
Abstract
The addition of a nitrogen-based functional group to alkenes via a direct catalytic method is an attractive way of synthesizing value-added amides. The regioselective hydroamidation of unactivated alkenes is considered one of the easiest ways to achieve this goal. Herein, we report the NiH-catalyzed anti-Markovnikov intermolecular hydroamidation of unactivated alkenes enabled by using 2,9-dibutylphenathroline (diBuphen) as the ligand. This protocol provides a platform for the direct synthesis of over 90 structurally diverse N-alkyl amides using dioxazolones, which can be easily derived from abundant carboxylic acid feedstocks. This method succeeds for both terminal and internal unactivated alkenes and some natural products. Mechanistic studies including DFT calculations reveal an initial reversible insertion/elimination of the [NiH] to the alkene, followed by the irreversible amidation to furnish the N-alkyl amides. By crossover experiments and deuterium labeling studies, the observed anti-Markovnikov regioselectivities are suggested to be controlled by the sterical environment of the coupling reaction.
Similar content being viewed by others
Introduction
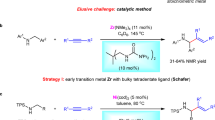
Amides are one of the most prevalent structures found in organic molecules, such as peptides, proteins, and DNA. It also constitutes an important motif in many functional materials and medicinal products1,2,3,4,5. While the conventional amide synthesis is performed by coupling carboxylic acids with amines, the production of stoichiometric amount unwanted side-products remains an environmental concern6. Recently, metal hydride-mediated hydrometalation using electrophilic aminating reagents received considerable attention for regioselective amines/amides synthesis (Metal = Fe7,8, Co9,10, Ni11,12, Cu13,14,15). Pioneered by Buchwald and Miura, CuH-catalyzed regioselective hydroamination of alkenes with hydroxy amine derivatives was achieved to give the amines/amide products. The reaction was suggested to proceed through some n-alkyl-Cu(I) complexes16,17. CuH-catalyzed hydroaminations of styrene usually show Markovnikov selectivity due to the formation of a more stable benzyl-Cu complex. In contrast, the analogous hydrometalation of aliphatic alkenes often exhibits anti-Markovnikov regioselectivity due to steric constraints (Fig. 1a, up)17. Another interesting example was reported by Hartwig et al. that an OBzCl3 group on alkenes would serve as a polarity-directing group for regioselective hydrometalation (Fig. 1a, down)18.

a [CuH] mediated regioselective hydroamination. b [NiH] mediated regioselective hydroamination/hydroamidation. c This work: Ligand enhanced regioselective hydroamidation.
Recently catalytic C–N bond formation was also enabled by the NiH-mediated hydroamidation of alkenes11,12,19,20,21,22. NiH hydrometalation of aliphatic alkenes is usually non-regioselective, giving both Markovnikov and anti-Markovnikov products. The regiocontrol of the NiH-mediated hydroamidation is further complicated by the propensity of the chain-walk isomerization via alkyl-Ni(II) species23. To achieve the proximal hydroamidation of alkenes, directing group can be used to suppressed the chain-walk isomerization, and the thermodynamic preferred five-membered nickelacycle further support the regioselective hydrometalation of some unactivated internal alkenes (Fig. 1b, up). Hong’s group utilized an aminoquinoline as a directing group to mediate the regioselective hydrometalation of some unactivated internal alkenes24. Yet, this strategy is limited by the necessity of a pre-installed functional group nearby the C=C bond, and thus, hydroamidation to access linear amides are spare. Direct regioselective hydroamination of directing group-free alkenes may be appealing to the synthetic industry. During the preparation of this manuscript, our group also reported an NiH-catalyzed remote C(sp3)−H hydroamidation of alkenes25. Also, in this period, Chang et al. demonstrated the NiH-catalyzed hydroamidation of alkynes with dioxazolones to give enamides in (E)-anti-Markonikov or Markonikov selectivity. The regioselectivities are controlled by the choice of ligands, which govern the initial hydrometalation step (Fig.1b, down)26.
Here we report the NiH-catalyzed anti-Markovnikov hydroamidation of alkenes with dioxazolones for the synthesis of structurally diverse linear amides (Fig. 1c). By employing 2,9-dibutyl-1,10-phenanthroline (diBuphen) as ligand, over 90 anti-Markovnikov N-alkyl amides were afforded in up to 92% yield. Mechanistic investigations combined with density functional theory (DFT) calculation suggested the observed selectivity can be understood by invoking Curtin-Hammett type equilibrium.
