
Abstract
Discrete block co-oligomers (BCOs) are gaining considerable attention due to their potential to form highly ordered ultrasmall nanostructures suitable for lithographic templates. However, laborious synthetic routes present a major hurdle to the practical application. Herein, we report a readily available discrete BCO system that is capable of forming various self-assembled nanostructures with ultrasmall periodicity. Click coupling of propargyl-functionalized sugars (containing 1–7 glucose units) and azido-functionalized terpenoids (containing 3, 4, and 9 isoprene units) afforded the discrete and monodisperse BCOs with a desired total degree of polymerization and block ratio. These BCOs microphase separated into lamellar, gyroid, and cylindrical morphologies with the domain spacing (d) of 4.2–7.5 nm. Considering easy synthesis and rich phase behavior, presented BCO systems could be highly promising for application to diverse ~4-nm nanofabrications.
Similar content being viewed by others


Introduction
Ordered nanostructures such as lamellar (LAM), gyroid (GYR), cylindrical, and spherical morphologies generated by block copolymer (BCP) microphase separation are highly promising nanotemplates for the low-cost, large-scale, and high-throughput production of sub-10 nm nanomaterials and nanodevices1,2,3,4. In particular, directed self-assembly (DSA)-assisted BCP lithography has attracted considerable attention as a next-generation nanopatterning technology because it is more cost-effective and scalable than competing approaches5,6,7,8. One of the key challenges in this field is the development of BCP systems for the formation of highly ordered ultrasmall microphase-separated structures, enabling nanopatterning of a few nanometers.
Previous studies have shown that decreasing the degree of polymerization (N) while increasing the Flory–Huggins interaction parameter (χ) is the most feasible and reliable method for achieving periodic microphase-separated structures with a domain spacing (d) <10 nm9,10,11,12,13,14,15,16,17. However, the high χ-low N BCPs are synthesized partially or entirely by polymerization reactions. As a result, it is virtually impossible for all growing polymer chains to terminate at the same length; therefore, the BCPs exhibit a range of molecular-weight distributions (typically, dispersity of 1.05–1.30)9,10,11,12,13,14,15,16. Such a dispersity is particularly detrimental for low-molecular-weight BCPs targeting a domain spacing of few nanometers (i.e., d ≈ 5 nm) since at that scale, a single monomer unit size rapidly approaches d. In other words, in such low-molecular-weight regimes, increasing or decreasing the number of BCP monomer units by only a single unit would impact the resulting self-assembly structural properties, such as the morphology, d, defects, roughness, and ordering18,19,20. Indeed, seminal works by Mejier et al.21 and Bates and Hawker et al.22 have demonstrated that d increased with increasing BCP dispersity. More importantly, in the low-molecular-weight (~2500 g mol−1) region, increasing the dispersity resulted in a loss of microphase separation, despite the fact that the monodisperse counterparts could form well-ordered nanostructures23. Another problem with conventional BCPs is synthesis reproducibility, whereby it is virtually impossible to reproduce BCPs showing exactly the same molecular weights and compositions, thereby causing batch-dependent variation in the resulting nanostructures. Given the importance of monodispersity in constructing ultrasmall nanostructures, a paradigm shift from conventional polydisperse BCPs to monodisperse block co-oligomer (BCO) systems is highly desirable.
Although several monodisperse BCP/BCO systems have been reported, their syntheses rely on either chromatographic fractionation of polydisperse polymers22 or iterative reactions to grow monomer units in a step-by-step manner21,23,24,25, both of which require laborious efforts and time-consuming processes, thereby posing a major hurdle to meaningful progress in the fundamental science of monodisperse BCPs/BCOs. Therefore, the preparation of a readily available monodisperse BCO system that can form various microphase-separated morphologies showing ultrasmall periodicity is particularly challenging.
