
Abstract
Much recent attention has been focused on the structure and reactivity of transition-metal superoxide complexes, among which mononuclear copper(II)-superoxide complexes are recognized as key reactive intermediates in many biological and abiological dioxygen-activation processes. So far, several types of copper(II)-superoxide complexes have been developed and their electrophilic reactivity has been explored in C–H and O–H bond activation reactions. Here we demonstrate that a mononuclear copper(II)-(end-on)superoxide complex supported by a N-[(2-pyridyl)methyl]-1,5-diazacyclooctane tridentate ligand can induce catalytic C–C bond formation reaction between carbonyl compounds (substrate) and the solvent molecule (acetone), giving β-hydroxy-ketones (aldol). Kinetic and spectroscopic studies at low temperature as well as DFT calculation studies support a nucleophilic reactivity of the superoxide species toward the carbonyl compounds, providing new insights into the reactivity of transition-metal superoxide species not only in biological oxidation reactions but also in synthetic organic chemistry.
Similar content being viewed by others

Introduction
Dioxygen activation by low-valent transition metal complexes is involved in many biological and abiological oxidation reactions, where the formation of superoxide complex is a fundamental common process.1,2 Such superoxide complexes can act as a direct oxidant or a precursor of other types of reactive intermediates for oxidative transformation reactions.3,4,5,6,7,8,9,10,11 In copper case, some end-on and side-on superoxide copper(II) complexes have been characterized and their reactivity has been examined mostly in hydrogen atom abstraction (HAA) from C–H and O–H bonds of organic compounds.12,13,14,15,16,17,18,19,20,21,22 In general, copper(II)-(end-on)superoxide complexes supported by neutral tridentate or tetradentate nitrogen-based ligands exhibit electrophilic reactivity in such HAA reactions.16,18,23,24 On the other hand, McDonald and co-workers investigated the reaction of copper(II)-(end-on)superoxide complex and some carbonyl compounds by using Tolman’s superoxide copper(II) complex supported by a deprotonated diamide-pyridine (dianionic) ligand.15,25 The superoxide complex exhibits formally nucleophilic reactivity toward the carbonyl compounds to induce the oxidative transformation reactions such as conversion of acid chlorides to carboxylic acids and Baeyer–Villiger oxidation and oxidative deformylation of aldehydes.25 They proposed that nucleophilic addition of the distal oxygen of superoxide moiety to the carbonyl carbon of the substrates is the key step in such reactions based on the reactivity study of the series of substrates.25 They suggested that such a nucleophilic reactivity is due to strong electron donor ability of the deprotonated dianionic supporting ligand.25
As our continuing research efforts in copper(I)-dioxygen chemistry, we have also developed a mononuclear copper(II)-(end-on)superoxide complex 2 using an N3-tridentate ligand LPye consisting of an eight-membered cyclic diamine with a 2-(2-pyridyl)ethyl sidearm (–CH2CH2Py; Py = 2-pyridyl) (Fig. 1).14,23 Our superoxide complex is a unique example that induces an benzylic C–H bond hydroxylation reaction, mimicking the reactivity of copper monooxygenases such as dopamine β-monooxygenases and peptidylglycine α-hydroxylating monooxygenases.14,23 Detailed mechanistic studies have indicated that the superoxide complex 2 exhibits an electrophilic reactivity in the C–H bond hydroxylation reaction.