Results
In this work, treating vinylcyclohexane 1 (0.2 mmol), 3-cyclobutyl-1,4,2-dioxazol-5-one 2 (0.4 mmol), pinacolborane (2.0 equiv) using [Ni(ClO4)2]∙6H2O (10 mol %) with diBuphen (12 mol %) in THF (2.0 ml) at room temperature for 12 h furnished the anti-Markovnikov hydroamidation product 3 in 95% yield. Notably, <5% Markovnikov hydroamidation product 4 was detected (Table 1, entry 1). Other nickel precursors, such as NiI2 and Ni(COD)2, are found to be ineffective catalysts (entries 2–3). The effect of some common organic solvents was then tested (entries 4–6). The reaction with DCE gave a better result (3: 90%), while running the reaction in MeCN and in DMA afforded 3 in 55% and 56% yields, respectively. To our delight, excellent regioselectivity and yield were achieved by employing a DMA/THF (1:9 v/v) mixture as the solvent system (entry 7); the desired 3 was obtained in 99% without any Markovnikov products formation. The effect of ligand was then studied, no desired products were obtained in the absence of the diBuphen ligand (entry 8). Employing related ligands such as L1 led to unselective hydroamidation: (3: 9% and 4: 10%) (entry 9). While L2 was found to be less effective, affording a moderate hydroamidation yield with poor regioselectivity: (3: 26% and 4: 35%) (entry 10). Reactions with L3 as ligand produced comparable results as diBuphen: 3 (75%; entry 11). Diphosphine ligands such as L5 and L6 were ineffective for this hydroamidation reaction (entries 12–13). Apparently, the presence of HBpin is critical for this transformation; no desired products were formed without HBpin (entry 14). Other common hydride sources such as Ph2SiH2 and (EtO)2SiMeH were found to be ineffective (entries15–16).
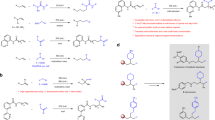
With the protocol of anti-Markovnikov alkene hydroamidation established, we studied the scope of dioxazolones (Fig. 2). Under the Ni-catalyzed conditions, dioxazolones containing primary alkyl groups were effectively coupled to the vinylcyclohexane 1 to form the corresponding N-alkyl amides (5–11) in good yields (~80%). Dioxazolones containing ethoxy, phthalimide, thiophene, ketone and indole groups were converted to their amides (12–16) in good yields. Moreover, the coupling reaction of 1 with dioxazolones derived from 2-chloropropanoic acid was also successful to give 17 in 71% yield. Similarly, the dioxazolones containing secondary alkyl groups were transformed to the corresponding linear N-alkyl amides (3, 18–28) in good yields (~80%). Presumably, due to steric reasons, transformations of the dioxazolones bearing tertiary alkyl groups were less successful (29: 25% and 30: 23%). The reactions of the dioxazolones derived from 3-(methoxycarbonyl)bicyclo[1.1.1]pentane-1-carboxylic acid and methyl 4-(5-oxo-1,4,2-dioxazol-3-yl)cubane1-carboxylate containing strained carbocycles afforded 31 and 32 in 92% and 70% yields, respectively.

aReaction conditions: 1 (0.2 mmol), dioxazolones (0.4 mmol), [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml) in N2 at room temperature for 12 h unless otherwise specified. bIsolated yield. cLarge scale reaction: 1 (10.0 mmol) was used, 3 was obtained in 1.45 g.
As anticipated, facile couplings with the dioxazolones containing aryl groups were also achieved, and 33–36 were formed in ~80% yields. For instance, dioxazolones derived from 6-fluoropicolinic acid and ferrocenecarboxylic acid produced 37 (76%) and 38 (47%). The analogous reactions with dioxazolones derived from some natural products gave the corresponding amides effectively, including 1-pyrenebutyric acid (39: 91%), Isoxepac (40: 63%), Citronellic acid (41: 78%) and Indomethacine (42: 40%). The molecular structure of 40 has been confirmed by X-ray crystallography. Less effective reactions were encountered for the dioxazolones bearing secondary or tertiary alkyl groups, such as Ibuprofen (43: 25%), Naproxen (44: 36%) and Gemfibrozil (45: 8%).
The synthetic versatility of this reaction was further explored with the alkenes scope (Fig. 3). With 3-cyclobutyl-1,4,2-dioxazol-5-one 2 as the nitrene source, we examined the reactivity of some terminal alkenes (46–52), and the corresponding amides were obtained in good yields (~80%). However, reaction of 3,3-dimethylbut-1-ene afforded the desired product 49 only in 33% yield. The relatively low yield could be attributed to steric interaction between the alkene and the Ni catalyst. Substrates with two C=C motifs, such as 4-vinylcyclohex-1-ene would undergo hydroamidation at the least hindered position, and 53 was formed in 47% yield. Hydroamidation of alkynes was also accomplished, and 54 was produced in 26% yield with hex-1-yne as substrate. Coupling reactions of 1,1-disubsituted alkenes gave 55–58 in moderate yields (~60%), indicative of the steric sensitivity of this reaction.

aReaction conditions: alkenes (0.2 mmol), 2/62 (0.4 mmol), [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml), in N2 at room temperature for 12 h unless otherwise specified. bIsolated yield. cRegioisomeric ratio (rr) is defined as linear/branched. dL3 instead of L4. e73% branched product. f70% branched product. g50% branched products. h53% branched products. i51% branched product.
