
Abstract
Natural products are well established as an important resource and play an important role in drug discovery. Here, two pyrrolinone-fused benzoazepine alkaloids, (+)-asperazepanones A (1) and B (2) with a 6/7/5 ring system, together with the artifact (−)-asperazepanone A (1), were isolated from the coral-derived Aspergillus candidus fungus. Their structures including absolute configurations were elucidated by extensive spectroscopic methods, single crystal X-ray diffraction, and ECD calculations. Furthermore, total syntheses of (±)-1 and (±)-2 have been achieved starting from the commercially L-aspartic acid diethyl ester hydrochloride and monoethyl malonate in 7 and 8 steps, respectively. The key step in the syntheses was an intramolecular Friedel-Crafts reaction to build the unique tricyclic skeleton. Interestingly, (+)-2 not only showed obviously inhibitory activity against NO production, but also inhibited potent LPS-induced expression of TNF-α and IL-6 at the concentration of 0.1 μM. It thus represents a potentially promising lead for anti-inflammatory drug discovery.
Similar content being viewed by others

Introduction
Inflammation is a natural biological response to infection and injury, directed towards eliminating harmful agents and restoration of the normal function of the affected tissue or organ1,2. Inflammatory diseases remain a major health problem worldwide. A large number of nonsteroidal anti-inflammatory drugs (NSAIDs)3,4 such as aspirin, diclofenac, and indomethacin have emerged in the past few decades, and the development of COXIBs5,6 has provided some relief to patients suffering from a wide spectrum of inflammatory diseases. However, these drugs often cause cardiovascular side effects which has subdued the medicinal applications of this class of anti-inflammatory drugs7,8,9,10. Therefore, new anti-inflammatory agents with higher efficacy and lower toxicity are urgently required.
Natural products have proven to be a reliable resource of new pharmaceutical agents11,12. Natural alkaloids and their derivatives with vast diversity of structures are a cornerstone of chemotherapeutic regimens worldwide11,13. Benzoazepines are a key structural motif in numerous pharmaceuticals and agrochemicals14,15,16. Particularly, a benzo[b]azepine moiety is a well-known pharmacophore that is present in several marketed tricyclic antidepressant drugs, including anafranil, imipramine, tienopramine, and amezepine (Fig. 1a)17. In addition, this core structure plays key roles in other biological activities including anticonvulsant, anti-HIV-1, and antiallergic properties18,19,20.

a Commercial benzoazepine drugs. b Chemical structures of (±)-1 and (+)-2.
During our ongoing search for new biologically active constituents from marine fungi, a series of bioactive natural products with antifungal, antimalarial, and antiviral activities have been discovered21,22,23,24,25. For example, plentifully functionalized p-terphenyl metabolites with attractive pharmacological activities were isolated from the gorgonian coral-derived Aspergillus candidus (CHNSCLM-0393) fungus25. The chemical constituents of the fungus were further studied under the guidance of LC-MS/MS-Based Molecular Networking and HPLC-UV-DAD, and two new benzoazepine alkaloids with an unprecedented tricyclic 6/7/5 skeleton, asperazepanones A (1) and B (2), were discovered (Fig. 1b). Herein, the isolation, structure elucidation, total syntheses, and anti-inflammatory activity of 1 and 2 are described.
Results
Structure elucidation of (±)-asperazepanone A (1) and (+)-asperazepanone B (2)
Asperazepanone A (1) was obtained as a yellow amorphous solid. Its molecular formula of C12H10N2O3 (nine degrees of unsaturation) was determined by the positive HRESIMS (m/z 231.0763 [M + H] +, calcd. 231.0764). The IR spectrum in KBr clearly suggested the presence of carbonyl group (1624 cm−1). The 1H NMR spectrum (see Supplementary Table S1) revealed these signals for three aromatic proton signals (δH 6.87, t, J = 7.9 Hz; 7.06, dd, J = 7.9, 1.2 Hz; 7.27, dd, J = 7.9, 1.2 Hz), one olefinic proton (δH 5.13, 1H, s), one methylene proton (δH 2.85, dd, J = 17.2, 13.1 Hz; 3.03, dd, J = 17.2, 2.8 Hz), and one methine proton (δH 4.51, dd, J = 13.1, 2.8 Hz). Furthermore, three exchangeable protons at δH 10.46, 9.13, and 7.22 were observed in DMSO-d6. Its 13C NMR and DEPT spectra exhibited a total of 12 carbon resonances divided into one methylene group, five methines, and six quaternary carbons.