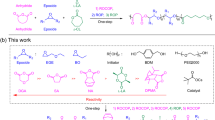
To develop such ideal BCO systems, we herein propose a combination of sugars and terpenoids. The previous studies into oligosaccharide-based BCPs synthesized via the “click” chemistry revealed the formation of small microphase-separated structures26,27,28,29,30 with the d of even down to 5.8 nm (in the bulk state)31,32. Therefore, strong segregation between hydrophilic saccharidic blocks and hydrophobic isoprene-based hydrocarbon chains would be expected, which would in turn allow microphase separation in the molecular-weight regime below 2000 g mol−1. In addition, recent reports33,34,35 by Lodge, Siepmann, and Hillmyer on sugar-based amphiphiles with a hydrocarbon chain, leading to a lamellar morphology with d of 3.5 nm (even down to d of 1.2 nm as revealed by molecular dynamic simulation), also supports that the combination of sugars and terpenoids is highly promising for realizing ultrasmall nanostructure formation. More importantly, oligosaccharide and terpenoid blocks showing defined and discrete degrees of polymerization (DPs) are commercially available, and a series of BCOs featuring monodispersity and discrete DPs can be synthesized readily from them, providing rapid access to various ultrasmall nanostructures showing diverse morphologies, features, and sizes. Thus, we herein employ a series of sugars from monosaccharide (i.e., glucose) to heptasaccharide (i.e., maltoheptaose) as oligosaccharide blocks, while farnesol (3,7,11-trimethyldodeca-2,6,10-trien-1-ol; mixture of isomers), dl-α-tocopherol (2,5,7,8-tetramethyl-2-[(4,8,12-trimethyltridecyl)]chroman-6-ol), and solanesol ((2E,6E,10E,14E,18E,22E,26E,30E)-3,7,11,15,19,23,27,31,35-nonamethylhexatriaconta-2,6,10,14,18,22,26,30,34-nonaen-1-ol)—which are a family of terpenoids containing three, four, and nine isoprene units, respectively—are selected as hydrophobic blocks. Using this BCO system, we attempt the construction of LAM, GYR, and hexagonally close-packed cylindrical (HEX) morphologies by adjusting the DP of each block, and the d values of the resulting organic-BCP/BCO-based microphase-separated structures are examined.
Results
Synthesis
Our BCO synthetic procedure relies on the “click” reaction between terpenoid (i.e., farnesol, tocopherol, and solanesol) and sugar units, as depicted in Fig. 1. Solanesol-based BCOs (Glcn-b-Sol; n = 1–7) were synthesized by the “click” reactions of the azido-functionalized solanesol (N3-Sol) with propargyl-functionalized glucose (Glc1-C≡CH), maltose (Glc2-C≡CH), maltotriose (Glc3-C≡CH), maltotetraose (Glc4-C≡CH), maltopentaose (Glc5-C≡CH), maltohexaose (Glc6-C≡CH), and maltoheptaose (Glc7-C≡CH) in the presence of a Cu catalyst in dimethylformamide (DMF) and tetrahydrofuran (THF) mixed solvent. Following the completion of the reaction, the Cu catalyst was removed by treating the reaction mixture with a cation-exchange resin, and the resulting residue was precipitated in a selective solvent to remove unreacted starting materials, thereby affording the desired BCOs in moderate to good yields. The reaction products did not exhibit any strong infrared (IR) absorption at 2100 cm−1, suggesting near-quantitative consumption of the azido group (Supplementary Fig. 1). Proton nuclear magnetic resonance (1H NMR) spectra were assigned to the expected BCO chemical structure (Supplementary Figs. 2–8), and the size-exclusion-chromatography (SEC; in DMF containing 0.01 M LiCl) elution peak clearly shifted toward the higher-molecular-weight region upon increasing the number of glucose units in the BCO (Fig. 2a). More importantly, matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF MS) analysis confirmed the presence of monodisperse and discrete products (Fig. 2b and Supplementary Fig. 9). It should be emphasized that successful multigram-scale synthesis could be attributed to the facile preparation of the starting materials and simple purification. Similarly, tocopherol- and farnesol-based BCOs (Glc1-b-Toc, Glc2-b-Toc, and Glc1-b-Far) were prepared and characterized (Supplementary Figs. 9–15).

Preparation of the monodisperse and discrete Glcn-b-Sol (n = 1–7), and Glcn-b-Toc (n = 1 and 2), and Glc1-b-Far.

a SEC traces (in DMF containing 0.01 M LiCl) and b MALDI-TOF mass spectra for Glcn-b-Sol (n = 1–7). Asterisks indicate peaks attributed to [M+K]+ or [M+Ag]+.
Nanostructural formation of solanesol-based BCOs
To gather fundamental information regarding the microphase separation behavior, a series of thermally annealed solanesol-based BCOs (Glcn-b-Sol; n = 1–7) was subjected to small-angle X-ray scattering (SAXS) (Fig. 3a). Prior to the SAXS experiment, we performed differential scanning calorimetry (DSC) analysis on the BCOs. Although all the BCO DSC thermograms did not exhibit any transitions due to crystallization and melting during the first cooling and second heating processes, they did exhibit two baseline shifts corresponding to glass transition temperatures (Tgs) for the solanesol and sugar segments (Supplementary Fig. 16). The fact that the Tgs of the two segments were independently observed suggested the microphase separation between the segments.

a SAXS profiles revealed the formation of ordered nanostructures, with the exceptions of Glc6-b-Sol and Glc7-b-Sol. b WAXS profiles of Glcn-b-Sol (n = 1–7) demonstrating amorphous BCOs, with the exception of Glc1-b-Sol. Glc2-b-Sol was annealed at 100 °C for 36 h while all other samples were annealed at 130 °C for 36 h. c qhkl versus (h2 + k2 + l2)1/2 plot based on the SAXS pattern for Glc2-b-Sol validated space group assignment, where h, k, and l are the Miller indices of the corresponding planes. The lattice parameter acub was calculated from the slope of the linear fit.