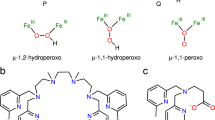
Structures of tridentate ligands LPym and LPye. LPym 1-phenethyl-5-(pyridin-2-ylmethyl)-1,5-diazocane. LPye 1-phenethyl-5-[2-(pyridin-2-yl)ethyl]-1,5-diazocane
In this study, we examine the reactivity of another mononuclear (end-on)superoxide complex 1 generated by using a similar N3-tridentate ligand LPym, which has a shorter pyridylmethyl sidearm (–CH2Py) instead of the pyridylethyl one (–CH2CH2Py) in LPye (Fig. 1). In this case, superoxide complex 1 exhibits completely different reactivity from that of 2 to induce catalytic C–C bond formation between carbonyl compounds (substrates) and the solvent molecule (acetone), giving β-hydroxy-ketones (aldol). Such a C–C bond formation reaction does not occur at all with superoxide complex 2. Thus, a subtle ligand modification (–CH2CH2Py to –CH2Py) greatly impacts the reactivity of the generated copper(II)-(end-on)superoxide complexes. Kinetic and spectroscopic studies at a low temperature (e.g. –95 °C) indicate that nucleophilic addition of the superoxide species to the carbonyl carbon of the substrate is involved as an initial step of the C–C bond formation reaction. Mechanistic details are further evaluated by DFT (density functional theory) calculation studies to provide new insights into the reactivity of the transition metal superoxide species. So far, a number of copper-catalyzed aldol reactions have been reported,26 but the role of copper has been simply considered as a Lewis acid catalyst. The present study demonstrates interesting synergistic effects of the superoxide ligand and Lewis acidic metal center for the catalytic C–C bond formation reaction, thus providing a new insight into the role of copper catalyst in synthetic organic chemistry.
Results
Synthesis and characterization of copper complexes
The precursor copper(I) complex of LPym was prepared by mixing an equimolar amount of the ligand and [CuI(CH3CN)4](PF6) in THF under anaerobic conditions (in a glovebox). Single crystals of the copper(I) complex suitable for X-ray crystallographic analysis were obtained as a BPh4– salt, [CuI(LPym)](BPh4), by treating the generated copper(I) complex with NaBPh4 (for synthetic procedures and characterization, see Supplementary Methods and Supplementary Fig. 1 and Supplementary Tables 1 and 2). The copper(I) complex exhibits a distorted three-coordinate T-shape structure involving the three nitrogen atoms of the ligand, which is stabilized by an intramolecular d–π interaction between the copper(I) ion and the ipso-carbon atom of the phenyl ring of the phenethyl sidearm (Cu–C, 2.936(4) Å, see Fig. 2 and Supplementary Fig. 1).

Generation of copper(II)-superoxide complex 1. The reaction of [CuI(LPym)]+ (0.10 mM) with O2 gas was conducted in acetone at –95 °C
To get insights into the ligand effect on the structure of copper(II) oxidation state, a copper(II)-chloride complex of LPym was also prepared by treating an equimolar amount of the ligand and CuIICl2 in CH3CN (see Supplementary Methods). Single crystals of the copper(II)-chloride complex suitable for X-ray crystallographic analysis were obtained as a BF4– salt, [CuII(LPym)(Cl)](BF4), by treating the generated copper(II)-chloride complex with NaBF4 (Supplementary Fig. 2 and Supplementary Tables 1 and 2). The copper(II) complex shows a square pyramidal structure, where the basal plane is occupied by the three nitrogen atoms of the ligand, N(1), N(2), and N(3), and the chloride counter anion, Cl(1), and the axial position is weakly coordinated by another chloride anion, Cl(2), of a neighboring copper(II) complex (Supplementary Fig. 2). Apparently, the square pyramidal structure of the copper(II) complex of LPym is different from that of the copper(II) complex of LPye, [CuII(LPye)(Cl)]+, which exhibits a tetrahedrally distorted four-coordinate structure as reported in our previous paper.23
In a cyclic voltammetric (CV) measurement, the copper(I) complex of LPym exhibits a quasi-reversible CuI/CuII redox couple at 0.17 V vs SCE in acetone, which is negative as compared to that of the copper(I) complex of LPye (0.40 V, see Supplementary Fig. 3). The result clearly indicates that electron donating ability of LPym is higher than that of LPye.
