
Abstract
Despite its electron deficiency, boron can form multiple bonds with a variety of elements. However, multiple bonds between boron and main-group metal elements are relatively rare. Here we report the observation of boron-lead multiple bonds in PbB2O– and PbB3O2–, which are produced and characterized in a cluster beam. PbB2O– is found to have an open-shell linear structure, in which the bond order of B☱Pb is 2.5, while the closed-shell [Pb≡B–B≡O]2– contains a B≡Pb triple bond. PbB3O2– is shown to have a Y-shaped structure with a terminal B = Pb double bond coordinated by two boronyl ligands. Comparison between [Pb≡B–B≡O]2–/[Pb=B(B≡O)2]– and the isoelectronic [Pb≡B–C≡O]–/[Pb=B(C≡O)2]+ carbonyl counterparts further reveals transition-metal-like behaviors for the central B atoms. Additional theoretical studies show that Ge and Sn can form similar boron species as Pb, suggesting the possibilities to synthesize new compounds containing multiple boron bonds with heavy group-14 elements.
Similar content being viewed by others
Introduction
Due to its electron deficiency, boron tends to form multicenter bonds in both its compounds and at nanoscales1,2,3,4,5. Boron is also capable of forming multiple chemical bonds with transition metals, such as in borylene (:BR) compounds, which usually involve a transition metal (M) with different ligands (Ln), LnMBR6. The bonding between the metal and borylene fragment is interpreted as B→M σ-donation and M(dπ)→B back-donation7,8,9,10. The similarities between borylenes and carbenes (:CR2) suggest the bonding between boron and metal should be a double bond. However, the B–M bond lengths vary in a wide range, depending on the ligands and the R group11. Since the syntheses of the first transition-metal borylene complexes12,13, considerable progresses have been achieved in this area14,15,16,17,18,19,20,21,22,23,24. Boron-metal triple-bond characters were first suggested in [(OC)5CrBSiH3] with the B–Cr bond length of 1.871 Å25, slightly shorter than the B=Cr double-bond length of 1.89 Å derived from Pyykkö’s covalent radii26. Several transition-metal complexes with B–M bond lengths shorter than B=M double bonds have been characterized in both solid compounds and gaseous molecules15,16,17,18,19, among which some have B–M bond lengths comparable to those computed from Pyykkö’s triple-bond covalent radii. The first electron-precise transition-metal-boron triple-bond complex was identified recently by combined photoelectron spectroscopy (PES) and quantum chemistry calculations, in the linear ReB2O– species with a B≡Re triple bond27. In fact, complexes with transition-metal-boron bond lengths shorter than B≡M triple bonds28,29 and even B≣M quadruple bonds30,31 have been characterized by joint gas-phase experimental and ab initio theoretical studies.
Compared with the transition-metal-boron multiple-bond complexes, compounds with multiple bonding between boron and main-group metal elements are rare24, even though multiple bonds of boron with light main-group elements are common. This is understandable because main-group metal elements have valence ns and np orbitals with large differences in orbital radii, decreasing the hybridization of these orbitals and making it difficult for heavy elements to form strong multiple bonds. The first molecules observed to contain main-group-metal-boron multiple bonds are the linear Bi2B– and BiB2O– species32, featuring two B=Bi double bonds in Bi2B– and a B≡Bi triple bond in BiB2O–. Besides these, boron has only been found to form double bonds with heavy main-group elements24,33,34,35.
Here we report the observation of B–Pb multiple bonds in two molecular anions, PbB2O– and PbB3O2–, by a joint PES and theoretical study. Well-resolved photoelectron spectra were obtained for these two species in the gas phase and used to elucidate their structures and bonding. Theoretical calculations and chemical bonding analyses showed that PbB2O– has an open-shell [Pb☱B–B≡O]– linear structure with a B–Pb bond order of 2.5, whereas the closed-shell [Pb≡B–B≡O]2– contains a B≡Pb triple bond. The PbB3O2– species has a Y-shaped structure, [Pb=B(B≡O)2]–, which consists of a B = Pb double bond coordinated by two boronyl ligands. Comparisons of the bonding in [Pb≡B–B≡O]2– and [Pb=B(B≡O)2]– with that in [Pb≡B−C≡O]– and [Pb=B(C≡O)2]+ also provide evidence for transition-metal-like properties for the central B atom.