For internal alkenes, effective coupling reactions of 2 with cyclohexene and bicyclo[2.2.1]hept-2-ene were achieved to furnish 59 (49%) and 60 (62%), respectively. Facile transformation with cholesterol as substrate furnished 61 in 43% yield. We then turned to expand the alkene scope using a bulkier 3-(heptan-4-yl)-1,4,2-dioxazol-5-one 62 for the reaction. Similar to our earlier examples, the hydroamidation of simple terminal alkenes produced 63–65 in good yields. Notably, the regioselectivity was reversed to branched amide products when but-3-enenitrile and allyltrimethylsilane were used. The desired linear products 66 and 67 were only obtained in 18 and 24% yields. Likewise, 2-allylcyclohexan-1-one reacted with 62 afforded the anti-Markovnikov product 68 (30%) accompanied with the branched products (50%). For the reaction with 1-allyl-N,N-diethylcyclopropane-1-sulfonamide, anti-Markovnikov product 69 was obtained exclusively in 74% yield.
Terminal alkenes with a four-carbon alkyl chain were also tested. The reactions of the alkenes with ester, phthalimide and epoxy substituents at the δ carbon displayed excellent selectivity for the formation of linear amides 70–72 albeit in moderate yields. However, ketone and amide substituted alkenes reacted with 62 afforded branched amides in 53% and 51% yields with the desired 73 and 74 being formed in 36 and 37%. For the terminal alkenes bearing a five-carbon alkyl chain such as 5-bromopent-1-ene, amide 75 was obtained in 44% yield. The reaction of hex-5-enenitrile gave the linear amide (76: 91%) in anti-Markovnikov selectivity; however, the analogous transformation of but-3-enenitrile only gave the anti-Markovnikov product 66 in 18% yield. Other alkenes with functional groups such as nitro, esters and alcohol derived motifs were effectively coupled to 62 to afford the corresponding linear amides 77–83 in 59–79% yields. The reactivities of terminal alkenes bearing a six-carbon alkyl chain were also examined. Substituents including hydroxyl, tosylate, 2-(trimethylsilyl)ethoxy and esters were found to be well tolerated: 84–89, 56–79%. Effective transformations for those alkenes containing natural product motifs such as Loxoprofen (90: 71%), Carprofen (91: 66%), Lithocholic acid (92: 67%) and Estrone (93, 62%) were proved to be successful.
During the substrate scope study, we found that the reaction of a disubstituted alkene 94 afforded chain-walked product 95 in 37% yield (Fig. 4a). Moreover, the hydroamidation reaction of 3-methoxyprop-1-ene 96 with 62 gave the desired linear amide product 97 in 43% yield along with two unexpected products 98 (30%) and 99 (7%). The 98 and 99 formation can be accounted for by the prior β-elimination of the OMe group to generate prop-1-ene, which then reacted with 62 (Fig. 4b). The feasibility of the analogous intramolecular hydroamidation reaction was also demonstrated with 3-cinnamyl-1,4,2-dioxazol-5-one 100 as substrate. To our delight, the cyclized β-lactam 4-benzylazetidin-2-one 101 was formed in 61% yield implied that the potential coordination of dioxazolones (Fig. 4c).

a Chain walking. b β-alkoxide elimination. c Intramolecular amidation. Conditions A: alkene (0.2 mmol), 62 (0.4 mmol), [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml), in N2 at room temperature for 12 h. Conditions B: alkene (0.2 mmol), [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), MeCN + DCE (1.0 + 1.0 ml), in N2 at room temperature for 12 h.
To study the uniqueness and the working principle of dioxazolones as amidating reagents, hydroamidation with some common aminating and amidating reagents were tested (Fig. 5). Benzoic acid morpholin-4-yl ester 102 and 4-nitrotoluene 105 were found to be ineffective reagents in the current reaction system. While cyclohexyl isocyanate 108 could furnish the amide products with branched amides as the major product. Notably, comparable yield and selectivity was observed when benzoyl azide 110 was employed as the coupling partner instead of dioxazolones, which implies dioxazolone could also function as a nitrene source27,28.

Standard conditions: alkene (0.2 mmol), aminating (amidating) reagents (0.4 mmol), [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml), in N2 at room temperature for 12 h. All yields were presented as NMR yield.
The reaction rate and selectivity has been probed by three sets of intermolecular competitive experiments. As depicted in Fig. 6a, upon treating an equimolar mixture of 3-cyclobutyl-1,4,2-dioxazol-5-one 2 and 3-ethyl-1,4,2-dioxazol-5-one 113 with 1 under the Ni-catalyzed conditions, the corresponding 3 and 5 were formed in 39% and 38%, respectively. This implies the reactivity of the dioxazolones bearing primary and secondary alkyl groups should be similar. Yet, the reaction with 2 and 3-(tert- butyl)-1,4,2-dioxazol-5-one 114 produced amide 3 predominantly in 69% vs. 9% for the 29 formation. Analogous crossover experiment using 1 and cyclohexene 115 with 2 as coupling partner was performed under the Ni-catalyzed conditions, amide 3 derived from the less hindered alkene 1 was formed in 75% yield vs. 23% yield for 59 formation. These findings support the steric sensitivity of the hydroamidation reactions.