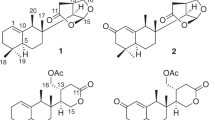
The HSQC spectrum clearly indicated the presence of a keto carbonyl (δC 195.1), and an amide (δC 173.9) in the molecule. The two carbonyl groups, and one olefinic carbon accounted for three degrees of unsaturation, the remaining six degrees of unsaturation indicated the presence of a tricyclic system in the structure of 1. The consecutive COSY correlation from H-6 to H-8, together with the HMBC correlations from aromatic H-6 (δH 7.06) and H-8 (δH 7.27) to C-4a (δC 129.5), from H-7 (δH 6.87) to C-8a (δC 125.5), and from hydroxy group signal at δH 11.49 (s) to C-5 (δC 147.2) and C-4a, indicating the presence of a disubstituted phenol ring A. The adjacent COSY correlation from H-10 (δH 2.85, dd, J = 17.2, 13.1 Hz; 3.03, dd, J = 17.2, 2.8 Hz) to H-10a (δH 4.51, dd, J = 13.1, 2.8 Hz) indicated the presence of –CH2CH– unit. The ring C was constructed on the basis of the following correlations in the COSY spectrum of H-1 (δH 7.22) to H-10a and H-3 (δH 5.13), and the HMBC correlations from H-1 to C-10a (δC 50.2), C-3 (δC 92.9), and C-3a (δC 162.8), H-3 to C-10a, and C-2 (δC 173.9), and H-10a to C-3a. The correlation of H-4 (δH 9.13) to H-10a and H-1, and HMBC correlations from H-4 to C-5 (δC 147.2), C-8a (δC 36.5), and C-10a, H-8 to C-9 (δC 195.1), C-6 (δC 118.6) and C-4a (δC 129.5), and H-10 to C-9, C-10a (δC 36.5), and C-3a, revealed the presence of a seven-membered ring B that was fused to the rings A and C. Thus, an unexpected pyrrolinone fused benzoazepine alkaloid with a tricyclic 6/7/5 skeleton was elucidated as shown in Fig. 2a.

a COSY and key HMBC correlations of 1 and 2. b X-ray structures of (±)-1 and (+)-2.
Asperazepanone B (2) was obtained as a pale yellow crystal with a molecular formula of C13H12N2O3 determined by HRESIMS (m/z 245.0917 [M + H] +, calcd. 245.0921). Both the 1H and 13C NMR data of 2 were closely related to those of 1, with the exception of the presence of an additional methyl (δH 3.16, δC 40.3). The position of the methyl was confirmed by the HMBC spectrum, which showed correlations from H3-4 to C-8a (δC 129.4), and C-3a (δC 166.4), and we could determine the planar structure of 2.
To unambiguously assign the unprecedented structure and absolute configuration, a single X-ray diffraction study was performed. However, compound 1 was initially isolated as an amorphous solid and it was difficult to get a suitable crystal for X-ray analysis, but after many attempts, X-ray quality crystals were obtained by slow evaporation of a solution of 1 in MeOH (Fig. 2b). Further analysis of the X-ray data revealed that 1 possessed a centrosymmetric space group P21/c, indicating its racemic nature (Supplementary Data 1). Fortunately, a recrystallization experiment also furnished a suitable crystal of 2 (Fig. 2b). To our surprise, Cu Kα X-ray crystallographic analysis confirmed the structure of 2 with absolute configuration of 10aS with a Flack parameter of −0.1(3) (Supplementary Data 2). And as shown in Fig. 3, the calculated ECD curve of 2 is also in agreement with the experimental one.

Experimental ECD spectra and calculated ECD spectra of (±)-1 and (+)-2.