Results and discussion
The PES of PbB2O–
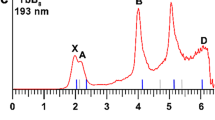
The photoelectron spectra of PbB2O– at three different photon energies are shown in Fig. 1. The spectrum at 355 nm revealed four detachment bands labeled as X, A, B, and C (Fig. 1a). The lowest binding energy band X corresponds to the detachment transition from the ground state of PbB2O– to that of neutral PbB2O, whereas the higher binding energy bands represent detachment transitions to excited states of neutral PbB2O. Band X yielded the first vertical detachment energy (VDE) of 2.26 eV and an adiabatic detachment energy (ADE) of 2.19 eV evaluated from its onset, which also represents the electron affinity (EA) of neutral PbB2O. Band A was observed at 2.40 eV with a short vibrational progression with the frequency of 890 cm−1. Band B consisting of a single peak was observed at 2.71 eV. Two weak features labeled as C were resolved near the detachment threshold at 355 nm and they turned out to be part of a broad vibrational progression fully observed at 266 nm (Fig. 1b). The ADE and VDE for band C were measured to be 3.20 eV and 3.34 eV, respectively, and the vibrational progression yielded a frequency of 970 cm−1. The 266 nm spectrum also displayed a sharp peak D at 4.04 eV, closely followed by two weak peaks E (at 4.13 eV) and F (at 4.18 eV). At 193 nm (Fig. 1c), a new peak F was observed at 4.95 eV, beyond which the signal-to-noise ratios were poor and no additional PES bands could be definitively identified. The VDEs of all the observed PES bands are given in Table 1, where they are compared with theoretical results.

a At 355 nm. b At 266 nm. c At 193 nm. The vertical lines represent resolved vibrational progressions.
The PES of PbB3O2 –
The PES spectra of PbB3O2– (Fig. 2) displayed a much simpler pattern compared to that of PbB2O–. The 355 nm spectrum revealed one PES band X with partially resolved vibrational structures (Fig. 2a). Band X gave rise to an ADE of 2.96 eV and a VDE of 3.00 eV. Two vibrational progressions were discernible for band X with a high-frequency mode of 2060 cm−1 and a low-frequency mode of ~350 cm−1. Following a large energy gap, a slightly broader band A at 4.07 eV was observed at 266 nm (Fig. 2b). No higher binding energy detachment transitions were observed in the 193 nm spectrum (Supplementary Fig. 1). The observed spectroscopic data for PbB3O2– are given in Table 2, where they are compared with the corresponding theoretical results.

a At 355 nm. b At 266 nm. The vertical lines represent resolved vibrational progressions.
Theoretical results
Figure 3 depicts the global minimum (GM) structures of PbB2O–, PbB2O2–, PbB2O, PbB3O2–, and PbB3O2 at the CCSD/AVTZ level of theory36,37,38,39, with alternative low-lying isomers within 2.5 eV at the PBE0/AVTZ level40 collectively shown in Supplementary Figs. 2, 3. The GM of PbB2O– possesses a highly stable linear structure (C∞v, 2Π), which lies 0.88 eV lower in energy than the second lowest-lying isomer PbB2O– (Cs, 4A”) at the PBE0/AVTZ level. It consists of a short B–Pb bond (rB–Pb = 2.122 Å) with a BO ligand coordinated to the central B atom (Fig. 3a), similar to the previously reported linear BiB2O– and ReB2O– systems27,32. The spin-orbit (SO) coupling effect41 was evaluated for PbB2O– using multi-reference configuration interaction (MRCI) calculations42. The SO coupling splits the 2Π state (electron configuration: 1σ22σ23σ24σ21π45σ22π3) into two sub-levels 2Π3/2 and 2Π1/2, with 2Π3/2 being lower in energy by 0.27 eV. The strong SO coupling quenches the Renner–Teller effect in the linear monoanion. Adding one electron to PbB2O– results in the closed-shell PbB2O2– (C∞v, 1Σ+), which has an even shorter B–Pb bond length (rB–Pb = 2.107 Å) (Fig. 3b). Removing an electron from PbB2O– leads to the triplet ground state of neutral PbB2O (C∞v, 3Σ–), as shown Fig. 3c.

a PbB2O–. b PbB2O2–. c PbB2O. d PbB3O2–. e PbB3O2. Bond lengths and bond angles are given in Å and degrees, respectively, at the CCSD/AVTZ level of theory. Cartesian coordinates of these structures are given in Supplementary Table 2.
PbB3O2– was found to have a closed-shell Y-shaped GM (C2v, 1A1) featuring a terminal Pb with two BO units coordinated to the central B atom (Fig. 3d). It is 0.53 eV more stable than the second lowest-lying triplet isomer (C2v, 3A2) at the PBE0/AVTZ level (Supplementary Fig. 3a). The GM of neutral PbB3O2 also possesses a similar Y-shaped structure (C2v, 2B1) with the second isomer lying 0.61 eV higher in energy at the PBE0 level/AVTZ (Fig. 3e). The valence molecular orbitals (MOs) of PbB2O– (C∞v, 2Π) and PbB3O2– (C2v, 1A1) are shown in Supplementary Fig. 4.