a Crossover experiments. b Hydroboration/amidation. Standard conditions: [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml), in N2 at room temperature for 12 h. The yields presented in part a are all isolated yield. The yield of 3 presented in part b is NMR yield. The yield of 116 presented in part b is GC yield.
A stepwise hydroboration/amidation pathway is improbable (Fig. 6b). Under the optimized conditions, the coupling reactions between 1 and 2 produced 3 in 88% yield. However, when the reaction was performed without the dioxazolone, ethylidenecyclohexane 116 (55%) was obtained exclusively without any hydroboration product 117 being formed (see Supporting Information)29. When subjecting a separately synthesized 117 to Ni-catalyzed conditions with dioxazolone 2, no amide 3 was not obtained30. These results are inconsistent with a stepwise hydroboration/amidation pathway.
Deuterium labeling studies were also performed to study the reaction mechanism (Fig. 7a). The Ni-catalyzed coupling reaction of 1 and 2 was performed under the standard conditions with DBpin instead of HBpin. HNMR and HRMS analysis revealed two deuterium exchanged products (122: 123 ~ 7: 3) were formed. Plausibly, alkene insertion by [NiD] should generate two regioisomeric d-alkylnickel intermediates 118 (Markovnikov) and 119 (anti-Markovnikov) in a 3: 7 ratio31,32,33,34,35. While 119 should react directly with 2 to give 122, the formation of 123 should proceed through reversible β-H elimination/insertion (via intermediate 120 and 121)36,37,38,39,40,41,42,43. Similar results were obtained when 124 was treated with 125 under the Ni-catalyzed conditions (126: 127 ~ 7: 3). Based on these findings, we postulated the Curtin-Hammett selectivity to be proceeded by the reversible elimination/insertion of nickel-hydride complexes, followed by an irreversible C–N bond coupling44. To scrutinize this hypothesis, we investigated the regioselectivity of this reaction by comparing the results of three different dioxazolones (2, 62, 103) (Fig. 7b). Apparently, the linear/branched regioisomeric ratios (rr = 2.4, 6.1, 15.8) were very much dependent on the dioxazolone reagents (i.e., higher linear selectivity for bulkier dioxazolones). This finding supports the operation of Curtin-Hammett equilibrium prior to the irreversible C–N coupling step.

a Deuterium labeling studies. b Trapping reagents experiments. Standard conditions: [Ni(ClO4)2]∙6H2O (10 mol %), L4 (12 mol %), HBpin/DBpin (2.0 equiv), THF + DMA (1.8 + 0.2 ml), in N2 at room temperature for 12 h. All the yields presented are NMR yield.
To gain further insight into the reaction mechanism and the origin of the anti-Markovnikov selectivity, we performed DFT calculations at the ωB97XD/SDD (Ni)/6-31G(d,p) level of theory (see Supporting Information for the computational details). Figure 8 depicts the energy profile calculated for the derived reaction mechanism. With [Ni(ClO4)2]∙6H2O as the precursor catalyst in the presence of propene, HBpin, and 2,9-disubsituted-1,10-phenanthroline as the ligand, a Ni(II) hydride intermediate (A) is formed as the active species of the reaction. Noted that the product molecule-coordinated Ni(II) hydride species G be the resting state according to the energetics we obtained, we took G as the energy reference point in the energy profile. Ligand exchange of the model propene substrate for the model product molecule transforms the resting state G to the Ni(II) hydride active species A, followed by alkene insertion to form the agostic species B, which is further converted to the square planar complex C after coordination of dioxazolone. The release of CO2 from the coordinated dioxazolone in the square planar Ni(II) complex C is rate-determining, with a see-saw shaped rate-determining transition state TSC–D, leading to a Ni(IV) complex D with an overall reaction barrier of 26.4 kcal mol−1. Here, it should be pointed out that although Ni(IV) species are rare, organometallic Ni(IV) complexes containing alkyl and N ligands have been reported45, and a few nickel-catalyzed reactions have proposed the intermediacy of Ni(IV)46,47,48. D is a four-coordinate d6 species. Such species are rare, but there are precedents with Ru(II), Rh(III) and Ir(III) centers49,50,51. HBpin acts as a Lewis acid and attaches to the oxygen in D to form the intermediate E, from which a very facile reductive elimination gives Ni(II) complex F involving a drop of the free energy of the system significantly. Finally, the intermediate F undergoes intramolecular hydride migration from the attached HBpin to Ni to form the resting state G that contains a coordinated product molecule. Subsequent ligand exchange should regenerate the active species A.

