised to identify monolayers prior to transfer91. We obtained monolayer flakes with lateral dimensions ~50 μm.
Substrates with arrays of silica nanopillars, 100 nm in diameter and 220–250 nm high, were prepared by direct-write lithography62. The substrates underwent wet cleaning (1 min ultrasonication in acetone and isopropanol) and were subsequently exposed to an oxygen-assisted plasma at 10 W for 60 s to remove impurities and contaminants from the surface. The WSe2 monolayer flakes were then stamped on the nanopillars with a micro-manipulator62,92. A WS2 monolayer was then deposited on the WSe2 monolayer following the same stamping procedure. In both steps the PDMS stamp was removed after depositing the monolayer. Raman spectroscopy (see Supplementary Fig. S3a), second-harmonic generation (see Supplementary Fig. S3b) and atomic force microscopy (see Supplementary Fig. S4) confirm the monolayer nature of the constituents, the twist angle and the interlayer spacing, respectively.
Photoluminescence measurements
All PL measurements were taken at 4 K in a 9 T closed-cycle cryostat (Attodry 1000 from Attocube Systems AG). Excitation and collection pass through a confocal setup with the device in reflection geometry. We used CW illumination from either a 2.33 eV laser (Ventus 532 from Laser Quantum Ltd.) or a Ti:Sapphire laser (Mira 900 from Coherent Inc.) at 1.58 or 1.50 eV. All spectra were corrected for the spectral transfer function of the setup, measured with a broadband incandescent source. The PL signal was sent to a 150-line grating spectrometer (Princeton Instruments Inc.) or an avalanche photodiode for PL maps (PerkinElmer Inc.).
Lifetime measurements
To perform lifetime measurements, we excite the samples every ~10 μs for the confined interlayer excitons, and up to ~1 ms for interlayer excitons, with ~3 ps pulses from a Ti:Sapphire laser (Mira 900 from Coherent Inc.) tuned to 785 nm (1.58 eV) or 825 nm (1.50 eV). An acousto-optic modulator (AOM) down-samples the 76 MHz laser repetition rate to the kHz–MHz range required to measure up to hundreds μs lifetimes. A time-to-digital converter (quTAU from qutools GmbH) with 81 ps timing resolution collects start-stop histograms with ‘start’ triggered by the AOM pulse-picking and ‘stop’ triggered by an avalanche photodiode (APD) output stemming from the single-photon detection of interlayer exciton PL. The converter remains idle until a subsequent ‘start’ signal arrives. To ensure there is no susceptibility to spurious artifacts from start-stop measurements, we determine the lifetime using photon time-tagging, and calculate the lifetimes in post-processing for a subset of locations. Both methods resulted in consistent lifetimes for confined interlayer excitons. Time-tagging is required for the measurement of delocalised interlayer exciton lifetimes, due to the count rate being comparable to, or even lower than, the APD dark counts, as well as being at least one order of magnitude smaller than the rate of pulsed excitations.
Data availability
The datasets generated and analysed during the current study are available from the corresponding authors upon reasonable request.
Code availability
The computer codes used for data analysis during the current study are available from the corresponding authors upon reasonable request.
References
Anderson, M. H. et al. Observation of Bose-Einstein Condensation in a Dilute Atomic Vapor. Science 269, 198–201 (1995).
Davis, K. B. et al. Bose-Einstein Condensation in a Gas of Sodium Atoms. Phys. Rev. Lett. 75, 3969–3973 (1995).
Greiner, M. et al. Quantum phase transition from a superfluid to a Mott insulator in a gas of ultracold atoms. Nature 415, 39–44 (2002).
Zwierlein, M. W. et al. Vortices and superfluidity in a strongly interacting Fermi gas. Nature 435, 1047–1051 (2005).
Schreiber, M. et al. Observation of many-body localization of interacting fermions in a quasirandom optical lattice. Science 349, 842–845 (2015).
Baranov, M. A. et al. Condensed matter theory of dipolar quantum gases. Chem. Rev. 112, 5012–5061 (2012).
Trefzger, C. et al. Ultracold dipolar gases in optical lattices. J. Phys. B Mol. Opt. Phys. 44, 193001 (2011).
Lahaye, T. et al. The physics of dipolar bosonic quantum gases. Rep. Prog. Phys. 72, 126401 (2009).
Schauss, P. et al. Observation of spatially ordered structures in a two-dimensional Rydberg gas. Nature 491, 87–91 (2012).
Schauss, P. et al. Crystallization in Ising quantum magnets. Science 347, 1455–1458 (2015).
Saffman, M. et al. Quantum information with Rydberg atoms. Rev. Mod. Phys. 82, 2313–2363 (2010).