Comparison between the experimental and computational results
Detaching one electron from the 2π SOMO of PbB2O– (Supplementary Fig. 4a) results in three final states in PbB2O, 3Σ–, 1Δ, and 1Σ+, among which the triplet 3Σ– state with a configuration of 1σ22σ23σ24σ21π45σ22π2 is the ground state. The computed VDE/ADE of 2.22/2.19 eV at CCSD(T)/AVTZ are in excellent agreement with the experimental values at 2.26/2.19 eV. To interpret the higher energy VDEs, we computed the term values of the neutral excited states relative to the triplet ground state using the SA-CASSCF method with the MRCI interaction and SO coupling effects considered simultaneously. In consequence, the triplet 3Σ– ground state splits into three closely spaced states 3Σ–0, 3Σ––1 and 3Σ–1 with the calculated VDEs of 2.22, 2.34, and 2.34 eV, respectively, in excellent agreement with the observed X and A bands (Table 1). The weak vibrational peak with a spacing of 890 cm–1 observed for band A is due to the B–B stretching mode (ν4) with a computed frequency of 1019 cm–1 (Supplementary Fig. 5a). Franck-Condon simulations suggest that there is unresolved low-frequency Pb–B stretching vibration in band X (Supplementary Fig. 6). The next two singlet final states 1Δ2 and 1Σ+0 with calculated VDEs of 2.73 and 3.17 eV agree well with bands B and C at 2.71 eV and 3.34 eV, respectively. The observed frequency of 970 cm−1 for the vibrational progression resolved for band C is likely due to the B–B stretching mode (ν4), which should have a similar frequency as the 3Σ– ground state (Supplementary Fig. 5). Detachment of one β-electron from the 5σ orbital (HOMO-1, Supplementary Fig. 4a) results in the 3Π neutral state, which gives rise to three SO states, 3Π2, 3Π1, and 3Π0, with calculated VDEs of 4.01, 4.13, and 4.33 eV, respectively, in agreement with the observed bands D at 4.04 eV, E at 4.13 eV, and F at 4.18 eV (Table 1). Removing one α-electron from the 5σ orbital gives rise to the 1Π final state with a calculated VDE of 5.07 eV, which accounts for band G at 4.95 eV. Overall, the theoretical results are in excellent agreement with the experiment, confirming the linear GM for PbB2O–.
For PbB3O2–, electron detachment from the 2b1 HOMO (Supplementary Fig. 4b) yields the 2B1 ground state of neutral PbB3O2. The calculated VDE/ADE of 3.06/3.01 eV at the CCSD(T)/AVTZ level are in good accord with the experimental VDE/ADE of 3.00/2.96 eV. The low-frequency vibrational progression (350 cm–1) corresponds to the B–Pb stretching mode with a calculated frequency of 318 cm–1 (a1 mode) for neutral PbB3O2 (C2v, 2B1), while the high-frequency mode (2060 cm−1) originates from the B–O stretching mode with a calculated frequency of 2026 cm–1 (a1 mode) (Supplementary Fig. 5b), as confirmed by the Franck-Condon simulations (Supplementary Fig. 7 and Supplementary Table 1). It should be noted that the observed frequency of 2060 cm–1 in PbB3O2 is similar to the previously reported B≡O symmetric stretching frequencies of 1950, 2040, 1980, and 1935 cm–1 in B3O2, B4O2, B4O3, and the bare BO, respectively43,44,45. The observed active vibrational modes are consistent with the geometry changes from the ground state of the anion to that of the neutral (Fig. 3) and the nature of the 2b1 HOMO that mainly involves B–Pb π bonding and weak B–O antibonding interactions (Supplementary Fig. 4b). The second VDE derived from electron detachment from the 6a1 HOMO-1 was calculated to be 3.84 and 3.97 eV at the PBE0 and CCSD(T) levels, respectively, consistent with band A at 4.07 eV. The 6a1 HOMO-1 is a strong B–Pb σ bonding orbital, consistent with the broad band A, which should contain an unresolved B–Pb stretching vibrational progression. There is a large energy gap between HOMO-2 and HOMO-1 (3.04 eV at PBE0/AVTZ level), agreeing well with the 193 nm spectrum which does not reveal any new spectral features between 4.5 and 6.4 eV (Supplementary Fig. 1). The excellent agreement between the theoretical and experimental results confirms firmly the Y-shaped GM for PbB3O2–.