The relative Gibbs energies and electronic energies (in parentheses) are given in kcal mol−1.
One may query why the dioxazolone would not react with the NiH species (prior to olefin insertion), leading to nitrene insertion into the Ni–H bond. To address this question, we calculated the CO2 extrusion transition state for the reaction of dioxazolone with the NiH species and found that this process is kinetically less favorable by 7.9 kcal mol−1 than the corresponding one with the Ni-alkyl species shown in Fig. 8. We reasoned that an alkyl ligand should be relatively more electron-releasing than a hydride ligand, thereby facilitating the CO2 extrusion process that raises the formal oxidation state of the nickel center. Based on this analysis, we concluded that alkene insertion to NiH should occur prior to the CO2 extrusion of the dioxazolones. We also examined the possible Curtius rearrangement from the species D. The rearrangement barrier was calculated to be higher by 0.8 kcal mol−1 than that calculated for D → TSE–F in Fig. 8. The barrier difference is expected to increase significantly if the experimentally-used dioxazolone containing a cyclobutyl substituent is employed in the calculations. The steric bulky metal fragment likely suppresses the rearrangement process.
We also briefly examined the possible involvement of Ni(I)–H species. Our additional calculation results indicated that the Ni(I)–H species shows extremely poor capability of coordinating the model alkene substrate. The alkene complex of Ni(I)–H was calculated to lie higher in free energy by 32.8 kcal mol−1 than its resting state [the Ni(I) analogue of G] (see Supporting Information). This implies that our system is unlikely to involve Ni(I)–H species.
Regarding to the finding that different ligands display different linear-to-branch selectivity, DFT calculations were carried out on the resting state G and the rate-determining transition state TSC–D with three different models: reaction of cyclohexylethene with 3-cyclobutyl-1,4,2-dioxazol-5-one employing the ligands 2-methyl-1,10-phenanthroline (L2), 2,9-dimethyl-1,10-phenanthroline (L3), and 2,9-di-n-butyl-1,10-phenanthroline (L4). Referring to Fig. 9, the calculated barrier differences between the Markovnikov hydroamidation transition state (branched) TS-BC–D and the anti-Markovnikov hydroamidation transition state (linear) TS-LC–D (ΔG(TS-BC–D)–ΔG(TS-LC–D)) are 0.3 kcal mol−1 (L2), −0.5 kcal mol−1 (L3) and −6.1 kcal mol−1 (L4), which agree qualitatively well with the experimentally observed trend (for L2, 26%: 38%; for L3, 57%: 22%; for L4, 99%: n.d.); the calculated overall reaction barriers are 35.6 kcal mol−1 (L2), 31.7 kcal mol−1 (L3) and 30.9 kcal mol−1 (L4) also agree qualitatively well with the experimentally observed trend in the reaction yields.

Rate-determining transition states TS-BL2–L4(C–D) and TS-LL2–L4(C–D) for Markovnikov hydroamidation (leading to branched product) and anti-Markovnikov hydroamidation (leading to linear product), respectively. The relative Gibbs free energies are given in kcal mol−1.
When the two substituents on the phenanthroline ligand were becoming bulkier as exemplified in L2 to L4, the barrier difference becomes more negative (i.e., more anti-Markovnikov), and the calculated energy barrier decreases. This selectivity should be originated from the difference of steric hindrance at rate-determining transition state TSC–D. In TS-LC–D, the cyclohexyl group is located further from the two substituents on the phenanthroline ligand when compared with the situation in TS-BC–D. In model L2, the steric effect becomes relatively unimportant. Then, the electronic effect dominates so that TS-BL2C–D becomes relatively more stable than TS-LL2C–D because the former has a stronger Ni–C bond, which contains a secondary alkyl ligand, whereas the latter contains a primary alkyl ligand. When the steric hindrance increases from L2 to L4, the steric effect becomes dominant, thus, the barrier differences change from positive to negative, causing a reversal of selectivity from Markovnikov to anti-Markovnikov. The energy barrier decreases from L2 to L4 should originate from the instability of the resting state G. G has a more sterically-hindered structure than TSC–D. Therefore, when the reactants are bulkier, the energy difference between G and TSC-D decreases. Clearly, the DFT results show that the proposed mechanism explains the experimentally observed trends in selectivity and reactivity when changing the size of substituents on the phenanthroline ligands.
Based on the mechanistic investigations and DFT calculations, the proposed mechanism is depicted in Fig. 10. The active nickel hydride I is generated by treating [Ni(ClO4)2]·6H2O with HBpin and 2,9-dibutylphenathroline (L4). A reversible 1,2-insertion would then take place to give an alkylnickel intermediates II. The bulky ligand plays an important role to facilitate the anti-Markovnikov selectivity. Coordination of dioxazolone to the alkylnickel II generates complex III, and the subsequent rate-determining irreversible amidation gives the nickel-nitrenoid species IV. Coordination of an additional HBpin to the carbonyl group affords intermediate V and assists the following Curtin-Hammett type hydroamidation to generate intermediate VII via VI. A final hydrolysis process would regenerate the active nickel hydride I and furnish the linear N-alkyl amides.