Griesmaier, A. et al. Bose-Einstein Condensation of Chromium. Phys. Rev. Lett. 94, 160401 (2005).
Lu, M. et al. Strongly dipolar Bose-Einstein Condensate of Dysprosium. Phys. Rev. Lett. 107, 190401 (2011).
Aikawa, K. et al. Bose-Einstein Condensation of Erbium. Phys. Rev. Lett. 108, 210401 (2012).
Baier, S. et al. Extended Bose-Hubbard models with ultracold magnetic atoms. Science 352, 201–205 (2016).
de Paz, A. et al. Nonequilibrium Quantum Magnetism in a Dipolar Lattice Gas. Phys. Rev. Lett. 111, 185305 (2013).
Truppe, S. et al. Molecules cooled below the Doppler limit. Nat. Phys. 13, 1173–1176 (2017).
Bohn, J. L. et al. Cold molecules: Progress in quantum engineering of chemistry and quantum matter. Science 357, 1002–1010 (2017).
Carr, L. D. Cold and ultracold molecules: science, technology and applications. New J. Phys. 11, 055049 (2009).
Deng, H. et al. Condensation of semiconductor microcavity exciton polaritons. Science 298, 199–202 (2002).
Kasprzak, J. et al. Bose-Einstein condensation of exciton polaritons. Nature 443, 409–14 (2006).
Balili, R. et al. Bose-Einstein condensation of microcavity polaritons in a trap. Science 316, 1007–1010 (2007).
Plumhof, J. D. et al. Room-temperature Bose-Einstein condensation of cavity exciton-polaritons in a polymer. Nat. Mater. 13, 247–252 (2013).
Byrnes, T. et al. Exciton-polariton condensates. Nat. Phys. 10, 803–813 (2014).
Wertz, E. et al. Spontaneous formation and optical manipulation of extended polariton condensates. Nat. Phys. 6, 860–864 (2010).
Amo, A. et al. Superfluidity of polaritons in semiconductor microcavities. Nat. Phys. 5, 805–810 (2009).
Chen, Y. J. et al. Effect of electric fields on excitons in a coupled double-quantum-well structure. Phys. Rev. B 36, 4562–4565 (1987).
Golub, J. E. et al. Type I-type II anticrossing and enhanced Stark effect in asymmetric coupled quantum wells. Appl. Phys. Lett. 53, 2584–2586 (1988).
Voros, Z. et al. Long-Distance Diffusion of Excitons in Double Quantum Well Structures. Phys. Rev. Lett. 94, 226401 (2005).
Hubert, C. et al. Attractive Dipolar Coupling between Stacked Exciton Fluids. Phys. Rev. X 9, 021026 (2019).
Hubert, C. et al. Attractive interactions, molecular complexes, and polarons in coupled dipolar exciton fluids. Phys. Rev. B 102, 045307 (2020).
Butov, L. V. et al. Macroscopically ordered state in an exciton system. Nature 418, 751–754 (2002).
High, A. A. et al. et al. Spontaneous coherence in a cold exciton gas. Nature 483, 584–588 (2012).
Shilo, Y. et al. Particle correlations and evidence for dark state condensation in a cold dipolar exciton fluid. Nat. Commun. 4, 2335 (2013).
Stern, M. et al. Exciton liquid in coupled quantum wells. Science 343, 55–57 (2014).
Rivera, P. et al. Observation of long-lived interlayer excitons in monolayer MoSe2-WSe2 heterostructures. Nat. Commun. 6, 6242 (2015).
Miller, B. et al. Long-Lived Direct and Indirect Interlayer Excitons in van der Waals Heterostructures. Nano Lett. 17, 5229–5237 (2017).
Rivera, P. et al. Interlayer valley excitons in heterobilayers of transition metal dichalcogenides. Nat. Nanotechnol. 13, 1004–1015 (2018).
Forg, M. et al. Cavity-control of interlayer excitons in van der Waals heterostructures. Nat. Commun. 10, 3697 (2019).
Jauregui, L. A. et al. Electrical control of interlayer exciton dynamics in atomically thin heterostructures. Science 366, 870–875 (2019).
Wang, G. et al. Colloquium: Excitons in atomically thin transition metal dichalcogenides. Rev. Mod. Phys. 90, 021001 (2018).
Kang, J. et al. Band offsets and heterostructures of two-dimensional semiconductors. Appl. Phys. Lett. 102, 012111 (2013).
Gong, C. et al. Band alignment of two-dimensional transition metal dichalcogenides: Application in tunnel field effect transistors. Appl. Phys. Lett. 103, 053513 (2013).
Fang, H. et al. Strong interlayer coupling in van der Waals heterostructures built from single-layer chalcogenides. Proc. Natl Acad. Sci. USA. 111, 6198–6202 (2014).