Multiple B–Pb bonding in PbB2O–/2– and PbB3O2 –
Adaptive natural density partitioning (AdNDP)46 analyses were performed on PbB2O–, PbB2O2–, and PbB3O2– to understand the nature of their chemical bonding. AdNDP basically transforms the MOs into more familiar bonding elements, such as lone pairs, two-center two-electron (2c-2e) bonds or multicenter delocalized bonds. As shown in Fig. 4a, the linear PbB2O– possesses one Pb 6 s and one O lone pairs and a B≡O triple bond in the first row. The second row depicts one 2c-2e B–B σ bond, one 2c-2e B–Pb σ bond, one 2c-2e B–Pb π bond, and one 2c-1e B–Pb π bond, giving rise to a B–Pb bond order of 2.5. Thus, there is no sp hybridization in Pb, which uses its three valence 6p orbitals to form a triple bond with the central B atom. Both B atoms undergo sp hybridization and form triple bonds. The B–Pb bond length of 2.122 Å lies between the B=Pb double-bond length (2.13 Å) and the B≡Pb triple-bond length (2.10 Å) predicted from Pyykkö’s covalent atomic radii25, consistent with the 2.5 bond order for the B–Pb multiple bond. Hence, the linear PbB2O– species can be described as [Pb☱B–B≡O]–. Adding one electron to PbB2O– yields the closed-shell and electron-precise PbB2O2– which contains an ideal Pb≡B triple bond including one 2c-2e Pb–B σ bond and two degenerate 2c-2e Pb–B π bonds, i.e. [Pb≡B–B≡O]2–, as detailed in the second row of Fig. 4b. The B–Pb bond length in PbB2O2– is reduced to 2.107 Å, in good accord with the B≡Pb triple-bond length (2.10 Å) derived from Pyykkö’s triple-bond covalent atomic radii26.

a PbB2O–. b PbB2O2–. c PbB3O2–. The occupation numbers (ONs) are indicated. The corresponding Lewis electronic structures are also depicted.
The central B atom in PbB3O2– (Fig. 3d) undergoes sp2 hybridization and the AdNDP bonding analyses of the electron-precise monoanion are straightforward (Fig. 4c). It contains one Pb 6 s lone pair, two O lone pairs, and two B≡O boronyl ligands in the first row. The second row displays two 2c-2e B–B σ bonds between the central B and two BO ligands, one 2c-2e B–Pb σ bond, and one 2c-2e B–Pb π bond, giving rise to a terminal Pb=B double bond. The B–Pb bond length of 2.152 Å is comparable to the B=Pb double-bond length of 2.13 Å from Pyykkö’s covalent radii26. PbB3O2– can thus be formulated as [Pb=B(B≡O)2]–. Natural resonance theory (NRT) analyses47,48 at the PBE0/AVTZ level give rise to a B–Pb bond order of 2.39 for PbB2O–, 2.84 for PbB2O2–, and 1.84 for PbB3O2–, consistent with the bond order designations of 2.5, 3, and 2 for these species on the basis of electron counts, respectively. The covalency of the Pb–B multiple bonds can be understood from the similar electronegativities of Pb (2.33) and B (2.04). The PbB2O–, PbB2O2– and PbB3O2– species are the first experimentally confirmed molecules with B–Pb multiple bonds. We have also calculated MB2O–/2– and MB3O2– for M=Ge and Sn and found that both the Ge and Sn species have similar GM structures and bonding patterns as their Pb counterparts (see Supplementary Figs. 8–10).
Transition-metal-like behaviors of the central boron in [Pb≡B−CO]− and [Pb=B(CO)2]+
The electron deficiency of boron has led to novel structures and bonding in various boron compounds. Recently, boron has been shown to exhibit “metallomimetic” properties49, such as the formation of stable borylene dicarbonyl complex, in which two CO ligands are coordinated to a monovalent boron via donor-acceptor bonds50, a prototypical transition-metal behavior. Another unexpected metallomimetic property of boron is its capability to form a half-sandwich complex with the aromatic B73− ligand in the recently observed [(η7-B7)-B-BO]− species51. Since BO− is isoelectronic with CO, we optimized the geometric and electronic structures of the linear [Pb≡B−C≡O]– and Y-shaped [Pb=B(C≡O)2]+ and found that these carbonyl complexes exhibit similar AdNDP bonding patterns as their boronyl counterparts [Pb≡B–B≡O]2– and [Pb=B(B≡O)2]–, respectively (see Fig. 4 and Supplementary Fig. 11), with the two 2c-2e B–Pb π bonds in [Pb≡B–B≡O]2– (Fig. 4b) changed to two 3c-2e Pb-B-C π bonds in [Pb≡B−C≡O]– (Supplementary Fig. 11a) and the 2c-2e B−Pb π bond in [Pb=B(B≡O)2]– (Fig. 4c) extended to a 4c-2e Pb-B-C2 π bond in [Pb=B(C≡O)2]+ (Supplementary Fig. 11b). These carbonyl complexes may be more viable for chemical syntheses if a suitable ligand can be found to coordinate to Pb.