Unless otherwise noted, bulk samples were annealed at 130 °C for 36 h and then quenched to room temperature. The SAXS profiles of Glc1-b-Sol, Glc2-b-Sol, Glc3-b-Sol, Glc4-b-Sol, and Glc5-b-Sol showed higher-order scattering peaks and a sharp primary peak (q*), demonstrating the formation of well-ordered periodic nanostructures (Table 1). It is rather surprising that even the BCO containing only one glucose unit, i.e., Glc1-b-Sol, had self-assembled into the HEX morphology with a domain spacing (d = 2π/q*) of 5.5 nm (cylinder-to-cylinder distance = 2d/√3 = 6.4 nm; cylinder diameter = [8fsugar ∙ d2/(√3∙π)]1/2 = 2.7 nm, where fsugar is the saccharide volume fraction calculated based on the densities of 1.36 and 0.95 for the saccharide and solanesol blocks, respectively)23, which is among the smallest ever obtained among structures that have been self-assembled from saccharide-based block BCPs. For example, d values of 5.8 and 6.6 nm were reported for LAM-forming cellobiose-block-polypropylene31 and HEX-forming maltotriose-block-polycaprolactone systems36, respectively, in the bulk states. In addition, a LAM morphology showing relatively large d = 12.7 nm had formed through self-assembly. Considering the multiple diffraction peaks present in the wide-angle X-ray scattering (WAXS) profiles (Fig. 3b), the coexisting LAM morphology should be a crystallization-driven rather than a microphase-separated structure (Supplementary Figs. 17 and 18). We thus denote this nanostructure as LAM_c to differentiate with the microphase-separated LAM structure. Indeed, in situ SAXS measurements obtained while heating the samples gave only the scattering pattern characteristic of HEX (q/q* = 1, √3, √4, and √7; Supplementary Fig. 18), confirming the absence of the LAM morphology at elevated temperatures.
The SAXS profile of Glc2-b-Sol thermally annealed at 100 °C for 36 h showed scattering peaks at q = 1.017, 1.179, 1.875, 1.961, 2.042, and 2.128 nm−1, which were assignable to the (211), (220), (420), (332), (422), and (431) crystallographic planes of the GYR phase, respectively (Fig. 3a)37. Although a similar scattering pattern assignable to the GYR phase was found in the sample after 130 °C for 36 h, the scattering peak width was broader, suggesting less ordering (Supplementary Fig. 19). Note that this is the first-ever example of GYR-forming saccharide-based BCPs. Since the maltose block is the minor component, the interwoven network and surrounding matrix should be composed of maltose and solanesol, respectively. The cubic lattice parameter acub was determined to be 15.1 nm from the slope of a plot of qhkl versus (h2 + k2 + l2)1/2 (acub = 2π(h2 + k2 + l2)1/2/qhkl), where h, k, and l are Miller indices (Fig. 3c). The lateral distance between interwoven networks was then calculated to be 6.5 nm (=√3acub/4)38, which is approximately the fully extended molecular length (ca. 7 nm). Thus, low BCO molecular weight should be responsible for the GYR nanomorphology.
SAXS profiles were then generated for the LAM structures showing q/q* ratios of 1:2:3:4 after increasing the number of glucose units to more than 3 (i.e., for Glc3-b-Sol, Glc4-b-Sol, and Glc5-b-Sol) (Fig. 3a); d was in the range 6.7–7.5 nm and increased in ca. 0.4 nm increments with each glucose unit added. It should be notable that Glc3-b-Sol formed the LAM despite the asymmetric composition (fsugar = 0.33). According to classical BCP theory, a volume fraction of ~0.3 should lead to HEX rather than LAM structural formations39. However, the rigid monodisperse oligosaccharide molecules encumbered BCO molecules from packing into higher-curvature morphologies, thus preferentially forming LAM structures even at such asymmetric volume ratios. For LAM structure, d is the sum of the lamellar microdomain thicknesses for the two constitutional blocks. Given the 1:1 volume ratio of Glc5-b-Sol as revealed by the SAXS analysis, the lamellar microdomain thickness for each block should be ~3.8 nm.
Further increasing the number of glucose units resulted in poor ordering (i.e., random two phase) in the bulk (Fig. 3a). Despite thermally annealing Glc6-b-Sol and Glc7-b-Sol up to 180 °C for 36 h, their SAXS profiles only exhibited a single broad peak without any higher-order scattering. The reduced chain mobility associated with high glass transition temperatures (Tgs) owing to longer oligosaccharide molecules (e.g., 145 and 150 °C for maltohexaose and maltoheptaose, respectively40) should be responsible for restricting the self-assembly process.