Copper(I)-O2 reactivity
Treatment of the copper(I) complex [CuI(LPym)](BPh4) (0.10 mM) with dry O2 gas in acetone at a low temperature (–95 °C) resulted in a spectral change, where an intense LMCT band at 386 nm (ε = 4430 M–1 cm–1) appeared together with broad bands at 558 nm (ε = 820) and 725 nm (ε = 1,160) as shown in Fig. 3 (red spectrum).27 The spectrum is similar to that of the mononuclear copper(II)-(end-on)-superoxide complex 2 generated by using LPye,14,23 indicating the formation of a similar end-on superoxide copper(II) complex 1 (Fig. 2). In fact, the oxygenated product showed isotope-sensitive resonance Raman bands at 1064 and 464 cm–1, which shifted to 997 cm–1 (ΔνO(16)–O(18) = 67 cm–1) and 451 cm–1 (ΔνO(16)–O(18) = 13 cm–1), respectively, upon using 18O2 instead of 16O2 (Supplementary Fig. 4a). Although interpretation is somewhat complicated by the presence of solvent peaks, the peak positions as well as the observed isotope shifts are similar to those of the O–O and the Cu–O stretching vibrations of the reported mononuclear copper(II)-(end-on)superoxide complexes.10 Furthermore, the Cu:O2 = 1:1 stoichiometry was confirmed by the titration of oxygenated product with ferrocene carboxylic acid (Supplementary Fig. 4b, c),28 which confirmed the formation of mononuclear copper-dioxygen adduct complex.

UV–vis spectrum of superoxide complex 1. UV–vis spectrum of copper(II)-(end-on)superoxide complex 1 (red line) obtained upon the treatment of [CuI(LPym)](BPh4) (0.10 mM, black) with dry O2 gas in acetone at –95 °C
Reactivity of copper(II)-superoxide complexes
Superoxide complex 1 gradually decomposed at a higher temperature (–60 °C; Supplementary Fig. 5a). In contrast to superoxide complex 2 supported by LPye, however, no aliphatic ligand hydroxylation took place after the reaction; 2 has been demonstrated to induce benzylic ligand hydroxylation of the phenethyl sidearm.14 The decay of 1 obeyed first-order kinetics (Supplementary Fig. 5b), and the first-order kinetics was further confirmed by the fact that rate constants (kobs) of the decay were virtually independent on the initial concentrations of 1 (0.10–0.50 mM) as shown in Supplementary Fig. 5c. These results confirm that the decay of 1 is a uni-molecular process with respect to the copper complex, and involvement of a dinuclear copper species such as a (μ-peroxido)dicopper(II) or a bis(μ-oxido)dicopper(III) complex or possibility of disproportionation of 1 during the decay process can be ruled out.
To our surprise, detailed product analysis of the final reaction mixture (work-upped at room temperature) indicated the formation of the aldol product (4-hydroxy-4-methylpentan-2-one; (CH3)2C(OH)CH2C(O)CH3) derived from two molecules of acetone (solvent) in a 560% based on 1. Thus, this is a catalytic C–C bond formation reaction of the carbonyl compounds. Such an aldol-type reaction of acetone did not proceed at all in the case of superoxide complex 2. To get insights into the reaction mechanism, we then examined the reaction of 1 and a series of carbonyl compounds in acetone.