In order to clarify the racemic mixture of 1, HPLC analysis of 1 on a chiral column was carried out. Two distinct chromatographic peaks with a ratio of 3:2 were found (Fig. 4). After chiral HPLC separation, compounds (+)-1 and (−)-1 were obtained. To determine the absolute configurations of (+)-1 and (−)-1, the experimental CD spectra and the calculated ones using the time-dependent DFT method of each enantiomer were compared. The results showed that the ECD curve of 10aS-1 was similar to that of (+)-1 and the ECD curve of 10aR-1 was similar to that of (−)-1 (Fig. 3). On the basis of the above evidence, the absolute configurations of (+)-1 and (−)-1 were established to be 10aS and 10aR, respectively.

Chiral-phase HPLC analysis of (±)-1 and (+)-2.
Differentiation and epimerization property of 1 and 2
With a systematic investigation on the structure elucidation of 1 and 2 completed, another question arose: why was compound 1 isolated as a racemate and compound 2 isolated as an optically active? Therefore, we wanted to determine whether the racemates of 1 were naturally occurring or obtained as artifacts during the isolation process. After 10 days of short-term fermentation, 1 was quickly separated by RP-HPLC and analyzed by chiral HPLC in 1 day. The results showed that (+)-1 was detected as the major one (95%; see Supplementary Fig. S7). Therefore, (+)-1 should be regarded as a true natural product, while (−)-1 is an artifact.
To explore the factors of the transformation from (+)-1 to (−)-1, the following experiments were carried out (see Supplementary Figs. S8 and S9). To our surprise, when (+)-1 was treated with 0.01% NaOH, 10% acetic acid, or 2% trifluoroacetic acid (TFA) in MeOH at room temperature, respectively, (+)-1 could be partially transformed into (–)-1 accompanied with its dehydrogenation product 3 (Fig. 5a). As we expected, 2 was relatively stable under the above conditions (see Supplementary Fig. S10). After eight months of fermentation, the pH value of the medium was close to 9, so we deduced that the racemization of (+)-1 was caused by long-time fermentation. Next, we deduced the mechanism of racemic reaction of 1 (Fig. 5b). At the beginning, enamine-imino tautomerism26 of (+)-1 might provide the intermediate A, which immediately underwent a second imino-enamine tautomerism to produce the intermediate B. Finally, it further underwent a racemization reaction to deliver (−)-1. As for 2, due to the lack of active hydrogen on the nitrogen atom, the corresponding intermediate A cannot be formed.

a The transformation from (+)-10aS-1 to (–)-10aR-1 and 3 in MeOH with 0.01% NaOH. b The plausible mechanism of racemization for 1.
Plausible biosynthetic pathway to 1 and 2
Structurally, compounds 1 and 2 are unprecedented benzoazepine alkaloids with a tricyclic 6/7/5 skeleton, which is the first examples of benzoazepine alkaloid, generating by the fusion of a benzoazepine scaffold with a pyrrolinone moiety. Plausible biogenetic pathways for 1 and 2 are postulated in Fig. 6. As shown, the unique biogenetic route could be plausibly traced back to L-tryptophan. L-Tryptophan has been known as a precursor of 3-hydroxykynurenine27,28. Then, 3-hydroxykynurenine reacted with acetyl-CoA to form intermediate A. Further, the intermediate A underwent an intramolecular nucleophilic addition to give the bicyclic intermediate B. Finally, (+)-1 was produced by the two-step dehydration of intermediate B. (+)-2 was further obtained by the methylation of (+)-1.

Plausible biosynthetic pathway to 1 and 2.
Retrosynthetic analysis of (±)-asperazepanones A (1) and B (2)
Benzoazepines are frequently encountered in pharmaceuticals and exhibit significant biological activities, and thus, they have attracted considerable interest from the synthetic community29,30,31,32. Therefore, the total syntheses of 1 and 2 were conducted. The retrosynthetic analysis for the total syntheses of 1 and 2 are delineated in Fig. 7. We assumed that 1 and 2 might be assembled from acid 4 through an intramolecular Friedel-Crafts reaction33. For the construction of the latter, the nucleophilic addition34 of the aromatic amine 5 to the 3-carbonyl of tetramic acid 6 was projected as the means to forge the skeleton of 4. The 3-carbonyl of tetramic acid 6 would be obtained through the decarboxylation of the substituted tetramic acid 7. Finally, the core of 7 could be constructed via Dieckmann cyclization35 of amidation intermediate, which could be derived from the commercially available L-aspartic acid diethyl ester hydrochloride (8) and monoethyl malonate (9).