BCO thin films were then prepared by spin-coating a toluene/DMF BCO solution onto silicon substrates. To confirm the ordered nanostructures in the BCO thin films, we investigated Glcn-b-Sol (n = 1–5) thin films by grazing-incidence small-angle X-ray scattering (GISAXS) measurements and atomic force microscopy (AFM). GISAXS analysis revealed that the thin films presented horizontally orientated LAM morphologies, regardless of the number of glucose units (Supplementary Fig. 20). In addition, the AFM images of the Glc3-b-Sol and Glc4-b-Sol thin films clearly showed terrace formations (Fig. 4), which agreed with the horizontally orientated LAM formations observed by GISAXS. The height difference between two consecutive layers (~6 and ~8 nm for Glc3-b-Sol and Glc4-b-Sol, respectively) roughly matched d, as determined by SAXS and listed in Table 1. The key morphological features of the Glcn-b-Sol thin films were consistent with horizontal orientations retained over large areas and relatively low distributions of LAM thickness. Such structural uniformity should be a direct result of the BCO monodispersity.

AFM height images (a, b) and corresponding cross-sectional profiles (c, d) indicating the formation of 6–8-nm-thick horizontal lamellae in Glc3-b-Sol (a, c) and Glc4-b-Sol thin films (b, d). Thin-film samples were prepared by spin-coating the BCO solution onto the hydrophilic surface of a silicon substrate followed by thermal annealing at 85 °C for 1 h.
Fine-tuning nanostructures by varying dispersity
The most important feature of the present BCO system is its monodispersity, which should allow high degrees of ordering in the bulk. Previous studies have suggested the possibility of tailoring BCP microphase separation—including size, morphology, and stability—by tuning the constitutional-block dispersities18,41,42,43,44,45. Thus, we hypothesized that BCO mixtures with different numbers of glucose units could further expand the morphologies, features, and sizes accessible from the current BCO system. To test this hypothesis, we investigated the effects of sugar-block dispersity (ÐGlc) on microphase-separated structures. By mixing different Glcn-b-Sol molecules (n = 1–5), polydisperse versions of Glc3-b-Sol and Glc4-b-Sol were artificially prepared through variation in ÐGlc while fixing the average number of glucose units. For example, Glc1-b-Sol and Glc5-b-Sol mixed in a 1:1 molar ratio showed the same average number of glucose units as Glc3-b-Sol but a different ÐGlc. All thermally annealed polydisperse samples underwent self-assembly into well-ordered microphase-separated structures regardless of ÐGlc, as evidenced by the multiple sharp scattering peaks in their SAXS profiles (Fig. 5). Compared to the microphase-separated morphology of monodisperse Glc3-b-Sol, that of polydisperse Glc3-b-Sol shifted from LAM (ÐGlc = 1.000) to GYR (ÐGlc = 1.190 and 1.244) and finally to HEX (ÐGlc = 1.301 and 1.333) with increasing ÐGlc (Fig. 5a–h, see Supplementary Fig. 21 for the GYR peak assignment). The coexistence of longer and shorter oligosaccharides facilitated the formation of curved microdomains, thereby allowing the morphology to shift from a lower to a higher curvature18. In addition to the morphological shift, d increased with increasing ÐGlc. Interestingly, for the GYR-forming samples, the full width at half maximum (Δq*) of the primary scattering peak increased with increasing ÐGlc (e.g., Δq* = 0.0326 nm−1 for ÐGlc = 1.069; Δq* = 0.0365 nm−1 for ÐGlc = 1.244). Since the Δq* is inversely proportional to the grain size according to the Scherrer’s equation46, this tendency implies the decrease in the GYR grain size with increasing ÐGlc. In the Glc4-b-Sol series, on the other hand, d increased from 7.1 nm (ÐGlc = 1.000) to 7.8 nm (ÐGlc = 1.165) with increasing ÐGlc, while the LAM morphology remained unchanged (Fig. 5i–m). Glc4-b-Sol shows a higher saccharide volume fraction (fsugar) than Glc3-b-Sol, and therefore, is deeply embedded in the LAM region of the phase diagram. This likely accounts for the fact that the Glc4-b-Sol series did not exhibit a morphological shift. In contrast, Glc3-b-Sol is likely located near the LAM–GYR phase boundary; therefore, slight variations in dispersity shifted the resulting morphology. Since the morphology and d value both correlated with ÐGlc, the morphology can be tailored, and d can be adjusted by a few angstroms simply by blending discrete BCO molecules. Conversely, however, these results imply that monodisperse and discrete natures are essential in fabricating highly reproducible ultrasmall nanostructures, which is particularly important in the context of nanolithographic applications.

SAXS profiles of the polydisperse (a–h) Glc3-b-Sol and (i–m) Glc4-b-Sol series revealed variation in the morphology and d upon increasing ĐGlc. Polydisperse samples were prepared by mixing different BCO molecules in given molar ratios. All samples were thermally annealed at 130 °C for 36 h. Blue, red, and green inverted triangles indicate peak assignments corresponding to the LAM, GYR, and HEX structures, respectively.