First, the reaction of 1 with benzaldehyde (3a, C6H5CHO) was examined (Fig. 4). Treatment of 1 (0.10 mM) with an excess amount 3a (1.0 mM) in acetone caused a significant increase in the decay rate of 1 even at a low temperature (–95 °C) as shown in Fig. 4a. The reaction also obeyed first-order kinetics (Fig. 4b), and the decay rates showed the first-order dependence on the benzaldehyde concentration, giving a second-order rate constant as 2.1 ± 0.2 M–1 s–1 (Supplementary Fig. 11). Then, the substituent effects of the substrate were examined using a series of p-substituted benzaldehyde derivatives (p-X-C6H4CHO; X = H: 3a, X = NO2: 3b, X = Br: 3c, X = Cl: 3d, X = CH3: 3e, and X = OCH3: 3f, Supplementary Figs. 6–10). As clearly seen in Fig. 4c, the second-order rate constants increase as the electron-withdrawing ability of the p-substituent increases to give a positive Hammett ρ value as 2.2 (R = 0.98). The result clearly demonstrates a nucleophilic reactivity of superoxide complex 1 toward benzaldehyde derivatives. On the other hand, superoxide complex 2 did not react with these benzaldehyde derivatives under the same experimental conditions. The nucleophilic reactivity of 1 was more prominent in the reaction with ketones. Namely, 1 did not react with acetophenone (C6H5C(O)CH3), whereas it did react with 2,2,2-trifluoroacetophenone (C6H5C(O)CF3, 3j) to provide a similar spectral change as shown in Supplementary Fig. 12 (the second-order rate constant was 7.9 ± 0.4 M–1 s–1). Nucleophilic and electrophilic reactivity of a nickel(II)-superoxide was recently reported.29

Kinetic analysis on the reaction of 1 and benzaldehyde derivatives. a UV–vis spectral change in the reaction of 1 (0.10 mM) with benzaldehyde (3a, 1.0 mM) in acetone at –95 °C. b The time course of the decay at 386 nm. c Hammett plot for the reaction of 1 with p-substituted benzaldehydes (3a–3f) in acetone at –95 °C
The intermediate generated in the reaction of 1 and 3a at the low temperature (blue spectrum in Fig. 4a) may be a superoxide adduct of benzaldehyde IM1 (detailed reaction mechanism is discussed below, Fig. 6). In fact, almost no kinetic deuterium isotope effect was observed, when deuterated benzaldehyde-d1 (C6H5CDO) was employed (kH/kD = 0.98 ± 0.08) (Supplementary Fig. 13). Formation of the copper(II)-alkylperoxido-type intermediate (IM1) was also supported by the resonance Raman spectrum shown in Supplementary Fig. 14. The intermediate showed isotope-sensitive resonance Raman bands at 876 and 508 cm–1, which shifted to 852 cm–1 (ΔνO(16)–O(18) = 24 cm–1) and 490 cm–1 (ΔνO(16)–O(18) = 18 cm–1), respectively, upon 18O-substitution. The peak positions as well as the observed isotope shifts are similar to those of the O–O and Cu–O stretching vibrations of the reported mononuclear copper(II)-alkylperoxide complexes.10
Catalytic C–C bond formation reaction
The reaction of benzaldehyde 3a was examined in a preparative scale (Table 1). Treatment of 3a (25 µmol) with a catalytic amount of [CuI(LPym)](BPh4) (10 mol%) in acetone (2.0 mL) under O2 at 30 °C for 5 h gave 4-hydroxy-4-phenyl butanone (4a) in a 74% yield (Entry 1) based on 3a; thus, turnover number of the copper complex was 7.4. Copper(I) complex [CuI(LPym)](PF6) gave a comparable yield (61%) to that of [CuI(LPym)](BPh4) (Entry 2). On the other hand, no reaction (NR) took place in the absence of 1 or under anaerobic conditions (Entries 3 and 4). KO2 itself was not effective, suggesting that free superoxide anion is not an active species (Entry 5). Neither copper(I) complex supported by LPye nor the copper(II) complex of LPym was sufficient catalyst (Entries 6 and 7). These results unambiguously demonstrate that superoxide complex 1 is essential for the catalytic C–C bond formation reaction, and that simple Lewis acid catalysis by the copper(I/II) complexes can be ruled out.

The aldol products (4a–4f) were obtained in the reaction with the series of benzaldehyde derivatives (3a–3f, Fig. 5), where higher yields were obtained with the substrates having stronger electron-withdrawing p-substituent (3b–3d; X = NO2, Br, Cl). The results are consistent with the kinetic data shown in Fig. 4c, where the rate constant increases with increasing the electron-withdrawing ability of the p-substituents. The catalytic reaction can be adapted to other aldehydes such as cyclohexanecarbaldehyde (3g, CCA), 2-phenylpropionaldehyde (3h, 2-PPA), and 2-naphtaldehyde (3i) to give the corresponding aldol products 4g, 4h, and 4i in 75%, 76%, and 61%, respectively. Product 4h was in a little favor of the syn product (syn: anti = 58:42). In these reactions, the oxidative deformylation products were not obtained from the final reaction mixture. Ketone with electron-withdrawing group such as 2,2,2-trifluoroacetophenone (3j) also gave the corresponding aldol product 4j in a 75% yield, which is also consistent with the kinetic result (Supplementary Fig. 12).