Heo, H. et al. Interlayer orientation-dependent light absorption and emission in monolayer semiconductor stacks. Nat. Commun. 6, 7372 (2015).
Alexeev, E. M. et al. Resonantly hybridized excitons in moiré superlattices in van der Waals heterostructures. Nature 567, 81–86 (2019).
Tran, K. et al. Evidence for moiré excitons in van der Waals heterostructures. Nature 567, 71–75 (2019).
Nayak, P. K. et al. Probing Evolution of Twist-Angle-Dependent Interlayer Excitons in MoSe2/WSe2 van der Waals Heterostructures. ACS Nano 11, 4041–4050 (2017).
Seyler, K. L. et al. Signatures of moiré-trapped valley excitons in MoSe2/WSe2 heterobilayers. Nature 567, 66–70 (2019).
Jin, C. et al. Observation of moiré excitons in WSe2/WS2 heterostructure superlattices. Nature 567, 76–80 (2019).
Wang, Z. et al. Evidence of high-temperature exciton condensation in two-dimensional atomic double layers. Nature 574, 76–80 (2019).
Tang, Y. et al. Simulation of Hubbard model physics in WSe2/WS2 moiré superlattices. Nature 579, 353–358 (2020).
Shimazaki, Y. et al. Strongly correlated electrons and hybrid excitons in a moiré heterostructure. Nature 580, 472–477 (2020).
Regan, E. C. et al. Mott and generalized Wigner crystal states in WSe2/WS2 moiré superlattices. Nature 579, 359–363 (2020).
Browaeys, A. et al. Many-body physics with individually controlled Rydberg atoms. Nat. Phys. 16, 132–142 (2020).
Blackmore, J. A. et al. Ultracold molecules for quantum simulation: rotational coherences in CaF and RbCs. Quantum Sci. Technol. 4, 014010 (2019).
Wang, J. et al. Optical generation of high carrier densities in 2D semiconductor heterobilayers. Sci. Adv. 5, eaax0145 (2019).
Sortino, L. et al. Enhanced light-matter interaction in an atomically thin semiconductor coupled with dielectric nano-antennas. Nat. Commun. 10, 5119 (2019).
Yu, H. et al. Anomalous Light Cones and Valley Optical Selection Rules of Interlayer Excitons in Twisted Heterobilayers. Phys. Rev. Lett. 115, 187002 (2015).
Jones, A. M. et al. Optical generation of excitonic valley coherence in monolayer WSe2. Nat. Nanotechnol. 8, 634–638 (2013).
Mitioglu, A. A. et al. Optical manipulation of the exciton charge state in single-layer tungsten disulfide. Phys. Rev. B 88, 245403 (2013).
Palacios-Berraquero, C. et al. Large-scale quantum-emitter arrays in atomically thin semiconductors. Nat. Commun. 8, 15093 (2017).
Barbone, M. et al. Charge-tuneable biexciton complexes in monolayer WSe2. Nat. Commun. 9, 3721 (2018).
Jin, C. et al. Identification of spin, valley and moiré quasi-angular momentum of interlayer excitons. Nat. Phys. 15, 1140–1144 (2019).
Branny, A. et al. Deterministic strain-induced arrays of quantum emitters in a two-dimensional semiconductor. Nat. Commun. 8, 15053 (2017).
Shamirzaev, T. S. et al. Millisecond fluorescence in InAs quantum dots embedded in AlAs. Physica E Low. Dimens. Syst. Nanostruct. 20, 282–285 (2004).
Jiang, C. et al. Microsecond dark-exciton valley polarization memory in two-dimensional heterostructures. Nat. Commun. 9, 1–8 (2018).
Kremser, M. et al. Discrete Interactions between a few Interlayer Excitons Trapped at a MoSe2-WSe2 Heterointerface. NPJ 2D Mater. Appl. 4, 8 (2020).
Nagler, P. et al. Interlayer exciton dynamics in a dichalcogenide monolayer heterostructure. 2D Mater. 4, 025112 (2017).
Li, W. et al. Dipolar interactions between localized interlayer excitons in van der Waals heterostructures. Nat. Mater. 19, 624–629 (2020).
Kurtsiefer, C. et al. Stable Solid-State Source of Single Photons. Phys. Rev. Lett. 85, 290–293 (2000).
He, Y.-M. et al. Single quantum emitters in monolayer semiconductors. Nat. Nanotechnol. 10, 497–502 (2015).
Aivazian, G. et al. Magnetic control of valley pseudospin in monolayer WSe2. Nat. Phys. 11, 148–152 (2015).
Srivastava, A. et al. Optically active quantum dots in monolayer WSe2. Nat. Nanotechnol. 10, 491–496 (2015).
Koperski, M. et al. Single photon emitters in exfoliated WSe2 structures. Nat. Nanotechnol. 10, 503–506 (2015).