Our proposed NiH-catalyzed ligand enabled Curtin-Hammett type hydroamidation mechanism.
Conclusion
In conclusion, we developed a ligand-controlled Ni-catalyzed intermolecular hydroamidation of unactivated alkenes, and N-alkyl amides in predominantly anti-Markovnikov selectivity were obtained in excellent yields. This protocol is applicable to a wide range of substrate scope including some architecturally unique natural products. Since dioxazolones could be prepared from abundant carboxylic acid feedstocks, this hydroamidation protocol would be a powerful tool for easy conversion of hydrocarbon feedstocks to functionally and structurally diverse amides to cater for specific applications. Further control experiments and DFT calculations implied that the Curtin-Hammett selectivity model should be in operation.
Methods
General procedure for Ni-catalyzed cross-coupling reaction between alkenes and 1,4,2-dioxazol-5-ones
In a nitrogen-filled glovebox, a 8 ml vial containing a magnetic stir bar was charged with Ni(ClO4)2.6H2O (7.3 mg, 10 mol %) and 2,9-dibutyl-1,10-phenanthroline (7.0 mg, 12 mol %). Anhydrous THF (1.8 ml) and DMA (0.2 ml) were then added to the mixture via syringes. The vial was then screw-capped and stirred for 10 min at room temperature to give a brownish yellow [Ni + L] standard solution. Alkene (0.20 mmol, 1.0 equiv) and 1,4,2-dioxazol-5-one (0.40 mmol, 2.0 equiv) were added to a separate 8 ml vial with a magnetic stirrer bar. The [Ni + L] standard solution was transferred to the alkene-dioxazolone mixture with vigorous stirring. 4,4,5,5-Tetramethyl-1,3,2- dioxaborolane [HBpin, (0.40 mmol, 2.0 equiv)] was then added dropwise to the mixture via a syringe. A dark green solution mixture would appear upon addition of HBpin. The vial was then capped and removed from the glovebox. The mixture was then stirred at room temperature for 12 h. The crude mixture was transferred to a 25 ml round bottom flask and concentrated in vacuo. The residue was then filtered through a short-packed column with silica gel with ethyl acetate as eluent. The residue was again concentrated in vacuo. The residue was purified by column chromatography (n-hexane: ethyl acetate = 5: 1 to 1: 1), and the desired amide product can be visualized by TLC using KMnO4 stain.
Supplementary Methods were provided in detail in the Supporting Information File.
Data availability
All data are available from the corresponding authors upon reasonable request. Supplementary Information contains cif file for crystal structure, DFT calculation and the NMR spectra. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2052631 (40). The cif files for crystal structure is available in Supplementary Data 1. The DFT calculation is provided in Supplementary Data 2. The NMR spectra of all the compounds are available in Supplementary Data 3. It can be declared that all the relevant data are provided in the article and its Supplementary Information files. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Neubauer, D. N. A review of Ramelteon in the treatment of sleep disorders. Neuropsychiatr. Dis. Treat. 4, 69–79 (2008).
Fitton, A., Faulds, D. & Goa, K. L. Moclobemide. A review of its pharmacological properties and therapeutic use in depressive illness. Drugs 43, 561–596 (1992).
Reshef, R. et al. Blockade of lymphocyte chemotaxis in visceral graft-versus-host disease. N. Engl. J. Med. 367, 135–145 (2012).
Sun, S. Y., Jia, Q. & Zhang, Z. H. Applications of amide isosteres in medicinal chemistry. BioOrg. Med. Chem. Lett. 29, 2535–2550 (2019).
Kumari, S., Carmona, A. V., Tiwari, A. K. & Trippier, P. C. Amide bond bioisosteres: strategies, synthesis, and successes. J. Med. Chem. 63, 12290–12358 (2020).
Smith, M. B. & March, J. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure 5th edn, 508 (John Wiley & Sons, 2000).
Villa, M. & Wangelin, A. J. V. Hydroaminations of alkenes: a radical, revised, and expanded version. Angew. Chem. Int. Ed. 54, 11906–11908 (2005).
Song, H., Yang, Z. Y., Tung, C.-H. & Wang, W. G. Iron-catalyzed reductive coupling of nitroarenes with olefins: intermediate of iron–nitroso complex. ACS Catal. 10, 276–281 (2020).
Yahata, K., Kaneko, Y. & Akai, S. Cobalt-catalyzed intermolecular Markovnikov hydroamination of nonactivated olefins: N2-selective alkylation of benzotriazole. Org. Lett. 22, 598–603 (2020).
Yang, D. D. et al. Regioselective intermolecular hydroamination of unactivated alkenes: “Co−H” enabled remote functionalization. ACS Catal. 11, 6602–6613 (2021).