Retrosynthesis of asperazepanones A (1) and B (2).
Total syntheses of (±)-asperazepanones A (1) and B (2)
As depicted in Fig. 8, our synthetic study began with the preparation of amide 10. Monoethyl malonate (9) reacted with commercially available L-aspartic acid diethyl ester hydrochloride (8) in the presence of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI), diisopropylethylamine, and a catalytic amount of 4-dimethylaminopyridine to provide amide 10 in 90% yield. Next, Dieckmann cyclization of the optical amide 10 delivered the racemic tetramic acid 7 in 70% yield. Not surprisingly, although the Dieckmann cyclization strategy is a convenient way of preparing tetramic acids, it is accompanied by non-negligible epimerization at position C-536,37. The attempt to obtain the desired 3-carbonyl of tetramic acid 6 through decarboxylation reaction of 7 was unsuccessful according to the literature procedure34. Fortunately, refluxing 7 in a mixture of acetonitrile and TFA resulted in decarboxylation and afforded the 3-carbonyl of tetramic acid 6.

Reagents and conditions: a DMAP, DIPEA, EDCI, CH2Cl2, 0 °C to RT, 12 h, 90%; b LiHMDS, THF, −20 °C to RT, 6 h, 70%; c TFA/MeCN (1: 10), reflux, 8 h; d 5, AcOH/EtOH (1: 10), 70 °C, 12 h, 30% over two steps; e LiOH, MeOH/H2O (1: 1), RT, 2 h, 85%. DMAP 4-dimethylaminopyridine, DIPEA diisopropylethylamine, EDCI 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, LiHMDS lithium hexamethyldisilazide. TFA trifluoroacetic acid, THF tetrahydrofuran, RT room temperature.
Without purification, the nucleophilic addition reaction of 6 was then carried out. Gratifyingly, by treating 5 and 6 with acetic acid in ethanol at 70 °C, the adduct 11 was obtained in 30% yield over two steps. Enantioselective synthesis of 11 starting from enantiomerically pure amino acids was not pursued because of reported partial racemization under the cyclization conditions36. With the successful synthesis of 11, we turned our attention to the construction of the unusual 6/7/5 ring system. After hydrolysis of 11, the resulting acid 4 was ready for the intramolecular Friedel–Crafts reaction.
Intramolecular Friedel−Crafts cyclization was found to be the crucial step in these syntheses. As summarized in Table 1, initial attempts to carry out the intramolecular Friedel−Crafts reaction of 4 using TFAA as cyclization reagent were unsuccessful, only byproduct 12a was obtained (entry 1). On the basis of the above experiments, different reagents (SOCl2/AlCl3, BF3·Et2O, POCl3, H2SO4, and PPA) were screened at this stage in different reaction conditions. Our various attempts to obtain the desired 12 through this coupling reaction, employing different solvents and Lewis acid, did not succeed (entries 2–5). Fortunately, upon treatment of 4 with PPA at 80 °C, we were able to successfully isolate the desired 12 in 3% yield (entry 6). The highest yield (22%) of 12 was obtained with PPA at 120 °C for 1 h (entry 8).
With the key intermediate 12 in hand, we now move to the stage for the completion of the total synthesis (Fig. 9). Finally, treatment of 12 with BBr3 in CH2Cl2 at room temperature furnished (±)-asperazepanone A (1) in 74% yield. Subsequent methylation of 12 and BBr3-mediated phenol demethylation under standard conditions furnished (±)-asperazepanone B (2) in 68% yield over two steps. The spectral and physical data of fully synthetic 1 and 2, including 1H NMR, 13C NMR, and HRMS data, matched those of naturally occurring 1 and 2 (see Supplementary Note 2, Tables S2 and S3, Figs. S25 and S26). (±)-Asperazepanone B (2) was resolved into the corresponding pure enantiomers (+)-2 and (−)-2 by HPLC using a Chiralpak IC chiral-phase column.

Reagents and conditions: a PPA, 120 °C, 1 h, 22%; b BBr3, CH2Cl2, RT, 2 h, 74%; c CH3I, Cs2CO3, acetone, RT, 6 h; d BBr3, CH2Cl2, RT, 2 h, 68% over two steps. PPA polyphosphoric acid, RT room temperature.