Further minimizing the feature size by tocopherol- and farnesol-based BCOs
Although solanesol-based BCOs underwent self-assembly into highly ordered nanostructures, our obtained results indicated that the lower limit of d achievable when combining solanesol with the monosaccharide was ~6 nm. We, therefore, examined the BCO nanostructures formed from shorter terpenoid blocks (i.e., Glc1-b-Toc, Glc2-b-Toc, and Glc1-b-Far) by SAXS analysis. Surprisingly, the SAXS profiles of the thermally annealed Glc1-b-Toc, Glc2-b-Toc, and Glc1-b-Far (80 °C for 6 h) exhibited multiple scattering peaks corresponding to GYR (d = 5.2 nm; acub = 12.6 nm), LAM (d = 6.3 nm), and LAM (d = 4.2 nm) nanostructures, respectively (Fig. 6a and Supplementary Fig. 22), despite their extremely low M.W. (580–964 g mol−1) and total DP (4–6) (Table 1). Such an ultrasmall d value of ~4 nm has rarely been reported among the organic-BCP-derived LAM microphase-separated structures9,14,23,31,34,47. Given the fsugar of Glc1-b-Far, the sugar microdomain thickness is approximately 1.2 nm, which shows promise to ultrahigh-resolution nanofabrication. In addition, we spin-coated a Glc1-b-Toc thin film, and the corresponding GISAXS image showed a strong scattering spot along the out-of-plane direction, indicating the formation of a horizontally orientated LAM morphology (Supplementary Fig. 23). Furthermore, the corresponding AFM height image clearly shows terrace formation corresponding to multilayer LAM stacking, which also supports the GISAXS result (Fig. 6b). The height difference between two consecutive layers (i.e., d for the LAM morphology) was ~4 nm (Fig. 6c), which again confirmed the astonishing capability of the present BCO system to form ultrasmall nanostructures.

a SAXS profiles for the bulk samples thermally annealed at 80 °C for 6 h revealed d = 4–6 nm microphase separation. b AFM height image, and c corresponding cross-sectional profiles of the as-cast Glc1-b-Far thin film confirmed the horizontally orientated LAM formation in ~4-nm-thick layers.
Phase behavior of sugar/terpenoid BCO Systems
One of the most important findings of our study is that the resulting BCO morphology varies from HEX to GYR and finally to LAM with increasing fsugar, as summarized in the conceptual phase diagram (Fig. 7). Note that we tentatively employed the total number of glucose and isoprene units (DPGlc + DPisoprene) as the phase-diagram vertical axis because it is highly challenging to determine χ and N for the present BCO system. The observed propensity is consistent with the conventional BCP microphase separation theory39. In other words, the desired morphology and feature size can both be achieved by simply designing the number of BCO glucose and isoprene units, which is analogous to the molecular design of conventional BCPs based on f and N. Unfortunately, however, a spherical morphology could not be obtained from the current combination of oligosaccharides and terpenoids. The fact that the most asymmetric Glc1-b-Sol formed a cylindrical morphology suggests the difficulty in obtaining spherical morphologies by the current molecular design.

Conceptual phase diagram was generated for the phase-separated Glcn-b-Sol (n = 1–5; blue), Glcn-b-Toc (n = 1 and 2; red), and Glc1-b-Far (green) bulk morphologies. Values in parentheses are the d values listed in Table 1. The vertical and horizontal axes represent the sum of glucose and isoprene DPs and the saccharide volume fraction, respectively.
Conclusion
We herein reported the development of a readily available monodisperse and discrete block co-oligomer (BCO) system consisting of hydrophilic sugars and hydrophobic terpenoids. Starting from readily available natural sugars and terpenoids with defined numbers of glucose and isoprene units, BCOs exhibiting the desired total degrees of polymerization (DPs) and compositions could be synthesized easily by connecting these blocks via a “click” reaction. The resulting BCOs self-assembled into well-ordered lamellar (LAM), gyroid (GYR), and hexagonally close-packed cylindrical (HEX) nanostructures showing 4–8 nm periodicity through simple thermal annealing. The microphase-separated morphology shifted from cylindrical to GYR and finally to LAM upon increasing the number of glucose units (i.e., the saccharide volume fraction) for a fixed hydrophobic segment length, which is analogous to well-established BCP phase behavior. Therefore, the desired BCO morphologies, features, and sizes can be achieved simply by designing the number of BCO glucose and isoprene units appropriately. Most importantly, we developed a well-ordered microphase-separated structure showing 4.2 nm periodicity and 1.2 nm microdomain thickness, which is among the smallest ever achieved by organic block copolymer (BCP)/BCO-based microphase-separated structures. Considering the commercial availability of all starting materials, the ease of BCO synthesis, and the facile BCO purification, we believe that presented BCOs strongly contribute to accelerating applied research of solid and solution state self-assembly of discrete and monodisperse BCOs, which will inevitably expand their application scopes in various fields of not only the nanolithography but also organic devices, separation materials, coatings, etc. We are currently working on the orientational control of the BCO nanostructures in the thin-film state to achieve nanolithography with a few-nanometer resolution.