In conclusion, we have characterized the first B–Pb multiple bonds in the PbB2O–/2– and PbB3O2– molecular anions, using photoelectron spectroscopy and ab initio calculations. Excellent agreement between the theoretical and experimental data confirms that the global minimum of PbB2O– has an open-shell linear structure with a B–Pb bond order of 2.5, whereas the closed-shell PbB2O2– contains a B≡Pb triple bond. The PbB3O2– species has been shown to have a Y-shaped structure with a terminal B=Pb double bond and two BO ligands coordinated to the central B atom. Theoretical calculations indicate that MB2O–/2– and MB3O2– with M=Ge and Sn display similar structures and bonding as the Pb counterparts. It is further revealed that [Pb≡B–B≡O]2– and [Pb=B(B≡O)2]– have similar bonding patterns with their isoelectronic carbonyl complexes [Pb≡B–C≡O]– and [Pb=B(C≡O)2]+, opening the door to design main-group-metal-boron complexes with multiple bonds, as well as new boron metallomemetic compounds.
Methods
Photoelectron spectroscopy
The PES experiments were carried out using a magnetic-bottle apparatus with a laser vaporization supersonic cluster source4,52. The PbB2O– and PbB3O2– clusters were produced by laser vaporization of a Pb/10B mixed target inside a clustering nozzle. The laser-induced plasma was cooled by a He carrier gas seeded with 5% Ar, initiating nucleation in the nozzle. The O-containing clusters were formed due to the residue oxygen impurity on the target surface. Clusters formed inside the nozzle were entrained in the carrier gas and underwent a supersonic expansion. Negatively charged clusters were extracted perpendicularly from the collimated cluster beam and analyzed by a time-of-flight mass spectrometer. The PbB2O– and PbB3O2– clusters were mass-selected and decelerated before photodetachment. Three photon energies were used in the current experiment, including 355 nm (3.496 eV) and 266 nm (4.661 eV) from a Nd:YAG laser, and 193 nm from an ArF excimer laser. The photoelectron spectra were calibrated using the known spectra of Bi–. The electron kinetic energy (Ek) resolution of the magnetic-bottle electron analyzer (ΔEk/Ek) was around 2.5%, that is, about 25 meV for photoelectrons with 1 eV kinetic energy.
Computational methods
The global minima of PbB2O–, PbB2O2–, PbB2O, PbB3O2–, and PbB3O2 were examined using the unbiased Coalescence Kick (CK) global search method53 at the PBE0/lanl2dz40,54,55,56 level of theory with different spin multiplicities. Low-lying isomers were further refined using the PBE0 functional with the aug-cc-pVTZ-pp basis set and the ECP60MDF relativistic effective core potential for Pb and the aug-cc-pVTZ basis sets for B and O (abbreviated as AVTZ)36,37. Vibrational analyses were performed at the same level to ensure that all the optimized structures were true minima. The lowest-lying isomer for each species was re-optimized at CCSD/AVTZ level38,39 to obtain more reliable structures and more accurate energies. The first ADEs and VDEs for the global minima of PbB2O– and PbB3O2– were calculated at the PBE0/AVTZ and CCSD(T)57,58,59/AVTZ//PBE0/AVTZ [abbreviated as CCSD(T)/AVTZ] levels. The ADE in each case was calculated as the energy difference between the anion and neutral species at their respective optimized geometries. For PbB2O–, the first VDE was calculated as the energy differences between the doublet ground state (2Π) of the anion and the lowest-lying triplet state (3Σ–) of the neutral species at the optimized anion geometry. The energies for higher excited states of neutral PbB2O were computed using the state-averaged (SA) complete active space self-consistent field (CASSCF) method60,61 with the AVTZ basis set. An active space with 8 electrons and 9 orbitals was chosen for PbB2O. Both multi-reference configuration interaction42 (MRCI) and spin-orbit (SO) coupling41 were considered in these calculations. The 3Σ–, 1Δ, 1Σ+, 3Π, and 1Π states of neutral PbB2O were included in the SA8-CASSCF(8,9) calculations. For PbB3O2–, the first VDE was calculated as the energy difference between the singlet ground state of the anion and the lowest doublet state of the neutral molecule at the anion geometry. Higher VDEs were calculated using the TD-DFT method62 at the PBE0/AVTZ level. Vibrational frequencies of the ground states of neutral PbB2O and PbB3O2 were calculated at the CCSD/AVTZ level. Chemical bonding analyses were done using the adaptive natural density partitioning (AdNDP) approach46 at the PBE0/AVTZ level of theory. Franck−Condon simulations were performed using ezSpectrum63 at the CCSD/AVTZ level of theory. All the PBE0, CCSD, and CCSD(T) calculations were performed using the Gaussian 09 program64, while the CASSCF and MRCI calculations were done employing the Molpro 2012 code65.