The anti-inflammatory activity of (±)-asperazepanone A (1), (+)- and (−)-asperazepanone B (2)
A macrophage stimulation assay is often used as a validated screening tool for anti-inflammatory compounds38,39. Excess nitric oxide (NO) generation is a sign of the inflammatory response40. Pro-inflammatory cytokines, such as IL-6 and TNF-α, have been recognized as critical regulators in inflammatory diseases41 such as rheumatoid arthritis42, inflammatory bowel disease43, asthma44, and diabetic complications45. Inhibiting the release of pro-inflammatory cytokines is an important mode of action for anti-inflammatory drugs46.
In current research, the effects of compounds (±)-1, (+)-2, and (−)-2 in vitro in lipopolysaccharide (LPS)-stimulated RAW 264.7 cells were evaluated (Fig. 10). Only (+)-2 did not affect the cell viability and showed obviously inhibitory activity against nitric oxide (NO) production with inhibition rate of 43 ± 4% at the concentration of 1 μM. However, Other compounds (±)-1 and (−)-2 did not show any inhibitory activity. L-NMMA was used as the positive control with the rate of 61 ± 11% at the concentration of 5 μM. To further measure the cytokine release level, pro-inflammatory cytokines TNF-α and IL-6 were detected by ELISA. The levels of TNF-α (p < 0.0001) and IL-6 (p < 0.01) were significantly decreased with 40 ± 2% and 77 ± 7% inhibition rates, respectively, by (+)-2 at the concentration of 0.1 μM (Fig. 10) without affecting cell viabilities (See Supplementary Note 3 Fig. S60). Very interestingly, there is obvious difference in the anti-inflammatory activity of each enantiomer of asperazepanone B (2). The results revealed that (+)-2 with S configuration exhibited potent inhibitory activity with these two cytokines, while (−)-2 with R configuration was inactive. The preliminary structure-activity relationships indicated that the absolute configuration of (S)-2 plays a key role in the observed anti-inflammatory activity. Moreover, (±)-(1) exhibited no inhibitory activity suggesting that the methyl group at the N-4 is also crucial to the anti-inflammatory activity. Our results suggest that the novel pyrrolinone-fused benzoazepine alkaloid, (+)-asperazepanone B (2) deserves further investigations as potential anti-inflammatory agents.

The cells were treated with the different concentrations and LPS (1 μg/mL) for 24 h. Data are presented as the mean of three experiments ± SD. ###p < 0.001, ####p < 0.0001 compared to the control group; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to the LPS-treated group.
Conclusion
In summary, we have isolated and characterized two novel natural products, (+)-asperazepanones A (1) and B (2), from the gorgonian derived fungus Aspergillus candidus. The artifact (−)-asperazepanone A (1) was confirmed and its transformation mechanism was also studied. Moreover, the total syntheses of (±)-1 and (±)-2 featuring intramolecular Friedel-Crafts reaction to construct the tricyclic 6/7/5 architecture were also accomplished. This synthetic process should provide routes for obtaining synthetic analogs. Biological studies revealed that (+)-asperazepanone B (2) showed potent anti-inflammatory activity, which confirmed it as promising new lead for developing anti-inflammatory agent. Further structural optimization, biological evaluation, and mechanism of action are ongoing and will be reported in due course.