Methods
Synthesis and characterization
See Supplementary Methods and Supplementary Figs. 1–15.
Typical synthesis procedure for Glcn-b-Sol
N3-Sol (6.00 g, 7.79 mmol), Glc2-C≡CH (3.80 g, 9.35 mmol), and copper(I) bromide (CuBr; 112 mg, 779 µmol) were placed into a Schlenk flask, which was evacuated and back-filled with argon three times. A solution of N,N,N′,N″,N″-pentamethyldiethylenetriamine (134 mg, 779 mmol) in a mixed solvent of DMF (25.0 mL) and THF (25.0 mL) was degassed by the argon bubbling, and the mixture was then transferred into the Schlenk flask under an argon atmosphere. The reaction mixture was stirred at 60 °C for 72 h and then treated with Dowex to remove Cu catalyst. The product was purified by reprecipitation from the DMF solution into a mixed solvent of acetonitrile and H2O (7/3 (v/v)) to give Glc2-b-Sol as a white solid (6.29 g, yield: 70.2%).
Small-angle X-ray scattering (SAXS) experiments
SAXS experiments for bulk samples were performed at BL-6A beamline of the Photon Factory of High Energy Accelerator Research Organization (KEK, Tsukuba, Japan). The X-ray wavelength and exposure time were 1.50 Å (8.27 keV) and 60 s, respectively. A PILATUS3 1M (Dectris Ltd., Switzerland) detector, with 981 × 1043 pixels at a pixel size of 172 × 172 μm, and a counter depth of 20 bits (1,048,576 counts), was used for data acquisition. The sample-to-detector distance was calibrated using the scattering patterns of silver behenate. The bulk samples were put into 1.5 mm diameter glass capillaries. After that, the bulk samples were annealed under vacuum at 70, 100, and 130 °C for 36 h. The SAXS data were acquired under ambient condition, and 1D profiles were obtained as plots of scattering intensity as functions of scattering vector (q), where q = (4π/λ) sin(θ/2) (λ, wavelength; θ, scattering angle).
Atomic force microscopy (AFM)
The AFM phase images were realized using a molecular imaging PicoPlus atomic force microscope operating in the tapping mode with a silicon cantilever (PPP-NCHR and SSS-NCHR, NANOSENSORS) having a resonant frequency of 330 kHz and a force constant of 42 N m−1. The AFM images were processed using Gwyddion software.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and Supplementary Information file, including experimental procedures and characterization data for all the new compounds.
References
Lazzari, M., Liu, G. & Lecommandoux, S. Block Copolymers in Nanoscience (Wiley-VCH, Weinheim, 2006).
Olson, D. A., Chen, L. & Hillmyer, M. A. Templating nanoporous polymers with ordered block copolymers. Chem. Mater. 20, 869–890 (2008).
Kim, J. K., Yang, S. Y., Lee, Y. & Kim, Y. Functional nanomaterials based on block copolymer self-assembly. Prog. Polym. Sci. 35, 1325–1349 (2010).
Hu, H., Gopinadhan, M. & Osuji, C. O. Directed self-assembly of block copolymers: a tutorial review of strategies for enabling nanotechnology with soft matter. Soft Matter 10, 3867–3889 (2014).
Jeong, S.-J., Kim, J. Y., Kim, B. H., Moon, H.-S. & Kim, S. O. Directed self-assembly of block copolymers for next generation nanolithography. Mater. Today 16, 468–476 (2013).
Stoykovich, M. P. et al. Directed self-assembly of block copolymers for nanolithography: fabrication of isolated features and essential integrated circuit geometries. ACS Nano 3, 168–175 (2007).
Tang, C., Lennon, E. M., Fredrickson, G. H., Kramer, E. J. & Hawker, C. J. Evolution of block copolymer lithography to highly ordered square arrays. Science 322, 429–432 (2008).
Cheng, J. Y., Ross, C. A., Smith, H. I. & Thomas, E. L. Templated self‐assembly of block copolymers: top‐down helps bottom‐up. Adv. Mater. 18, 2505–2521 (2006).
Sinturel, C., Bates, F. S. & Hillmyer, M. A. High χ–low N block polymers: how far can we go? ACS Macro Lett. 4, 1044–1050 (2015).