Catalytic C–C bond formation in the reaction of 1 with aldehydes or ketone. Reaction conditions: substrate (25 µmol), catalyst (2.5 µmol), in acetone (2.0 mL) under O2 (1 atm) at 30 °C for 5 h. The yields of products were determined by 1H NMR based on substrates (carbonyl compounds) using 1,1,2,2-tetrachloroethane as an internal standard
Computational study
DFT calculations have been carried out to get detailed insights into the reaction mechanism of the catalytic C–C bond formation reaction (Fig. 6). As experimentally observed, superoxide complex 1 attacks the carbonyl carbon of benzaldehyde to give an alkylperoxido/alkoxyl radical intermediate (IM1). The DFT calculation suggests that association of an acetone molecule to the cupric ion stabilizes the intermediate IM1 by 6.0 kcal mol–1 (Supplementary Fig. 15). Existence of the copper(II)-alkylperoxido substructure in IM1 was also confirmed by the resonance Raman spectra as described above (Supplementary Fig. 14). Furthermore, the DFT results indicated that IM1X (X = NO2, Cl, H, CH3, OCH3) generated by the reaction of complex 1 with a series of p-substituted benzaldehyde derivatives (3a, 3b, 3d, 3e, and 3f) becomes more stable as the electron-withdrawing ability of the p-substituent increases (Supplementary Fig. 16). This computational result is completely consistent with the experimental results (Hammett analysis) shown in Fig. 4c.

Energy profile for the C–C bond formation by the reaction of 1 with benzaldehyde. a The relative energies with respect to IM1 in the triplet ground state are in kcal mol−1. b The transition state structures of TS1P and TS1S are shown together with their representative bond lengths (Å) and spin density
In the next step, we found two types of transition states (TS1S and TS1P) for the hydrogen migration process from the associated acetone molecule. TS1S is a transition state, in which a proton (H+) transfer from the acetone molecule to the benzylic oxygen O(4) takes place with an activation barrier of 7.3 kcal mol–1, where a superoxido-type canonical form is dominant (O(1)–O(2):1.29 Å; spin density: O(1) = 0.60, O(2) = 0.46) with an alkoxide character on O(4) (spin density: 0.00). Another transition state TS1P is consisting of a peroxido-type canonical form (O(1)–O(2):1.44 Å; spin density: O(1) = 0.14, O(2) = 0.00) with an alkoxyl radical character on O(4) (spin density: 0.55), which induces hydrogen atom (H•) transfer with an activation barrier of 14.4 kcal mol–1. Therefore, the intramolecular electron transfer from the peroxido moiety O(1)–O(2) to the alkoxyl radical group O(4) in IM1 takes place to increase the basicity of benzylic oxygen O(4), which enable to induce the proton (H+) abstraction from the acetone molecule. After this step, the generated superoxido-type intermediate IM2S having a shorter O(1)–O(2) bond (1.30 Å) with an anionic character on C(1) (spin density: 0.01) returns to the peroxido-type intermediate IM2P with a longer O(1)–O(2) bond (1.43 Å) with a radical character on C(1) (spin density: 0.81), since the latter is more stable compared to the former by 9.9 kcal mol–1 (Fig. 6; Supplementary Fig. 17). Then, IM2P is converted to a superoxide complex with the enolate of acetone (IM3), which is more stable compared to IM2P by 3.4 kcal mol–1. We examined the proton transfer process from IM1 to IM3 kinetically at –65 °C to obtain apparent kinetic deuterium isotope effect kH/kD as 2.7 by using acetone-d6 as the solvent (kH = 5.6 × 10–3 s–1 for acetone-h6 and kD = 2.1 × 10–3 s–1 for acetone-d6, see Supplementary Fig. 18). Such a relatively small KIE value (2.7) may be consistent with the proton (H+) transfer rather than hydrogen atom (H•) transfer. An analogous mechanism involving keto–enol tautomerization has been recently reported for the manganese(III)-peroxo catalyzed deformylation of aldehydes.30,31 Then, C–C bond formation takes place through TS2 to provide the product 1-PC with an activation barrier of 12.8 kcal mol–1, completing the catalytic cycle.