Data availability
The data that support the findings of this study are available within the article and the associated Supplementary Information. Any other data are available from the corresponding authors upon request.
Code availability
All codes used in this study are commercially available or available from the authors cited in the references.
References
Lipscomb, W. N. The boranes and their relatives. Science 196, 1047–1055 (1977).
Alexandrova, A. N., Boldyrev, A. I., Zhai, H. J. & Wang, L. S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 250, 2811–2866 (2006).
Sergeeva, A. P. et al. Understanding boron through size-selected clusters: structure, chemical bonding, and fluxionality. Acc. Chem. Res. 47, 1349–1358 (2014).
Wang, L. S. Photoelectron spectroscopy of size-selected boron clusters: from planar structures to borophenes and borospherenes. Int. Rev. Phys. Chem. 35, 69–142 (2016).
Jian, T. et al. Probing the structures and bonding of size-selected boron and doped-boron clusters. Chem. Soc. Rev. 48, 3550–3591 (2019).
Braunschweig, H., Kollann, C. & Rais, D. Transition-metal complexes of boron—new insights and novel coordination modes. Angew. Chem. Int. Ed. 45, 5254–5274 (2006).
Pandey, K. K., Braunschweig, H. & Dewhurst, R. D. DFT study on alkyl- and haloborylene complexes of manganese and rhenium: structure and bonding energy analysis in [(η5‐C5H5)(CO)2M(BR)] and [(η5‐C5H5)(CO)2M(BX)] (M=Mn, Re; R=Me, Et, iPr, tBu; X=F, Cl, Br, I). Eur. J. Inorg. Chem. 2045–2056 (2011).
Pandey, K. K., Braunschweig, H. & Lledos, A. Nature of bonding in thermal borylene, alylene, and gallylene complexes of vanadium and niobium [(η5-C5H5)(CO)3M(ENR2)] (M=V, Nb; E=B, Al, Ga; R=CH3, SiH3, CMe3, SiMe3): a DFT study. Inorg. Chem. 50, 1402–1410 (2011). .
Pandey, K. K. & Aldridge, S. A theoretical study of bonding and charge distribution in cationic group 8 metal borylene and alylene complexes: consequences for complex stability and reactivity. Polyhedron 43, 131–139 (2012).
Frenking, G., Hermann, M., Andrada, D. M. & Holzmann, N. Donor-acceptor bonding in novel low-coordinated compounds of boron and group-14 atoms C-Sn. Chem. Soc. Rev. 45, 1129–1144 (2016).
Vidovic, D., Pierce, G. A. & Aldridge, S. Transition metalborylene complexes: boron analogues of classical organometallic systems. Chem. Commun. 1157–1171 (2009).
Braunschweig, H., Kollann, C. & Englert, U. Synthesis and structure of the first terminal borylene complexes. Angew. Chem. Int. Ed. 37, 3179–3180 (1998).
Cowley, A. H., Lomelí, V. & Voigt, A. Synthesis and characterization of a terminal borylene (boranediyl) complex. J. Am. Chem. Soc. 120, 6401–6402 (1998).
Braunschweig, H., Colling, M., Kollann, C., Stammler, H. G. & Neumann, B. Terminal borylene complexes as a source for the borylene B−N(SiMe3)2: alternative synthesis and structure of [(OC)5Cr=B=N(SiMe3)2]. Angew. Chem. Int. Ed. 40, 2298–2300 (2001).
Coombs, D. L., Aldridge, S., Jones, C. & Willock, D. J. Cationic terminal borylenes by halide abstraction: synthesis and spectroscopic and structural characterization of an Fe=B double bond. J. Am. Chem. Soc. 125, 6356–6357 (2003).
Coombs, D. L., Aldridge, S., Rossin, A., Jones, C. & Willock, D. J. Fe=B double bonds: synthetic, structural, and reaction chemistry of cationic terminal borylene complexes. Organometallics 23, 2911–2926 (2004).