Kennemur, J. G., Yao, L., Bates, F. S. & Hillmyer, M. A. Sub-5 nm domains in ordered poly(cyclohexylethylene)-block-poly(methyl methacrylate) block polymers for lithography. Macromolecules 47, 1411–1418 (2014).
Luo, Y. et al. Poly(dimethylsiloxane-b-methyl methacrylate): a promising candidate for sub-10 nm patterning. Macromolecules 48, 3422–3430 (2015).
Kanimozhi, C. et al. Isomeric effect enabled thermally driven self-assembly of hydroxystyrene-based block copolymers. ACS Macro Lett. 5, 833–838 (2016).
Jeong, G. et al. Realizing 5.4 nm full pitch lamellar microdomains by a solid-state transformation. Macromolecules 50, 7148–7154 (2017).
Kwak, J. et al. Fabrication of sub-3 nm feature size based on block copolymer self-assembly for next-generation nanolithography. Macromolecules 50, 6813–6818 (2017).
Azuma, K. et al. Self-assembly of an ultrahigh-χ block copolymer with versatile etch selectivity. Macromolecules 51, 6460–6467 (2018).
Katsuhara, S. et al. Metallopolymer-block-oligosaccharide for sub-10 nm microphase separation. Polym. Chem. 11, 2995–3002 (2020).
Yu, X. et al. Giant surfactants provide a versatile platform for sub-10-nm nanostructure engineering. Proc. Natl Acad. Sci. USA 110, 10078–10083 (2013).
Lynd, N. A., Meuler, A. J. & Hillmyer, M. A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 33, 875–893 (2008).
Jiao, G.-S., Li, Y., Qian, H.-J. & Lu, Z.-Y. Computer simulation study of polydispersity effect on the phase behavior of short diblock copolymers. Polymer 96, 6–12 (2016).
Peters, A. J., Lawson, R. A., Dation, B. D., Ludovice, P. J. & Henderson, C. L. Simulation study of the effect of molar mass dispersity on domain interfacial roughness in lamellae forming block copolymers for directed self-assembly. Nanotechnology 26, 385301 (2015).
van Genabeek, B. et al. Dispersity under scrutiny: phase behavior differences between disperse and discrete low molecular weight block co-oligomers. ACS Macro Lett. 6, 674–678 (2017).
Oschmann, B. et al. Effects of tailored dispersity on the self-assembly of dimethylsiloxane–methyl methacrylate block co-oligomers. ACS Macro Lett. 6, 668–673668–673 (2017).
van Genabeek, B. et al. Synthesis and self-assembly of discrete dimethylsiloxane–lactic acid diblock co-oligomers: the dononacontamer and its shorter homologues. J. Am. Chem. Soc. 138, 4210–4218 (2016).
Jiang, Y. et al. Iterative exponential growth synthesis and assembly of uniform diblock copolymers. J. Am. Chem. Soc. 138, 9369–9372 (2016).
Golder, M. R. et al. Stereochemical sequence dictates unimolecular diblock copolymer assembly. J. Am. Chem. Soc. 140, 1596–1599 (2018).
Otsuka, I. et al. 10 nm Scale cylinder–cubic phase transition induced by caramelization in sugar-based block copolymers. ACS Macro Lett. 1, 1379–1382 (2012).
Isono, T., Ree, B. J., Tajima, K., Borsali, R. & Satoh, T. Highly ordered cylinder morphologies with 10 nm scale periodicity in biomass-based block copolymers. Macromolecules 51, 428–437 (2018).
Liao, Y., Chen, W.-C. & Borsali, R. Carbohydrate‐based block copolymer thin films: ultrafast nano‐organization with 7 nm resolution using microwave energy. oligosaccharide/silicon-containing block copolymers with 5 nm features for lithographic applications. Adv. Mater. 20, 1701645 (2017).
Cushen, J. D. et al. Oligosaccharide/silicon-containing block copolymers with 5 nm features for lithographic applications. ACS Nano 6, 3424–3433 (2012).
Otsuka, I. et al. Sub-10 nm scale nanostructures in self-organized linear di- and triblock copolymers and miktoarm star copolymers consisting of maltoheptaose and polystyrene. Macromolecules 48, 1509–1517 (2015).
Nowak, S. R., Hwang, W. & Sita, L. R. Dynamic sub-10-nm nanostructured ultrathin films of sugar–polyolefin conjugates thermoresponsive at physiological temperatures. J. Am. Chem. Soc. 139, 5281–5284 (2017).
Thomas, T. S., Hwang, W. & Sita, L. R. End-group-functionalized poly(α-olefinates) as non-polar building blocks: self-assembly of sugar-polyolefin hybrid conjugates. Angew. Chem. Int. Ed. 55, 4683–4687 (2016).
Shen, Z. et al. From order to disorder: computational design of triblock amphiphiles with 1 nm domains. J. Am. Chem. Soc. 142, 9352–9362 (2020).