Chakraborty, C. et al. Voltage-controlled quantum light from an atomically thin semiconductor. Nat. Nanotechnol. 10, 507–511 (2015).
Wozniak, T. et al. Exciton g factors of van der Waals heterostructures from first-principles calculations. Phys. Rev. B 101, 235408 (2020).
Deilmann, T. et al. Ab initio Studies of Exciton g Factors: Monolayer Transition Metal Dichalcogenides in Magnetic Fields. Phys. Rev. Lett. 124, 226402 (2020).
Koperski, M. et al. Orbital, spin and valley contributions to Zeeman splitting of excitonic resonances in MoSe2, WSe2 and WS2 Monolayers. 2D Mater. 6, 015001 (2019).
Glazov, M. M. et al. Exciton fine structure and spin decoherence in monolayers of transition metal dichalcogenides. Phys. Rev. B 89, 201302(R) (2014).
Nagler, P. et al. Zeeman Splitting and Inverted Polarization of Biexciton Emission in Monolayer WS2. Phys. Rev. Lett. 121, 057402 (2018).
Feierabend, M. et al. Impact of strain in the optical fingerprint of monolayer transition-metal dichalcogenides. Phys. Rev. B 96, 045425 (2017).
Citrin, D. S. Radiative lifetimes of excitons in quantum wells: Localization and phase-coherence effects. Phys. Rev. B 47, 3832–3841 (1993).
Kuc, A. et al. The electronic structure calculations of two-dimensional transition-metal dichalcogenides in the presence of external electric and magnetic fields. Chem. Soc. Rev. 44, 2603–2614 (2014).
Kuhlmann, A. V. et al. Transform-limited single photons from a single quantum dot. Nat. Commun. 6, 8204 (2015).
Matthiesen, C. et al. Subnatural Linewidth Single Photons from a Quantum Dot. Phys. Rev. Lett. 108, 093602 (2012).
Trusheim, M. E. et al. Transform-Limited Photons From a Coherent Tin-Vacancy Spin in Diamond. Phys. Rev. Lett. 124, 023602 (2020).
Kumar, S. et al. Resonant laser spectroscopy of localized excitons in monolayer WSe2. Optica 3, 882–886 (2016).
Zhang, X. et al. Flux method growth of bulk MoS2 single crystals and their application as a saturable absorber. Cryst. Eng. Commun. 17, 4026–4032 (2015).
Novoselov, K. S. et al. Two-dimensional atomic crystals. Proc. Natl Acad. Sci. USA. 30, 10451–10453 (2005).
Casiraghi, C. et al. Rayleigh Imaging of Fraphene and Graphene Layers. Nano Lett. 7, 2711–2717 (2007).
Purdie, D. G. et al. Cleaning interfaces in layered materials heterostructures. Nat. Commun. 9, 5387 (2018).
Acknowledgements
We acknowledge funding from the EU Quantum and Graphene Flagships; ERC grants PEGASOS, Hetero2D and GSYNCOR; EPSRC Grants EP/K01711X/1, EP/K017144/1, EP/N010345/1 and EP/L016057/1; the Faraday Institution FIRG001; and DOE, NSF DMR 1552220, DMR 1904716, and NSF CMMI 1933214. D. M. K. acknowledges support of a Royal Society University Research Fellowship URF\R1\180593.
Author information
Authors and Affiliations
Contributions
A.R.-P.M., D.M.K., A.C.F. and M.A. conceived and managed the project; Y.Q., M.B. and S.T. provided the bulk WS2 and WSe2 crystals and performed the XRD and STEM measurements; P.L. and M.L. provided the nanopillar substrates; I.P., A.R.C. and G.W. fabricated the devices; A.R.-P.M., D.M.K., I.P., C.P., M.F., E.M.A., L.S. and M.A. performed the optical measurements and analysed the results. All authors participated in the discussion of the results and the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Montblanch, A.RP., Kara, D.M., Paradisanos, I. et al. Confinement of long-lived interlayer excitons in WS2/WSe2 heterostructures. Commun Phys 4, 119 (2021). https://doi.org/10.1038/s42005-021-00625-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42005-021-00625-0
This article is cited by
-
Engineering interlayer hybridization in van der Waals bilayers
Nature Reviews Materials (2024)
-
Probing the optical near-field interaction of Mie nanoresonators with atomically thin semiconductors
Communications Physics (2023)
-
Interlayer donor-acceptor pair excitons in MoSe2/WSe2 moiré heterobilayer
Nature Communications (2023)
-
Layered materials as a platform for quantum technologies
Nature Nanotechnology (2023)
-
Electrical control of quantum emitters in a Van der Waals heterostructure
Light: Science & Applications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