Braunschweig, H., Burzler, M., Kupfer, T., Radacki, K. & Seeler, F. Synthesis and electronic structure of a terminal alkylborylene complex. Angew. Chem. Int. Ed. 46, 7785–7787 (2007).
Braunschweig, H., Radacki, K. & Uttinger, K. Synthesis and structure of a cationic platinum borylene complex. Angew. Chem. Int. Ed. 46, 3979–3982 (2007).
Braunschweig, H., Forster, M., Kupfer, T. & Seeler, F. Borylene transfer under thermal conditions for the synthesis of rhodium and iridium borylene complexes. Angew. Chem. Int. Ed. 47, 5981–5983 (2008).
Alcaraz, G., Helmstedt, U., Clot, E., Vendier, L. & Sabo-Etienne, S. A terminal borylene ruthenium complex: from B−H activation to reversible hydrogen release. J. Am. Chem. Soc. 130, 12878–12879 (2008).
Bertsch, S. et al. Towards homoleptic borylene complexes: incorporation of two borylene ligands into a mononuclear iridium species. Angew. Chem. Int. Ed. 49, 9517–9520 (2010).
Braunschweig, H., Dewhurst, R. D. & Schneider, A. Electron-precise coordination modes of boron-centered ligands. Chem. Rev. 110, 3924–3957 (2010).
Soleilhavoup, M. & Betrand, G. Borylenes: an emerging class of compounds. Angew. Chem. Int. Ed. 56, 10282–10292 (2017).
Borthakur, R. & Chandrasekhar, V. Boron-heteroelement (B–E; E=Al, C, Si, Ge, N, P, As, Bi, O, S, Se, Te) multiply bonded compounds: recent advances. Coord. Chem. Rev. 429, 213647 (2021).
Blank, B. et al. Aminoborylene complexes of group 6 elements and iron: a synthetic, structural, and quantum chemical study. Chem. Eur. J. 13, 4770–4781 (2007).
Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: A summary. J. Phys. Chem. A 119, 2326–2337 (2015).
Chen, T. T., Cheung, L. F., Chen, W. J., Cavanagh, J. & Wang, L. S. Observation of transition‐metal–boron triple bonds in IrB2O− and ReB2O−. Angew. Chem. Int. Ed. 59, 15260–15265 (2020).
Drance, M. J. et al. Terminal coordination of diatomic boron monofluoride to iron. Science 363, 1203–1205 (2019).
Xu, B. et al. Boron-transition-metal triple-bond FB≡MF2 complexes. Inorg. Chem. 58, 13418–13425 (2019).
Chi, C. et al. Quadruple bonding between iron and boron in the BFe(CO)3− complex. Nat. Commun. 10, 4713 (2019).
Cheung, L. F. et al. Observation of four-fold boron–metal bonds in RhB(BO–) and RhB. J. Phys. Chem. Lett. 11, 659–663 (2020).
Jian, T., Cheung, L. F., Chen, T. T. & Wang, L. S. Bismuth–boron multiple bonding in BiB2O− and Bi2B−. Angew. Chem. Int. Ed. 56, 9551–9555 (2017).
Hofmann, A., Legare, M. A., Wust, L. & Braunschweig, H. Heterodiatomic Multiple bonding in group 13: a complex with a boron–aluminum π bond reduces CO2. Angew. Chem. Int. Ed. 58, 9776–9781 (2019).
Raiser, D., Sindlinger, C. P., Schubert, H. & Wesemann, L. Ge=B π‐bonding: synthesis and reversible [2+2]cycloaddition of germaborenes. Angew. Chem. Int. Ed. 59, 3151–3155 (2020).
Rao, B. & Kinjo, R. Crystalline boragermenes. Angew. Chem. Int. Ed. 59, 3147–3150 (2020).
Peterson, K. A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 119, 11099–11112 (2003).
Kendall, R. A., Dunning, T. H. & Harrison, R. J. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806 (1992).
Scuseria, G. E., Janssen, C. L. & Schaefer, H. F. An efficient reformulation of the closed‐shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 89, 7382–7387 (1988).
Scuseria, G. E. & Schaefer, H. F. Is coupled cluster singles and doubles (CCSD) more computationally intensive than quadratic configuration interaction (QCISD)? J. Chem. Phys. 90, 3700–3703 (1989).
Adamo, C. & Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999).
Berning, A., Schweizer, M., Werner, H. J., Knowles, P. J. & Palmieri, P. Spin-orbit matrix elements for internally contracted multireference configuration interaction wavefunctions. Mol. Phys. 98, 1823–1833 (2000).