Barreda, L. et al. Synthesis, simulation, and self-assembly of a model amphiphile to push the limits of block polymer nanopatterning. Nano Lett. 19, 4458–4462 (2019).
Chen, Q. P. et al. Computational design of high-χ block oligomers for accessing 1 nm domains. ACS Nano 12, 4351–4361 (2018).
Isono, T. et al. Microphase separation of carbohydrate-based star-block copolymers with sub-10 nm periodicity. Polym. Chem. 10, 1119–1129 (2020).
Gan, K. P., Yoshio, M., Sugihara, Y. & Kato, T. Guanine–oligothiophene conjugates: liquid-crystalline properties, photoconductivities and ion-responsive emission of their nanoscale assemblies. Chem. Sci. 9, 576–585 (2018).
Poppe, M., Chen, C., Liu, F., Poppe, S. & Tschierske, C. Formation of a cubic liquid crystalline nanostructure with π‐conjugated fluorinated rods on the gyroid minimal surface. Chem. Eur. J. 23, 7196–7200 (2017).
Bates, F. S. & Fredrickson, G. H. Block copolymers—designer soft materials. Phys. Today 52, 32–38 (1999).
Imamura, K. et al. Temperature scanning FTIR analysis of hydrogen bonding states of various saccharides in amorphous matrixes below and above their glass transition temperatures. J. Phys. Chem. B 110, 15094–15099 (2006).
Schmitt, A. K. & Mahanthappa, M. Order and disorder in high χ/low N, broad dispersity ABA triblock polymers. Macromolecules 50, 677–6787. (2017).
Gentekos, D. T., Sifri, R. J. & Fors, B. P. Controlling polymer properties through the shape of the molecular-weight distribution. Nat. Rev. Mater. 4, 761–774 (2019).
Matsen, M. W. Polydispersity-induced macrophase separation in diblock copolymer melts. Phys. Rev. Lett. 99, 148304 (2007).
Matse, M. W. Comparison of A-block polydispersity effects on BAB triblock and AB diblock copolymer melts. Eur. Phys. J. E Soft Matter 36, 44 (2013).
Kim, I. & Li, S. Recent progress on polydispersity effects on block copolymer phase behavior. Poly. Rev. 59, 561–587 (2019).
Smilgies, D.-M. Scherrer grain-size analysis adapted to grazingincidence scattering with area detectors. J. Appl. Crystallogr. 42, 1030–1034 (2009).
Hancox, E. et al. Microphase separation of highly amphiphilic, low N polymers by photoinduced copper-mediated polymerization, achieving sub-2 nm domains at half-pitch. Polym. Chem. 10, 6254–6259 (2019).
Acknowledgements
This work was financially supported by a JSPS Grant-in-Aid for Scientific Research (B) (No. 19H02769, T.S.; No. 20H02792, T.I.), a JSPS Grant-in-Aid for Young Scientists (No. 18K14268, T.I.), the Photoexcitonix Project (Hokkaido University, T.S.), the Frontier Chemistry Center (Hokkaido University, T.S. and T.I.), the Creative Research Institute (CRIS; Hokkaido University, T.S.), the Tonen General Sekiyu Research/Development Encouragement & Scholarship Foundation (T.I.), the Shorai Foundation for Science and Technology (T.I.), the Iketani Science and Technology Foundation (T.I.), and the Asahi Glass Foundation (T.I.). This work was performed in part under the approval of the Photon Factory Program Advisory Committee (Proposals Nos. 2017G589 and 2019G579). T.I. gratefully acknowledges the Nanotech CUPAL NRP program. B.J.R., K.W., and K.Y. were funded by the JSPS Fellowship for Young Scientists. We thank Prof. Hajime Ito (Hokkaido University) for his assistance with AFM experiments.
Author information
Authors and Affiliations
Contributions
T.I. and T.S. designed the experiment. T.I. wrote the manuscript. T.I., R.K., C.L., and N.K. synthesized and characterized the BCOs and analyzed the X-ray data. B.J.R., K.W., K.Y., and M.H. helped in X-ray measurements and analysis. K.W., K.Y., and K.T. helped in AFM measurements. R.B. and T.Y. contributed to the discussion. The manuscript was written through the contributions of all authors. All authors have given their approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Isono, T., Komaki, R., Lee, C. et al. Rapid access to discrete and monodisperse block co-oligomers from sugar and terpenoid toward ultrasmall periodic nanostructures. Commun Chem 3, 135 (2020). https://doi.org/10.1038/s42004-020-00385-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-00385-y
This article is cited by
-
Self-assembly of carbohydrate-based block copolymer systems: glyconanoparticles and highly nanostructured thin films
Polymer Journal (2022)
-
Enzyme-catalyzed propagation of cello-oligosaccharide chains from bifunctional oligomeric primers for the preparation of block co-oligomers and their crystalline assemblies
Polymer Journal (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