Materials and methods
General
All reactions were performed under nitrogen or argon in vacuum-dried glassware using dry solvents and standard syringe techniques. Reagents were purchased at the highest commercial quality (>95%) and used without further purification, unless otherwise stated. Anhydrous tetrahydrofuran (THF) was distilled from sodium-benzophenone, dichloromethane (CH2Cl2) was distilled from calcium hydride. Column chromatography was carried out by using silica gel (200–300 mesh). Yields refer to chromatographically, unless otherwise specified. Optical rotations were measured on a JASCO P-1020 digital polarimeter (JASCO Ltd., Tokyo, Japan). ECD spectra were acquired on a JASCO J-715 (JASCO) or Chirascan CD (Applied Photophysics) spectropolarimeter. IR spectra were obtained on a Nicolet Nexus 470 spectrophotometer (Perkin Elmer Ltd., Boston, MA, USA) in KBr discs. NMR spectra were recorded on a JEOL JEM-ECP NMR spectrometer (JEOL Ltd., Tokyo, Japan; 500 MHz for 1H and 125 MHz for 13C), using TMS as an internal standard. HRESIMS spectra were performed on a Thermo Scientific LTQ Orbitrap XL spectrometer. Single-crystal data were obtained on an Agilent Gemini Ultra diffractometer (Cu Kα radiation) (Agilent Technologies Inc., California, America). HPLC analysis was performed on a Hitachi L-2000 system (Hitachi Ltd., Tokyo, Japan) using a C18 column [(YMC Co., Ltd., Tokyo, Japan) YMC-Park, ODS-A, 250 × 4.6 mm, S-5 μm, 12 nm, 1.0 mL/min]. Semi-preparative HPLC was performed on a Hitachi L-2000 system (Hitachi Ltd., Tokyo, Japan) using a C18 column [(Eka Ltd., Bohus, Sweden) Kromasil 250 × 10 mm, 5 μm, 2.0 mL/min]. Racemic mixtures were resolved on a Chiralpak IC column (5 μm, 4.6 × 150 mm, hexane−ethanol eluent, 0.6 mL/min) and Chiralpak IA column (5 μm, 4.6 × 250 mm, hexane–ethanol eluent, 0.6 mL/min). Silica gel ((Qingdao Haiyang Chemical Group Co., Qingdao, China; 200–300 mesh), octadecylsilyl silica gel (YMC Co., Ltd., Tokyo, Japan; 45–60 μm), macroporous resin (H&E Co., Ltd., Connecticut, America) and Sephadex LH-20 (GE Ltd., Connecticut, America) were used for column chromatography. Precoated silica gel plates (Yantai Zhifu Chemical Group Co., Yantai, China; G60, F-254) were used for thin layer chromatography.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the article and its supplementary information files, and from the corresponding author on request. For 1H and 13C NMR spectra of compounds, see Supplementary Note 1 Figs. S11–S24 and Supplementary Note 2 Figs. S46–S59. For the spectroscopic and physical data of compounds, see Supplementary Note 1 Figs. S1–S6 and Supplementary Note 2 Figs. S27–S45. The X-ray crystallographic coordinates for the structure of 1 and 2 have been deposited at the Cambridge Crystallographic Data Center (CCDC), under deposition number 2143832 and 2143833. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. Other data are available from the authors upon reasonable request. The CIF files of CCDC 2143832 and CCDC 2143833 are also included as Supplementary Data 1 and 2.
References
Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 140, 771–776 (2010).
Kishore, N., Kumar, P., Shanker, K. & Verma, A. K. Human disorders associated with inflammation and the evolving role of natural products to overcome. Eur. J. Med. Chem. 179, 272–309 (2019).
Patrono, C. & Baigent, C. Low-dose aspirin, coxibs, and other NSAIDS: A clinical mosaic emerges. Mol. Interventions 9, 31–39 (2009).
Skoutakis, V. A. et al. Review of diclofenac and evaluation of its place in therapy as a nonsteroidal antiinflammatory agent. Drug Intell. Clin. Pharm. 22, 850–859 (1988).
Shi, S. & Klotz, U. Clinical use and pharmacological properties of selective COX-2 inhibitors. Eur. J. Clin. Pharmacol. 64, 233–252 (2008).
Moore, R. A., Derry, S., Phillips, C. J. & McQuay, H. J. Nonsteroidal anti-inflammatory drugs (NSAIDs), cyxlooxygenase-2 selective inhibitors (coxibs) and gastrointestinal harm: review of clinical trials and clinical practice. BMC Musculoskelet. Disord. 7, 79–92 (2006).
Dogne, J. M., Supuran, C. T. & Pratico, D. Adverse cardiovascular effects of the coxibs. J. Med. Chem. 48, 2251–2257 (2005).
White, W. B. et al. Risk of cardiovascular events in patients receiving celecoxib: A meta-analysis of randomized clinical trials. Am. J. Cardiol. 99, 91–98 (2007).
Solomon, S. D. et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 352, 1071–1080 (2005).
Cairns, J. A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: A current perspective on cardiovascular risks. Can. J. Cardiol. 23, 125–131 (2007).
Newman, D. J. & Cragg, G. M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83, 770–803 (2020).
Clardy, J. & Walsh, C. Lessons from natural molecules. Nature 432, 829–837 (2004).