Discussion
In this study, we have found that the mononuclear copper(II)-(end-on)superoxide complex 1 supported by a simple tridentate ligand LPym shows an unprecedented, to our knowledge, reactivity toward carbonyl compounds (substrate) to induce catalytic C–C bond formation with the solvent molecule (acetone), giving an β-hydroxy-ketones (aldol). Based on the detailed kinetic and DFT studies, we suggest that the superoxide complex exhibits nucleophilic reactivity toward carbonyl compounds to give the copper(II)-alkylperoxido/alkoxyl radical intermediate IM1, from which keto–enol tautomerization of the solvent molecule takes place via proton transfer through TS1S and subsequent C–O bond homolysis giving IM3, regenerating the superoxide species. Then, the generated enol attacks the carbonyl group of substrates giving the aldol products (Fig. 6).
Such a catalytic C–C bond formation reaction did not occur at all, when the superoxide complex 2 supported by LPye was employed instead of complex 1 of ligand LPym. We have already reported that the copper(II) complexes of LPye favor four-coordinate distorted tetrahedral geometry.23,32 On the other hand, the copper(II) complex of LPym favors a five-coordinate square pyramidal geometry (Supplementary Fig. 2). Therefore, the association of acetone molecule to the copper(II) center may occur more easily in the LPym ligand system as compared to the LPye ligand system (Supplementary Fig. 15). This may be a reason for such a difference in the reactivity between 1 and 2. To examine this interpretation, we also performed DFT calculation on the reaction of copper(II)-superoxide complex 2 with benzaldehyde (Supplementary Fig. 19). Apparently, the energy gap between 2 and IM1′ in the LPye ligand system is larger than that between 1 and IM1 in the LPym ligand system (16.2 kcal mol–1 for LPye and 12.3 kcal mol–1 for LPym, see Supplementary Fig. 19a and Fig. 6). Moreover, the distance between the copper(II) ion and the oxygen atom of acetone in IM1′ (LPye) is significantly longer (3.15 Å; Supplementary Fig. 19b) as compared to that in IM1 (LPym) (2.34 Å; Supplementary Fig. 20a). Taken together, these preliminary computational results are consistent with our interpretation regarding to the ligand effects (LPym vs LPye) on the reactivity.
So far, copper/O2 systems have been frequently employed in the oxidative C–C bond formation reactions in synthetic organic chemistry.33 In most cases, the copper ion is simply considered as an electron transfer catalyst from a reduced transition metal catalyst to O2, regenerating a reactive catalyst in a higher oxidation state or a simple Lewis acid catalyst to enhance the reactivity of the carbonyl compounds.26 However, the present results suggest that there may be an alternative catalytic role of transition metal superoxide species in such C–C bond formation reactions.
Methods
Synthetic procedures
See Supplementary Methods.
X-ray structure determination
See Supplementary Figs. 1 and 2, Supplementary Tables 1 and 2, and Supplementary Data 1.
Electrochemical measurements
See Supplementary Fig. 3.
Resonance Raman measurements
See Supplementary Figs. 4a and 14.
Kinetic measurements
See Supplementary Figs. 4b, 4c, 5–13 and 18.
Computational methods
See Supplementary Figs. 15–17 and 19–25, and Supplementary Data 2.
General procedure for the catalytic C–C bond formation reactions
In a vial (4.0 mL), a copper complex (2.5 µmol) was dissolved in acetone (2.0 mL), and substrate (25 µmol) was added to the solution under N2 atmosphere. Then, O2 was bubbled to the solution and an O2 balloon (1 atm) was attached at top of the vial. After the reaction, the solvent was removed under reduced pressure, and the products were confirmed by comparing their 1H NMR spectra to those of the authentic samples (see Supplementary Methods). The product yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane (δ 5.97) as an internal standard.