Werner, H. J. & Knowles, P. J. An efficient internally contracted multiconfiguration–reference configuration interaction method. J. Chem. Phys. 89, 5803–5814 (1988).
Zhai, H. J., Wang, L. M., Li, S. D. & Wang, L. S. Vibrationally-resolved photoelectron spectroscopy of BO– and BO2–: a joint experimental and theoretical study. J. Phys. Chem. A 111, 1030–1035 (2007).
Zhai, H. J., Li, S. D. & Wang, L. S. Boronyls as key structural units in boron oxide clusters: B(BO)2− and B(BO)3−. J. Am. Chem. Soc. 129, 9254–9255 (2007).
Li, S. D., Zhai, H. J. & Wang, L. S. B2(BO)22– — Diboronyl diborene: A linear molecule with a triple boron-boron bond. J. Am. Chem. Soc. 130, 2573–2579 (2008).
Zubarev, D. Y. & Boldyrev, A. I. Developing paradigms of chemical bonding: adaptive natural density partitioning. Phys. Chem. Chem. Phys. 10, 5207–5217 (2008).
Glendening, E. D. & Weinhold, F. Natural resonance theory: I. General formalism. J. Comput. Chem. 19, 593–609 (1998).
Glendening, E. D., Landis, C. R. & Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 34, 1429–1437 (2013).
Legaré, M. A., Pranckevicius, C. & Braunschweig, H. Metallomimetic chemistry of boron. Chem. Rev. 119, 8231–8261 (2019).
Braunschweig, H. et al. Multiple complexation of CO and related ligands to a main-group element. Nature 522, 327–330 (2015).
Tian, W. J. et al. S, Transition-metal-like bonding behaviors of a boron atom in a boron-cluster boronyl complex [(η7-B7)-B-BO]. Chem. Sci. 12, 8157–8164 (2021).
Wang, L. S., Cheng, H. S. & Fan, J. Photoelectron spectroscopy of size-selected transition metal clusters: Fen–, n = 3−24. J. Chem. Phys. 102, 9480–9493 (1995).
Sergeeva, A. P., Averkiev, B. B., Zhai, H. J., Boldyrev, A. I. & Wang, L. S. All-boron analogues of aromatic hydrocarbons: B17− and B18−. J. Chem. Phys. 134, 224304 (2011).
Wadt, W. R. & Hay, P. J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284–298 (1985).
Hay, P. J. & Wadt, W. R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985).
Hay, P. J. & Wadt, W. R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 82, 270–283 (1985).
Čížek, J. The full CCSD(T) model for molecular electronic structure. Adv. Chem. Phys. 14, 35–89 (1969).
Purvis, G. D. & Bartlett, R. J. A full coupled‐cluster singles and doubles model: the inclusion of disconnected triples. J. Chem. Phys. 76, 1910–1918 (1982).
Raghavachari, K., Trucks, G. W., Pople, J. A. & Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 157, 479–483 (1989).
Werner, H. J. & Knowles, P. J. A second order multiconfiguration SCF procedure with optimum convergence. J. Chem. Phys. 82, 5053–5063 (1985).
Celani, P. & Werner, H. J. Multireference perturbation theory for large restricted and selected active space reference wave functions. J. Chem. Phys. 112, 5546–5557 (2000).
Bauernschmitt, R. & Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 256, 454–464 (1996).
Mozhayskiy, V. A. & Krylov, A. I. ezSpectrum. http://iopenshell.usc.edu/downloads (accessed March 21, 2019).
Frisch, M. J. et al. Gaussian 09, Revision A.1; Gaussian, Inc., Wallingford, CT, 2009.
Werner, H. J. et al. MOLPRO, version 2012.1.
Acknowledgements
The authors would like to thank Professor Jun Li for valuable discussions. The experimental work at Brown University was supported by the National Science Foundation (CHE-2053541). The theoretical work was performed at Shanxi University supported by the National Natural Science Foundation of China (21720102006, 21973057, 22003034). Q.C. gratefully acknowledges the start-up fund from Shanxi University for support.
Author information
Authors and Affiliations
Contributions
W.J.C. and T.T.C. did the experiment; Q.C., H.G.L., X.Y.Z., Y.Y.M., Q.Q.Y., and R.N.Y. did the calculations; L.S.W. and S.D.L. guided the work; W.J.C., T.T.C., Q.C., S.D.L., and L.S.W. co-wrote the manuscript, which was read and commented on by all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Yanli Zeng and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, WJ., Chen, TT., Chen, Q. et al. Boron-lead multiple bonds in the PbB2O– and PbB3O2– clusters. Commun Chem 5, 25 (2022). https://doi.org/10.1038/s42004-022-00643-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-022-00643-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