Kochanowska-Karamyan, A. J. & Hamann, M. T. Marine indole alkaloids: Potential new drug leads for the control of depression and anxiety. Chem. Rev. 110, 4489–4497 (2010).
Kondo, K. et al. Novel design of nonpeptide AVP V2 receptor agonists: Structural requirements for an agonist having 1-(4-aminobenzoyl)-2,3,4,5-tetrahydro-1H-1-benzazepine as a template. J. Med. Chem. 43, 4388–4397 (2000).
Diguarher, T. L. et al. Synthesis of N, N′-disubstituted 3-aminobenzo [c] and [d] azepin-2-ones as potent and specific farnesyl transferase inhibitors. Bioorg. Med. Chem. Lett. 14, 767–771 (2004).
Failli, A. A. et al. Pyridobenzodiazepines: A novel class of orally active, vasopressin V2 receptor selective agonists. Bioorg. Med. Chem. Lett. 16, 954–959 (2006).
Singh, H., Gupta, N., Kumar, P., Dubey, S. K. & Sharma, P. K. A new industrial process for 10-methoxyiminostilbene: Key intermediate for the synthesis of oxcarbazepine. Org. Process Res. Dev. 13, 870–874 (2009).
Shalini, M., Yogeeswari, P., Sriram, D. & Stables, J. P. Cyclization of the semicarbazone template of aryl semicarbazones: Synthesis and anticonvulsant activity of 4, 5-diphenyl-2H-1, 2, 4-triazol-3 (4H)-one. Biomed. Pharmacother. 63, 187–193 (2009).
Seto, M. et al. Orally active CCR5 antagonists as anti-HIV-1 agents. Part 3: Synthesis and biological activities of 1- benzazepine derivatives containing a sulfoxide moiety. Bioorg. Med. Chem. 13, 363–386 (2005).
Roszkowski, P., Maurin, J. K. & Czarnocki, Z. The enantioselective synthesis of (S)-(+)-mianserin and (S)-(+)-epinastine. Beilstein J. Org. Chem. 11, 1509–1513 (2005).
Guo, F. W. et al. Scalable total synthesis of (+)-aniduquinolone A and its acid-catalyzed rearrangement to aflaquinolones. Commun. Chem. 5, 35 (2022).
Hai, Y. et al. Trends of antimalarial marine natural products: progresses, challenges, and opportunities. Nat. Prod. Rep. 39, 969–990 (2022).
Hai, Y., Wei, M. Y., Wang, C. Y., Gu, Y. C. & Shao, C. L. The intriguing chemistry and biology of sulfur-containing natural products from marine microorganisms (1987–2020). Mar. Life. Sci. Tech. 3, 488–518 (2021).
Hou, X. M., Liang, T. M., Guo, Z. Y., Wang, C. Y. & Shao, C. L. Discovery, absolute assignments, and total synthesis of asperversiamides A–C and their potent activity against Mycobacterium marinum. Chem. Commun. 55, 1104–1107 (2019).
Zhang, X. Q. et al. Design, semisynthesis, α-glucosidase inhibitory, cytotoxic, and antibacterial activities of p-terphenyl derivatives. Eur. J. Med. Chem. 146, 232–244 (2018).
Mukano, Y. et al. Imino–enamine tautomerism and dynamic prototropy in 1-imino-3-amino-1H-indens. Tetrahedron 66, 605–611 (2010).
Sanna, D. et al. Role of ligands in the uptake and reduction of V(V) complexes in red blood cells. J. Med. Chem. 62, 654–664 (2019).
Tran, T. D., Pham, N. B., Ekins, M., Hooper, J. N. & Quinn, R. J. Isolation and Total Synthesis of Stolonines A-C, Unique Taurine Amides from the Australian Marine Tunicate Cnemidocarpa stolonifera. Mar. Drugs 13, 4556–4575 (2015).
Reamtong, O. et al. Synthesis of benzoazepine derivatives via azide rearrangement and evaluation of their antianxiety activities. ACS Med. Chem. Lett. 12, 1449–1458 (2021).
Zhang, K., Cai, L., Hong, S. & Kwon, O. Phosphine-catalyzed α-umpolung–aldol reaction for the synthesis of benzo [b] azapin-3-ones. Org. Lett. 21, 5143–5146 (2019).