Data availability
The X-ray crystallographic data (CIF format) of [CuI(LPym)](BPh4) and [CuII(LPym)](Cl)](BF4) are shown in Supplementary Data 1 and deposited in Cambridge Crystallographic Data Centre (CCDC); CCDC1882971 and CCDC1882972, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: +44 1223 336033. The authors declare that all the other data supporting the findings of this study are available within the paper and its supplementary information files.
References
Que, L. & Tolman, W. B. Biologically inspired oxidation catalysis. Nature 455, 333–340 (2008).
Tolman, W. B. & Solomon, E. I. Preface: forum on dioxygen activation and reduction. Inorg. Chem. 49, 3555–3556 (2010).
van der Donk, W. A., Krebs, C. & Bollinger, J. M. Jr. Substrate activation by iron superoxo intermediates. Curr. Opin. Struct. Biol. 20, 673–683 (2010).
Ray, K., Pfaff, F. F., Wang, B. & Nam, W. Status of reactive non-heme metal–oxygen intermediates in chemical and enzymatic reactions. J. Am. Chem. Soc. 136, 13942–13958 (2014).
Nam, W. Synthetic mononuclear nonheme iron–oxygen intermediates. Acc. Chem. Res. 48, 2415–2423 (2015).
Sahu, S. & Goldberg, D. P. Activation of dioxygen by iron and manganese complexes: a heme and nonheme perspective. J. Am. Chem. Soc. 138, 11410–11428 (2016).
Peck, S. C. & van der Donk, W. A. Go it alone: four-electron oxidations by mononuclear non-heme iron enzymes. J. Biol. Inorg. Chem. 22, 381–394 (2017).
Fiedler, A. T. & Fischer, A. A. Oxygen activation by mononuclear Mn, Co, and Ni centers in biology and synthetic complexes. J. Biol. Inorg. Chem. 22, 407–424 (2017).
Itoh, S. Mononuclear copper active-oxygen complexes. Curr. Opin. Chem. Biol. 10, 115–122 (2006).
Elwell, C. E. et al. Copper–oxygen complexes revisited: structures, spectroscopy, and reactivity. Chem. Rev. 117, 2059–2107 (2017).
Quist, D. A., Diaz, D. E., Liu, J. J. & Karlin, K. D. Activation of dioxygen by copper metalloproteins and insights from model complexes. J. Biol. Inorg. Chem. 22, 253–288 (2017).
Würtele, C. et al. Crystallographic characterization of a synthetic 1:1 end-on copper dioxygen adduct complex. Angew. Chem. Int. Ed. 45, 3867–3869 (2006).
Maiti, D. et al. A 1:1 Copper-dioxygen adduct is an end-on bound superoxo copper(II) complex which undergoes oxygenation reactions with phenols. J. Am. Chem. Soc. 129, 264–265 (2007).
Kunishita, A. et al. Mononuclear copper(II)-superoxo complexes that mimic the structure and reactivity of the active centers of PHM and DβM. J. Am. Chem. Soc. 131, 2788–2789 (2009).
Donoghue, P. J., Gupta, A. K., Boyce, D. W., Cramer, C. J. & Tolman, W. B. An anionic, tetragonal copper(II) superoxide complex. J. Am. Chem. Soc. 132, 15869–15871 (2010).
Peterson, R. L. et al. Cupric superoxo-mediated intermolecular C-H activation chemistry. J. Am. Chem. Soc. 133, 1702–1705 (2011).
Kobayashi, Y. et al. Copper(I)-dioxygen reactivity in a sterically demanding tripodal tetradentate tren ligand: formation and reactivity of a mononuclear copper(II) end-on superoxo complex. Eur. J. Inorg. Chem. 2012, 4574–4578 (2012).
Lee, J. Y. et al. Mechanistic insights into the oxidation of substituted phenols via hydrogen atom abstraction by a cupric–superoxo complex. J. Am. Chem. Soc. 136, 9925–9937 (2014).
Kim, S. et al. A N3S(thioether)-ligated cuII-superoxo with enhanced reactivity. J. Am. Chem. Soc. 137, 2796–2799 (2015).