Xiao, J., He, Y., Ye, F. & Zhu, S. L. Remote sp3 C−H amination of alkenes with nitroarenes. Chem 4, 1645–1657 (2018).
Meng, L. P., Yang, J. J., Duan, M., Wang, Y. & Zhu, S. L. Facile synthesis of chiral arylamines, alkylamines and amides by enantioselective NiH-catalyzed hydroamination. Angew. Chem. Int. Ed. 60, 23584–23589 (2021).
Yang, Y., Shi, S.-L., Niu, D. W., Liu, P. & Buchwald, S. L. Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 349, 62–66 (2015).
Guo, S., Yang, J. C. & Buchwald, S. L. A practical electrophilic nitrogen source for the synthesis of chiral primary amines by copper-catalyzed hydroamination. J. Am. Chem. Soc. 140, 15976–15984 (2018).
Yuan, Y. et al. Copper-catalyzed carbonylative hydroamidation of styrenes to branched amides. Angew. Chem. Int. Ed. 59, 22441–22445 (2020).
Miki, Y., Hirano, K., Satoh, T. & Miura, M. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew. Chem. Int. Ed. 52, 10830–10834 (2013).
Zhu, S. L., Niljianskul, N. & Buchwald, S. L. Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J. Am. Chem. Soc. 135, 15746–15749 (2013).
Xi, Y., Butcher, T. W., Zhang, J. & Hartwig, J. F. Regioselective asymmetric formal hydroamination of unactivated internal alkenes. Angew. Chem. Int. Ed. 55, 776–780 (2016).
Lee, C., Seo, H., Jeon, J. & Hong, S. γ-Selective C(sp3)–H amination via controlled migratory hydroamination. Nat. Commun. 12, 5657–5665 (2021).
Du, B. N., Ouyang, Y. X., Chen, Q. S. & Yu, W. Y. Thioether-directed NiH-catalyzed remote γ-C(sp3)−H hydroamidation of alkenes by 1,4,2-dioxazol-5-ones. J. Am. Chem. Soc. 143, 14962–14968 (2021).
Gao, Y. et al. Nickel-catalyzed hydroamination of olefins with anthranils. J. Org. Chem. 86, 12107–12118 (2021).
Choi, H., Lyu, X., Kim, D., Seo, S. & Chang, S. Endo-selective intramolecular alkyne hydroamidation enabled by NiH catalysis incorporating alkenylnickel isomerization. J. Am. Chem. Soc. 144, 10064–10074 (2022).
Zhang, Y. L., He, J., Song, P. H., Wang, Y. & Zhu, S. L. Ligand-enabled NiH-catalyzed migratory hydroamination: chain walking as a strategy for regiodivergent/regioconvergent remote sp3 C–H amination. CCS Chem. 2, 2259–2268 (2020).
Jeon, J., Lee, C., Seo, H. & Hong, S. NiH-catalyzed proximal-selective hydroamination of unactivated alkenes. J. Am. Chem. Soc. 142, 20470–20480 (2020).
Du, B. N., Ouyang, Y. X., Chen, Q. S. & Yu, W.-Y. Thioether-directed NiH-catalyzed remote γ-C(sp3)–H hydroamidation of alkenes by 1,4,2-dioxazol-5-ones. J. Am. Chem. Soc. 143, 14962–14968 (2021).
Lyu, X., Zhang, J., Kim, D., Seo, S. & Chang, S. Merging NiH catalysis and inner-sphere metal-nitrenoid transfer for hydroamidation of alkynes. J. Am. Chem. Soc. 143, 5867–5877 (2021).
Vliet, K. M. & Bruin, B. D. Dioxazolones: stable substrates for the catalytic transfer of acyl nitrenes. ACS Catal. 10, 4751–4769 (2020).
Vliet, K. M. et al. Efficient copper-catalyzed multicomponent synthesis of N-acyl amidines via acyl nitrenes. J. Am. Chem. Soc. 141, 15240–15249 (2019).
Cramer, R. Jr. & Lindsey, R. V. The mechanism of isomerization of olefins with transition metal catalysts. J. Am. Chem. Soc. 88, 3534–3544 (1966).
Wang, Y., Guan, R., Sivaguru, P., Cong, X. & Bi, X. H. Silver-catalyzed anti-Markovnikov hydroboration of C–C multiple bonds. Org. Lett. 21, 4035–4038 (2019).
Eberhardt, N. A. & Guan, H. Nickel hydride complexes. Chem. Rev. 116, 8373–8426 (2016).
Zhang, Y., Xu, X. & Zhu, S. L. Nickel-catalysed selective migratory hydrothiolation of alkenes and alkynes with thiols. Nat. Commun. 10, 1752–1761 (2019).
Nguyen, J., Chong, A. & Lalic, G. Nickel-catalyzed anti-Markovnikov hydroarylation of alkenes. Chem. Sci. 10, 3231–3236 (2019).