Wales, S. M. et al. Benzoazepine-fused isoindolines via intramolecular (3+2)-cycloadditions of azomethine ylides with dinitroarenes. Org. Lett. 21, 4703–4708 (2019).
Stockerl, S., Danelzik, T., Piekarski, D. G. & Mancheno, O. G. Mild, metal-free oxidative ring-expansion approach for the synthesis of benzo[b]azepines. Org. Lett. 21, 4535–4539 (2019).
Gore, P. H. The Friedel–Crafts acylation reaction and its application to polycyclic aromatic hydrocarbons. Chem. Rev. 55, 229–281 (1955).
Liu, Y. X., Zhao, H. P., Song, H. B., Gu, Y. C. & Wang, Q. M. Studies on the synthesis and bioactivities of 4‐amino derivatives of tetramic acid. J. Heterocycl. Chem. 51, E25–E33 (2014).
Davis, B. R. & Garratt, P. J. Acylation of esters, ketones, and nitriles. Comp. Org. Syn. 2, 795–863 (1991).
Peukert, S. et al. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): Tetramic, tetronic acids and dihydropyridin-2-ones. Bioorg. Med. Chem. Lett. 18, 1840–1844 (2008).
Poncet, J. et al. Tetramic acid chemistry. Part I. Reinvestigation of racemisation during the synthesis of tetramic acids via Dieckmann cyclisation. J. Chem. Soc., Perkin Trans. 1, 611–616 (1990).
Buscher, K. et al. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat. Commun. 8, 1–10 (2017).
Shapouri‐Moghaddam, A. et al. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 233, 6425–6440 (2018).
Inamdar, A. A. & Bennett, J. W. A common fungal volatile organic compound induces a nitric oxide mediated inflammatory response in Drosophila melanogaster. Sci. Rep. 4, 1–9 (2014).
Hanada, T. & Yoshimura, A. Regulation of cytokine signaling and inflammation. Cytokine Growth F. R. 13, 413–421 (2002).
Xing, Z. et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Invest. 101, 311–320 (1998).
Kolesov, S. A. et al. S-nitrosothiols, nitric oxide and proinflammatory cytokines in children with inflammatory bowel disease. Clin. Lab. 59, 953–957 (2013).
Brown, P. H., Crompton, G. K. & Greening, A. P. Proinflammatory cytokines in acute asthma. Lancet 338, 590–593 (1991).
Jeffcoate, W. J., Game, F. & Cavanagh, P. R. The role of proinflammatory cytokines in the cause of neuropathic osteoarthropathy (acute Charcot foot) in diabetes. Lancet 366, 2058–2061 (2005).
Fang, B. et al. Synthesis and anti-inflammatory evaluation of (R)-, (S)-, and (±)-sanjuanolide isolated from Dalea frutescens. J. Nat. Prod. 82, 748–755 (2019).
Acknowledgements
We thank Syngenta for the fellowship to Feng-Wei Guo. This work was supported by the Fundamental Research Funds for the Central Universities (No. 202264001), the Program of National Natural Science Foundation of China (Nos. U1706210 and 41322037), AoShan Talents Program Supported by Pilot National Laboratory for Marine Science and Technology (Qingdao) (No. 2015ASTP-ES11), Key Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education, Hainan Normal University (Nos. RDZH2021003 and RDZH2022002), the Program of Natural Science Foundation of Shandong Province of China (No. JQ201510), and the Taishan Scholars Program, China (No. tsqn20161010).
Author information
Authors and Affiliations
Contributions
C.L.S. supervised and designed the project. L.X., F.W.G., X.Q.Z., and C.J.W. performed chemical experiments. T.Y.Z. contributed to biological experiments. L.X., F.W.G., X.Q.Z., and C.L.S. co-wrote the manuscript. C.L.S., L.X., F.W.G., M.Y.W., Y.C.G., and C.Y.W. discussed the results and assisted in the preparation of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, L., Guo, FW., Zhang, XQ. et al. Discovery, total syntheses and potent anti-inflammatory activity of pyrrolinone-fused benzoazepine alkaloids Asperazepanones A and B from Aspergillus candidus. Commun Chem 5, 80 (2022). https://doi.org/10.1038/s42004-022-00696-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00696-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