Fujisawa, K., Tanaka, M., Morooka, Y. & Kitajima, N. A monomeric side-on superoxocopper(II) complex: Cu(O2)(HB(3-tBu-5-iPrpz)3. J. Am. Chem. Soc. 116, 12079–12080 (1994).
Sanchez-Eguia, B. N., Flores-Alamo, M., Orio, M. & Castillo, I. Side-on cupric-superoxo triplet complexes as competent agents for H-abstraction relevant to the active site of PHM. Chem. Commun. 51, 11134–11137 (2015).
Iovan, D. A. et al. Reactivity of a stable copper-dioxygen complex. Chem. Commun. 53, 10306–10309 (2017).
Kunishita, A. et al. Active site models for the CuA site of peptidylglycine α-Hydroxylating monooxygenase and dopamine β-monooxygenase. Inorg. Chem. 51, 9465–9480 (2012).
Tano, T. et al. Redox properties of a mononuclear copper(II)-superoxide complex. Inorg. Chem. 52, 10431–10437 (2013).
Pirovano, P. et al. Nucleophilic reactivity of a copper(II)–superoxide complex. Angew. Chem., Int. Ed. 53, 5946–5950 (2014).
Allen, S. E., Walvoord, R. R., Padilla-Salinas, R. & Kozlowski, M. C. Aerobic copper-catalyzed organic reactions. Chem. Rev. 113, 6234–6458 (2013).
Woertink, J. S. et al. Spectroscopic and computational studies of an end-on bound superoxo-Cu(II) complex: geometric and electronic factors that determine the ground state. Inorg. Chem. 49, 9450–9459 (2010).
Herres-Pawlis, S. et al. Phenolate hydroxylation in a Bis(mu-oxo)dicopper(III) complex: lessons from the guanidine/amine series. J. Am. Chem. Soc. 131, 1154–1169 (2009).
Panda, C. et al. Nucleophilic versus electrophilic reactivity of bioinspired superoxido nickel(II) complexes. Angew. Chem., Int. Ed. 57, 14883–14887 (2018).
Prasenjit, B. et al. Deformylation reaction by a nonheme manganese(III)–peroxo complex via initial hydrogen-atom abstraction. Angew. Chem., Int. Ed. 55, 11091–11095 (2016).
Cantú Reinhard, F. G. et al. Keto–enol tautomerization triggers an electrophilic aldehyde deformylation reaction by a nonheme manganese(III)-peroxo complex. J. Am. Chem. Soc. 139, 18328–18338 (2017).
Abe, T. et al. Geometric control of nuclearity in copper(I)/dioxygen chemistry. Inorg. Chem. 53, 8786–8794 (2014).
Evano, G., Blanchard, N. & Toumi, M. Copper-mediated coupling eactions and their applications in natural products and designed biomolecules synthesis. Chem. Rev. 108, 3054–3131 (2008).
Acknowledgements
The present research work was financially supported by the JST-CREST (JPMJCR16P1) and a Grant-in-Aid for challenging Exploratory Research (# 16K13963) from JSPS. This work was also supported by Grant-in-Aid (# JP15K13710 and JP17H03117) from JSPS and MEXT and by the MEXT Projects of “Integrated Research Consortium on Chemical Sciences”, “Elements Strategy Initiative to Form Core Research Center”, and “Network Joint Research Center for Materials and Devices”. The computational study was mainly carried out using the computer facilities at Research Institute for Information Technology, Kyushu University.
Author information
Authors and Affiliations
Contributions
The manuscript was achieved through contributions of all authors. T.A. and S.I. designed the project, conducted the experiments, and wrote the manuscript. Y.H., Y.S., and K.Y. performed DFT calculations. T. Ohta and T. Ogura contributed to the measurements of resonance Raman spectra. Y.M. and H.S. contributed to discussions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abe, T., Hori, Y., Shiota, Y. et al. Cupric-superoxide complex that induces a catalytic aldol reaction-type C–C bond formation. Commun Chem 2, 12 (2019). https://doi.org/10.1038/s42004-019-0115-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-019-0115-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