Bera, S. & Hu, X. Nickel-catalyzed regioselective hydroalkylation and hydroarylation of alkenyl boronic esters. Angew. Chem. Int. Ed. 58, 13854–13859 (2019).
Li, Z. Q. et al. Ligand-controlled regiodivergence in nickel-catalyzed hydroarylation and hydroalkenylation of alkenyl carboxylic acids. Angew. Chem. Int. Ed. 59, 23306–23312 (2020).
Juliá-Hernández, F., Moragas, T., Cornella, J. & Martin, R. Remote carboxylation of halogenated aliphatic hydrocarbons with carbon dioxide. Nature 545, 84–88 (2017).
Chen, F. et al. Remote migratory cross-electrophile coupling and olefin hydroarylation reactions enabled by in situ generation of NiH. J. Am. Chem. Soc. 139, 13929–13935 (2017).
Janssen-Müller, D., Sahoo, B., Sun, S. & Martin, R. Tackling remote sp3 C–H functionalization via Ni-catalyzed “Chain-Walking” reactions. Isr. J. Chem. 60, 195–206 (2020).
Qian, D. & Hu, X. Ligand-controlled regiodivergent hydroalkylation of pyrrolines. Angew. Chem. Int. Ed. 58, 18519–18523 (2019).
Li, Y. et al. Nickel-catalyzed 1,1-alkylboration of electronically unbiased terminal alkenes. Angew. Chem. Int. Ed. 58, 8872–8876 (2019).
Li, Y., Wu, D., Cheng, H. & Yin, G. Difunctionalization of alkenes involving metal migration. Angew. Chem. Int. Ed. 59, 7990–8003 (2020).
Kumar, G. S. et al. Nickel-catalyzed chain-walking cross-electrophile coupling of alkyl and aryl halides and olefin hydroarylation enabled by electrochemical reduction. Angew. Chem. Int. Ed. 59, 6513–6519 (2020).
Dhungana, R. K., Sapkota, R. R., Niroula, D. & Giri, R. Walking metals: catalytic difunctionalization of alkenes at nonclassical sites. Chem. Sci. 11, 9757–9774 (2020).
Seeman, J. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett/Winstein-Holness kinetics. Chem. Rev. 83, 83–134 (1983).
Camasso, N. M. & Sanford, M. S. Design, synthesis, and carbon-heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 347, 1218–1220 (2015).
Tasker, S. Z., Standley, E. A. & Jamison, T. F. Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014).
Terao, J. & Kambe, N. Cross-coupling reaction of alkyl halides with Grignard reagents catalyzed by Ni, Pd, or Cu complexes with π-carbon ligand(s). Acc. Chem. Res. 41, 1545–1554 (2008).
Aihara, Y. & Chatani, N. Nickel-catalyzed direct arylation of C(sp3)–H bonds in aliphatic amides via bidentate-chelation assistance. J. Am. Chem. Soc. 136, 898–901 (2014).
Huang, D. et al. 14-electron four-coordinate Ru(II) carbyl complexes and their five-coordinate precursors: synthesis, double agostic interactions, and reactivity. J. Am. Chem. Soc. 121, 8087–8097 (1999).
Pike, S. D., Kramer, T., Rees, N. H., Macgregor, S. A. & Weller, A. S. Stoichiometric and catalytic solid−gas reactivity of rhodium bisphosphine complexes. Organometallics 34, 1487–1497 (2015).
Scott, N. M., Pons, V., Stevens, E. D., Heinekey, D. M. & Nolan, S. P. An electron-deficient iridium(III) dihydride complex capable of intramolecular C-H activation. Angew. Chem. Int. Ed. 44, 2512–2515 (2005).
Acknowledgements
The authors thank The Hong Kong Research Grants Council (153152/16P, 153023/17P, C5023-14G and HKUST16300021) for financial support. B.D and C.-M.C. are grateful for the Postdoctoral Fellowships generously supported by State Key Laboratory of Chemical Biology and Drug Discovery.
Author information
Authors and Affiliations
Contributions
B.D. and Y.O. performed the experiments. W.-Y.Y. and C.-M.C. directed the project. K.C. and Z.L. conducted the density functional theory calculations. All authors contributed to the preparation of the manuscript and the Supplementary Information.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, B., Chan, CM., Ouyang, Y. et al. NiH-catalyzed anti-Markovnikov hydroamidation of unactivated alkenes with 1,4,2-dioxazol-5-ones for the direct synthesis of N-alkyl amides. Commun Chem 5, 176 (2022). https://doi.org/10.1038/s42004-022-00791-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00791-4
This article is cited by
-
Intramolecular hydroamidation of alkenes enabling asymmetric synthesis of β-lactams via transposed NiH catalysis
Nature Catalysis (2023)
-
Ni-catalyzed highly regio- and stereo-selective diborylative cyclization of 1,6-enynes with diboron reagent
Science China Chemistry (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
